
Abstract
In our juvenile traumatic brain injury (jTBI) model, emergence of cognitive dysfunctions was observed up to 6 months after trauma. Here we hypothesize that early brain injury induces changes in the neurovascular unit (NVU) that would be associated with amyloid-beta (Aβ) accumulation. We investigated NVU changes for up to 6 months in a rat jTBI model, with a focus on the efflux protein P-glycoprotein (P-gp) and on the basement membrane proteins perlecan and fibronectin, all known to be involved in Aβ clearance. Rodent-Aβ staining is present and increased after jTBI around cerebral blood microvessels, and the diameter of those is decreased by 25% and 34% at 2 and 6 months, respectively, without significant angiogenesis. P-glycoprotein staining in endothelium is decreased by 22% and parallels an increase of perlecan and fibronectin staining around cerebral blood vessels. Altogether, these results strongly suggest that the emergence of long-term behavioral dysfunctions observed in rodent jTBI may be related to endothelial remodeling at the blood–brain barrier alongside vascular dysfunction and altered Aβ trafficking. This study shows that it is important to consider jTBI as a vascular disorder with long-term consequences on cognitive functions.
INTRODUCTION
Traumatic brain injury (TBI) is an acute injury resulting from a direct or indirect biomechanical force on the brain. It represents the highest cause of disability and mortality in developed countries, accounting for 30.5% of all injury-related deaths in the USA. 1 Traumatic brain injury affects a broad range of the population, and juveniles are more vulnerable than adults. In fact, juvenile TBI (jTBI) has a poor prognosis and worse symptom severity than a comparable injury occurring in adult patients. In the clinic, young TBI patients show long-term impairment of cognitive functions including memory deficits and alteration of attention. 2 Similar long-term impairments are seen after concussions and mild TBI in adults; however, the pediatric population remains more vulnerable.
At the cellular level, TBI is associated with a broad profile of damage in the neurovascular unit (NVU). The primary lesion occurs at the moment of brain impact, affecting not only the neurons and glia, but also the blood vessels. 3 Then, secondary injuries occur that include decreased cerebral blood flow, hypometabolism, blood–brain barrier (BBB) disruption, edema formation, increased intracranial pressure, hypoxia, ischemia, and a related cascade of molecular events like excitotoxicity, inflammation, and oxidative stress. Usually, BBB disruption normalizes within the first week in a juvenile rat model of controlled cortical impact, which is consistent with some clinical observations. 3 Although the BBB is no longer physically disrupted, BBB function may continue to be compromised. In this jTBI model, long-term phenotypic changes were observed in endothelial cells up to 2 months after jTBI, with increased levels of the tight-junction protein claudin-5 and decreased levels of P-glycoprotein (P-gp) compared with noninjured animals. 4 P-glycoprotein is an endothelial efflux pump known to expel several proteins from endothelial cells to the extracellular space, mostly into the blood compartment. P-glycoprotein has been proposed by several groups to be implicated in amyloid-beta (Aβ) clearance from brain tissue into the blood circulation. 5 It has been shown that P-gp expression decreases during normal aging as well as in Alzheimer's disease. 6 Decreased levels of P-gp 2 months after jTBI highlight long-term consequences of jTBI on neuropathology like those found in Alzheimer's disease and during the aging process. 7 This decrease is paralleled with an accumulation of Aβ in the brain 2 months after a jTBI. 4 Similar to clinical observations, we recently described a development in long-term behavioral changes after a single jTBI event, suggesting jTBI evolves into a chronic brain disorder. Persistent behavioral and motor deficits were observed in our P17-old controlled cortical impact rats for up to 6 months, primarily in spatial memory measured with Morris Water maze. 8 We originally proposed a possible link between the BBB phenotypic changes including decreased P-gp expression, and resulting in increased brain Aβ content, to explain part of the emergence of cognitive dysfunctions.
Transport of Aβ across the endothelium mediated via P-gp is only one of several complex vascular routes for brain Aβ clearance from the brain. For example, basement membrane proteins, which are essential components of the BBB, were recently implicated in perivascular drainage of Aβ 9 showing that changes in basement membrane composition and thickness may contribute to brain deposition of Aβ. Heparan sulfate proteoglycans like agrin and perlecan promoted Aβ fibrillization,10,11 whereas other basement membrane proteins like type IV collagen, laminin and nidogen/entactin promoted disaggregation of Aβ fibrils. 12 Perlecan and another heparan sulfate proteoglycans, fibronectin, had a neuroprotective role at the NVU during brain injury affecting the vascular and neuronal level.13–18 Fibronectin was neuroprotective after focal brain ischemia and spinal cord injury in rats by decreasing lesion size, improving functional outcome, and promoting antiapoptotic pathways.15,18 Likewise, domain V (DV), a proteolytic fragment of perlecan, was shown to maintain NVU integrity after stroke, perhaps by inducing neuroprotection and angiogenesis through increased production and release of vascular endothelial growth factor (VEGF).14,16 Domain V further inhibited neurotoxic pathways by competing with Aβ binding to α2 integrin, 17 and it also modulated astrocyte functions and reduced glial scar formation. 13 Interestingly, DV inhibited Aβ-induced endothelial cell toxicity by interacting with α5β1 integrin receptor, inducing its internalization, degradation, and clearance. 19
The emergence of behavioral dysfunctions up to 6 months after jTBI 8 and the presence of Aβ accumulation and vascular dysfunctions at 2 months after jTBI 4 led us to the following hypothesis: jTBI induces long-term vascular phenotypic changes in endothelial proteins, like P-gp, and in the basement membrane proteins perlecan and fibronectin, which may work together to affect the process of Aβ clearance and promote chronic brain dysfunction commonly observed during brain injury and neurodegenerative diseases. To address this hypothesis, we evaluated Aβ accumulation and P-gp levels at 6 months, and changes in the basement membrane proteins fibronectin and perlecan, as well as the α5β1 integrin receptor, at both 2 and 6 months after jTBI.
MATERIALS AND METHODS
Animals
Experiments and manuscript comply with the Animal Research: Reporting of
Juvenile Traumatic Brain Injury Model
Controlled cortical impact was induced in rats as previously described. 8 Briefly, rats were anesthetized with isoflurane and given a 5-mm diameter craniotomy over the right frontoparietal cortex (1mm posterior and 2mm lateral from Bregma) and controlled cortical impact was delivered to jTBI animals using a 3-mm impactor at a 20° angle to cortex, 1.5mm depth, 200 millisecond impact duration, and 6m/s velocity. Body temperature was maintained at 37°C during surgery. A subcutaneous buprenorphine injection was administered for pain relief (0.01 mg/mL per kg at 1 and 24 hours after surgery). Naive animals were under anesthesia but did not receive any surgery or buprenorphine. Buprenorphine and its metabolite norbuprenorphine are known to affect P-gp at the BBB. 20 In our previous study, sham and jTBI groups both received buprenorphine and jTBI animals showed a difference in the level of P-gp expression. 4 It is therefore very unlikely that a single injection 6 months before would have any effect on P-gp levels by itself.
Brain Tissue Processing for Western Blot and Immunohistochemistry
At 2 or 6 months after injury, rats were anesthetized with a combination of ketamine and xylazine at the appropriate dose/body weight. For western blot studies, brains were extracted and fresh frozen on dry ice. For immunohistochemistry studies, rats were transcardially perfused with 4% paraformaldehyde. Brains were excised and cryoprotected in 30% sucrose solution for 48 hours, and frozen on dry ice.
Coronal cryostat free-floating sections (50
Immunolabeling of Blood-Brain Barrier Proteins
For P-gp staining, sections were pretreated for antigen retrieval using 33% acetic acid+66% ethanol solution for 10 minutes at — 20°C. For perlecan, fibronectin, and α5 integrin, sections were blocked for 1 hour in 1% bovine serum albumin in phosphate-buffered saline (PBS) before overnight primary antibody incubation at 4°C. All antibody incubations were in 0.25% bovine serum albumin with 0.25% Triton X-100 made in PBS, pH 7.4. For immunolabeling, we used mouse anti-P-gp (1:100, Calbiochem, EMD Chemicals, Merck KGaA, Darmstadt, Germany), rabbit anti-perlecan (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-fibronectin (1:250, Sigma-Aldrich, Saint Louis, MO, USA), and rabbit anti-α5 integrin (1:300, Millipore, Temecula, CA, USA) antibodies. After PBS rinses, sections were incubated in secondary antibody for 2 hours at room temperature at 1:1,000 as appropriate for each primary antibody (all secondary antibodies from Invitrogen, Grand Island, NY, USA). After washes in PBS, sections for classic immunofluorescence were mounted on glass slides and coverslipped with vectashield antifading medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA).
Fluorescein-labeled tomato-lectin (t-lectin, 1:200, Vector Laboratories) was used as a marker of blood vessels. T-lectin was incubated overnight at 4°C in 0.25% bovine serum albumin with 0.25% Triton X-100 made in PBS, pH 7.4.
Immunolabeling of Amyloid-beta
Sections for amyloid analysis were pretreated for 4 minutes in 88% formic acid at room temperature and all other immunostaining procedures were identical to the procedure described above. We used a monoclonal antibody raised in mouse against rodent pan-Aβ (recognizing both Aβ1–40 and 1–42) at the N-terminal amino acids 1–16 (1:1,000, from Dr M. Paul Murphy) and visualized staining with goat anti-mouse secondary antibody Alexa-Fluor-488 (1:1,000; Invitrogen).
Quantification of Immunohistochemistry
For quantification of immunolabeling of P-gp, perlecan, fibronectin, t-lectin, and Aβ, images were evaluated and collected using an epifluo-rescent microscope (BX41, Olympus, Center Valley, PA, USA). The threshold and morphologic user-defined parameters were selected to maximize visualization of positive staining in the region of interests for each protein-staining pattern. These parameters were kept consistent for all animals during image acquisition. Images were taken in the parietal and temporal cortices, both above and below the rhinal fissure on each hemisphere ipsilateral and contralateral to the lesion (or right and left hemispheres) using a × 20 objective.
For quantification of P-gp, perlecan, fibronectin, and t-lectin immunoreactivity, 12 images per animal were acquired (
For quantification of rodent-Aβ immunoreactivity, we used eight whole serial coronal slices per animal spaced 1.2 mm apart, from bregma levels + 3.2 to —5.2 mm (
For analyses of microvessel density and diameter measurements, we used the Mercator software with the t-lectin images. For the microvessel density, 60 boxes (7 × 7
Brain Tissue Processing for Western Blotting
Fresh frozen tissue (ipsilateral cortex) was homogenized in radio immunoprecipitation assay extraction buffer containing a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) and centrifuged 10 minutes at 10000 g. Samples were assayed for total protein concentration by bicinchoninic assay (Pierce Biotechnology, Rockford, IL, USA). Samples (30
Statistical Analyses
All data are presented as mean ± s.e.m., statistical analyses were done using SPSS (New York, NY, USA), and graphs were obtained using SigmaPlot (San Jose, CA, USA). For the Aβ analyses, we used a repeated-measures analysis of variance with group (jTBI, naive) × bregma level (eight serial coronal sections) and a conservative Huyhn–Feldt adjustment to the degrees of freedom was used to protect against any violations of the sphericity and compound symmetry assumptions underlying this analysis of variance model. All other histologic data between naive and jTBI animals met statistical assumptions and were analyzed using Student's
RESULTS
Accumulation of Amyloid-beta and Modifications of Endothelial Cell Phenotype 6 Months after Juvenile Traumatic Brain Injury Accumulation of Aβ combined with a decrease of P-gp has been previously shown in rat brain 2 months after a single impact on juvenile rats, suggesting long-term consequences of jTBI on neurodegenerative processes. 4 Neurovascular phenotypic changes are still present 6 months after jTBI. In fact, P-gp immunolabeling intensity remained significantly decreased after jTBI compared with naive animals at 6 months after injury onset (Figures 1A–1C). P-glycoprotein immunoreactivity is primarily observed in the vascular compartment in the naive animals, whereas staining is absent in the jTBI animals (Figures 1A and 1B).
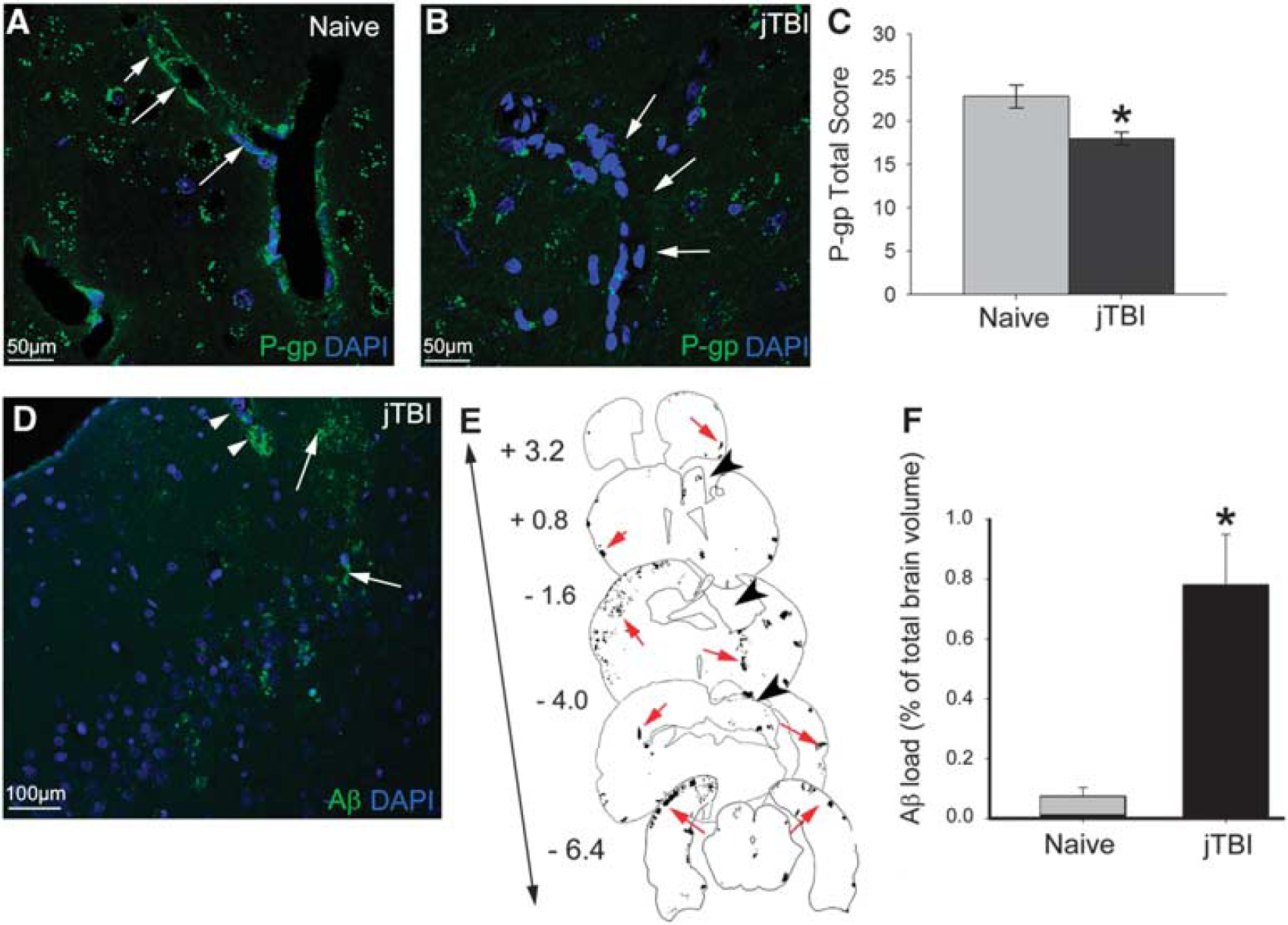
Juvenile traumatic brain injury (jTBI) induces amyloid-beta (Aβ) accumulation and decrease of P-glycoprotein (P-gp) levels in the brain 6 months after injury. (
In parallel to the phenotypic changes in endothelial cells, Aβ immunohistochemical studies demonstrated Aβ staining in jTBI animals after 6 months (Figure 1D) with a similar extracellular compartment and perivascular distribution as previously observed after 2 months. The injured animals show a higher staining compared with naive animals (Figures 1F,
Remodeling of Basement Membrane Composition 2 and 6 Months after Juvenile Traumatic Brain Injury
Perlecan and fibronectin, two major basement membrane proteins, were studied via immunohistochemistry and western blot in both naive and jTBI animals at 2 and 6 months after jTBI. Perlecan immunostaining was observed around cerebral blood vessels (revealed by t-lectin) in both naive and jTBI animals at 2 and 6 months after injury (Figure 2). However, jTBI animals showed higher intensity of perlecan staining around all cerebral blood vessels compared with naive controls (Figures 2A–2L) at both time points. These qualitative observations were confirmed by quantification of images from the ipsilateral and contralateral cortices, showing a general increase of total perlecan in jTBI animals compared with naive at 2 and 6 months, respectively, ~2.4-fold (
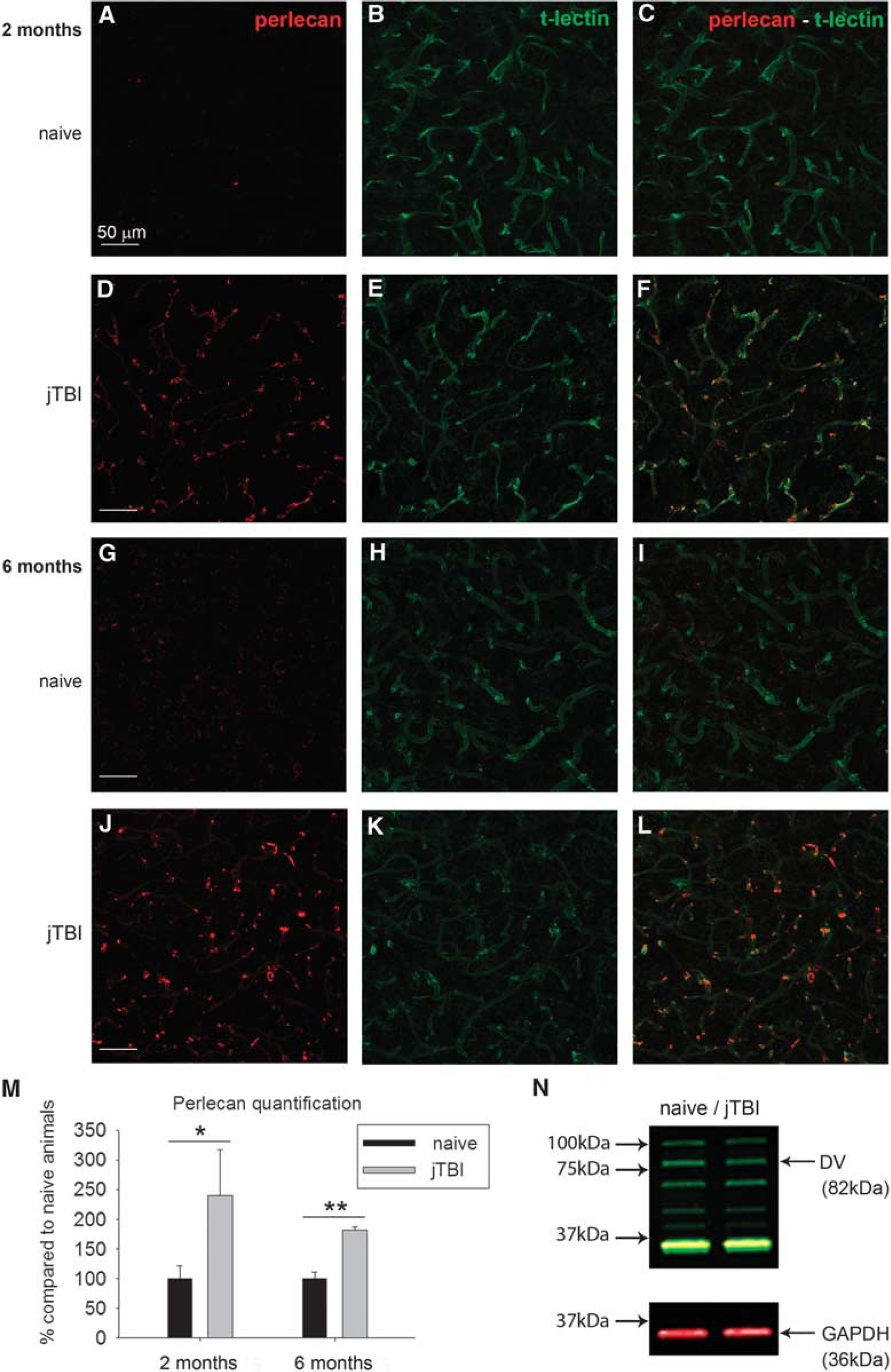
Juvenile traumatic brain injury (jTBI) induces increase of perlecan vascular staining 2 and 6 months after injury. The basement membrane protein perlecan was observed (red) in both naive (
Fibronectin immunoreactivity was almost absent in the whole cortex of naive rats at both time points (Figures 3A–3G) as previously described.
21
However, fibronectin immunoreactivity was increased for jTBI animals compared with naive, revealing all cerebral blood vessels stained with t-lectin at 2 and 6 months (Figures 3A–3L). Quantification of fibronectin immunoreactivity from images of the ipsilateral and contralateral cortices confirmed qualitative observations in jTBI compared with naive animals with a 3.7-fold increase at 2 months and a 2.7-fold increase at 6 months (
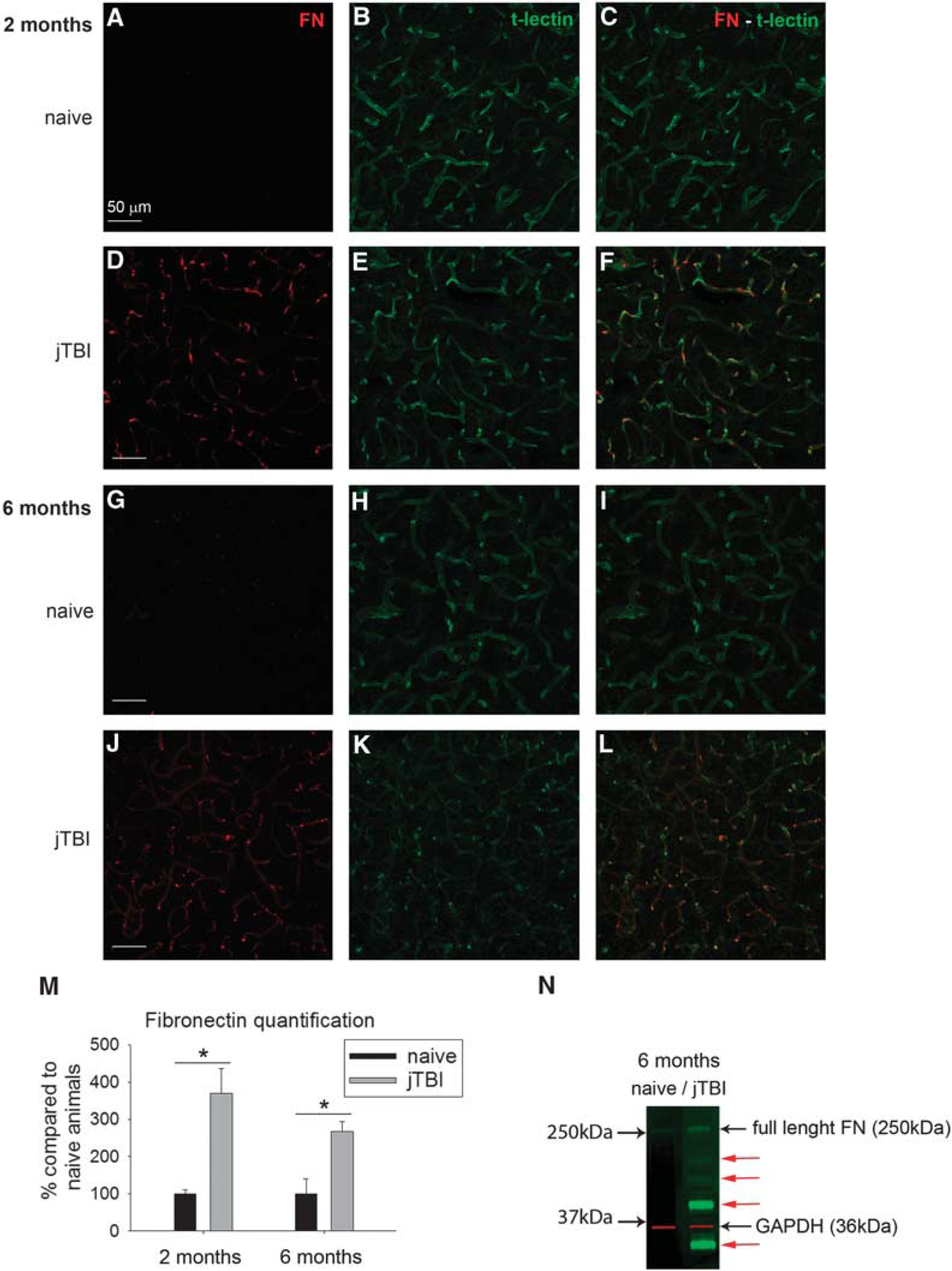
Juvenile traumatic brain injury (jTBI) induces increase of fibronectin vascular staining. The basement membrane protein fibronectin was almost absent in naive animals (
α5β1 Integrin Protein Levels and Microvessel Numbers are not Modified Long Term after Juvenile Traumatic Brain Injury α5β1 integrin is known to be a receptor for perlecan DV and fibronectin, and is implicated in angiogenesis.16,22 This receptor and its ligand fibronectin are highly expressed during development and angiogenesis, and are then downregulated during adulthood. 23 However, α;5β1 integrin and fibronectin expression levels have been shown to be acutely increased in cerebral blood vessels after a model of focal transient cerebral ischemia or a model of hypoxia. 24 We performed α5 immunostaining to determine whether, like its ligand, the expression of this receptor was modified after jTBI. These studies showed α5 staining in the brain of both groups, mainly in cortical and hippocampal neurons, and in astrocytes of the corpus callosum (Figure 4). Surprisingly, no blood vessels were positive for the staining and no difference of expression was detected between naive and jTBI animals at 2 or 6 months.
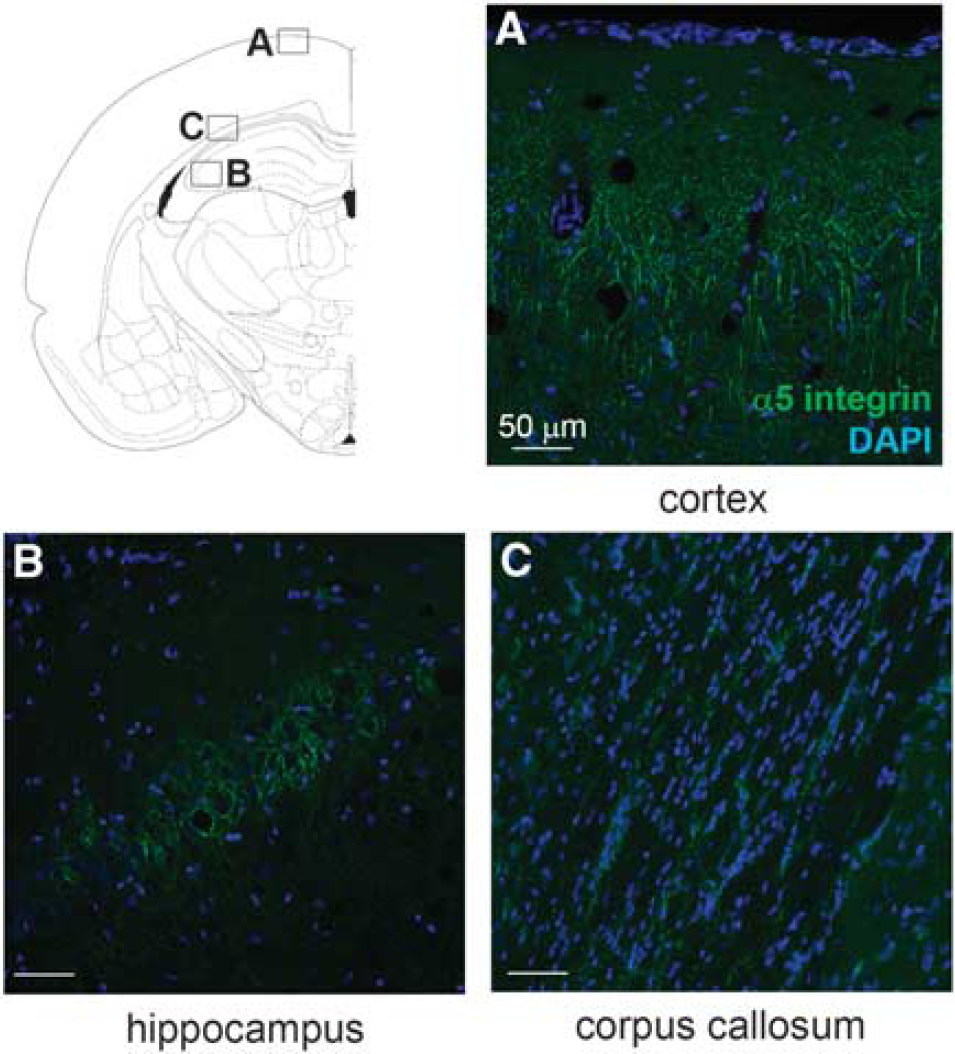
α5β1 integrin levels are not modified long term after a juvenile traumatic brain injury (jTBI). α5 integrin staining is present in cortical (
These increases of perlecan and fibronectin staining around the blood vessels are not due to an increase in the number of blood vessels. In fact, we did not observe any differences between the conditions when the brain vasculature is outlined using a t-lectin staining. Quantifications using t-lectin vascular staining revealed no difference in intensity (data not shown) or in the number of microvessels between naive and jTBI animals, at 2 months and 6 months (Figure 5). However, the vascular modifications of the matrix were associated with some structural changes in the blood vessels. Specifically, we observed that the diameter of microvessels in the cortex was significantly smaller in jTBI animals compared with naive at both time points (— 25% at 2 months and — 34% at 6 months; Figure 5E).
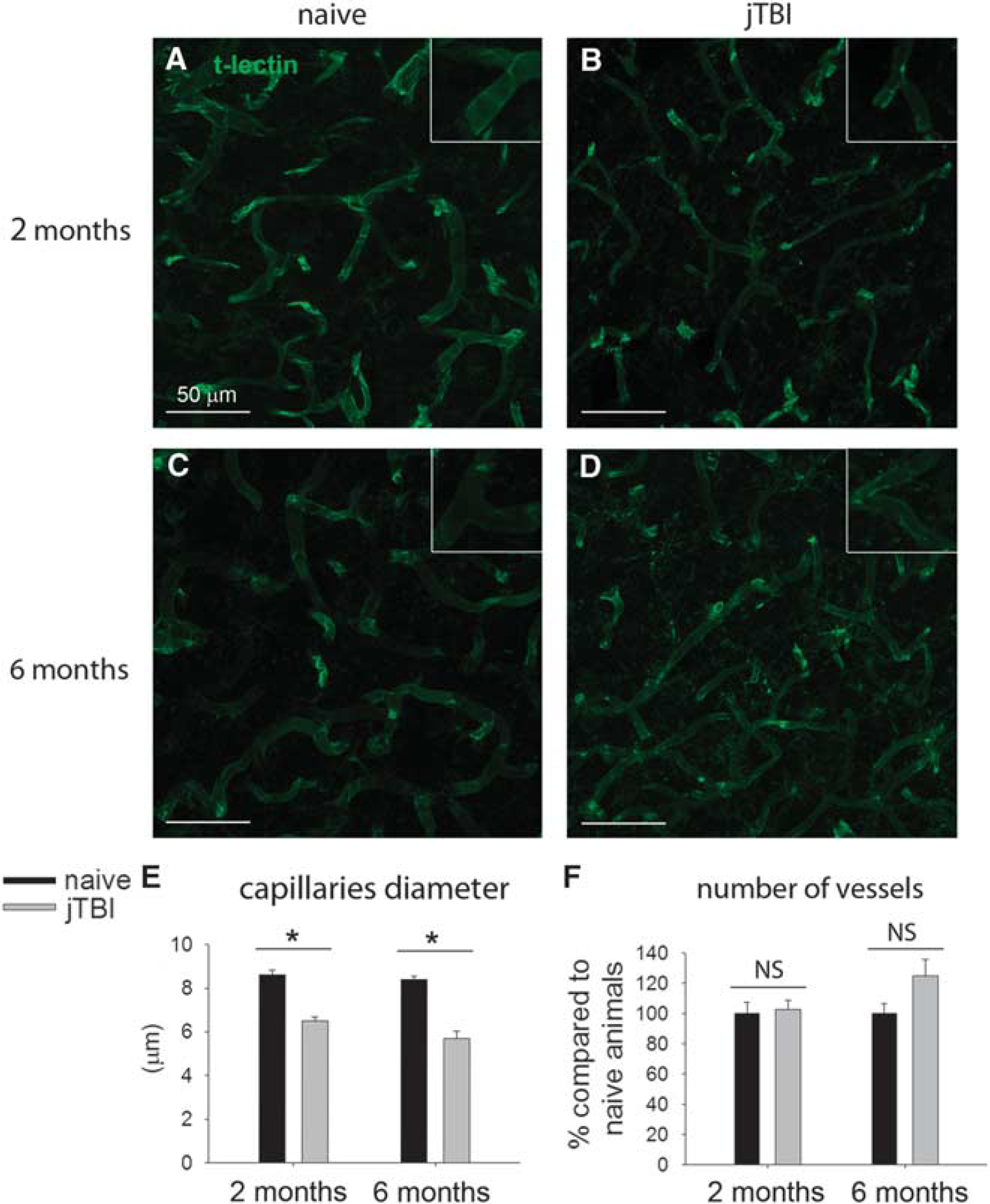
Quantification of t-lectin staining does not reveal angiogenesis in the cerebral cortex of juvenile traumatic brain injury (jTBI) rats after 2 or 6 months, but a decrease in microvessel diameter. (
DISCUSSION
We previously showed that when the BBB is no longer physically disrupted after jTBI, endothelial cells still carry phenotypic changes including a decrease of P-gp levels in microvessels and an increase of the tight-junction protein claudin-5 in large intracortical vessels. 4 These phenotypic changes were described along with Aβ accumulation and cognitive dysfunctions such as a lack of spatial memory use in jTBI animals during a water maze test and an inability to improve performance over time. Interestingly, various investigators have hypothesized that vascular dysfunctions are implicated in cognitive impairment during aging and neurodegenerative disease.25–27 Therefore, we hypothesized that long-term vascular phenotypic transformations after early-life injury contribute to the long-term cognitive dysfunctions observed in our jTBI model. In fact, in parallel to Aβ accumulation in the brain of injured animals up to 6 months after the impact, cerebral microvessels show major phenotypic changes with a decrease in diameter, a decrease in endothelial P-gp expression, and an increase in two major basement membrane proteins, perlecan and fibronectin. All these structural and molecular changes are very likely implicated in the decreased perivascular drainage of Aβ, leading to the observed Aβ accumulation (Figure 6). To the best of our knowledge, our study is the first to show that the brain vasculature exhibits long-term modifications after jTBI, suggesting that the brain does not go back to the same preinjury steady state. These observations might be different for a TBI occurring during adult life, leading to different therapeutic strategies between pediatric and adult population.
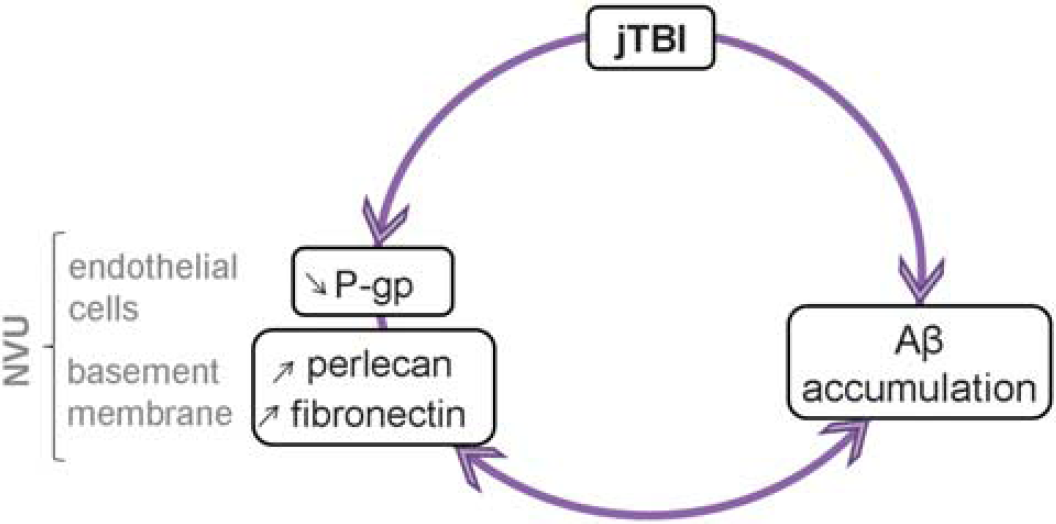
Schematic summary of the neurovascular unit (NVU) changes after juvenile traumatic brain injury (jTBI). An early traumatic brain injury (TBI) induces long-term changes within the NVU with a decrease of P-glycoprotein (P-gp) expression in endothelium associated with an increase of perlecan and fibronectin staining. Collectively, these changes within the NVU may contribute to the decreased amyloid-beta (Aβ) clearance observed after TBI and accelerate the neurodegenerative process. The presence of Aβ may also contribute to the phenotypic transformation of the NVU, ending in a vicious circle.
Long-Term Accumulation of Amyloid-beta after Juvenile Traumatic Brain Injury and Link with Basement Membrane Proteins
The significant increase of Aβ deposition between 2 and 6 months suggests a progression in the pathophysiology. Interestingly, our previous behavioral studies in this model also showed spatial memory deficits emergence at 3 months while some other cognitive traits normalized. 8 This accumulation of Aβ may be responsible for or contribute to the cognitive dysfunctions. In fact, the Aβ staining is not only observed around the lesion but in various areas, not necessarily involving the hippocampus but also temporal and frontal cortices.
Although several publications have described the potential involvement of P-gp in Aβ clearance,5,6, Aβ is cleared across the endothelium by additional mechanisms. For example, low-density lipoprotein-related protein receptor 1 (LRP1) is involved in Aβ transport from the brain to the blood. 6 However, in our previous study, LRP1 did not show a significant difference at 2 months between noninjured and jTBI animals. 4 Interestingly, several other proteins have been proposed to participate in Aβ clearance like other transporters (ABCA1 and ABCA2)28,29 or like the receptor α5β1 integrin. 30 In addition, various basement membrane proteins, including perlecan and fibronectin, have been implicated in the perivascular drainage of Aβ.9–11,31
Basement membrane proteins are mainly known to be the target of various proteases, like matrix metalloproteases, that are massively released after an acute brain injury, leading to alteration of the BBB. 32 They are key components of the BBB and they have been shown to be implicated in drainage of Aβ.9,31 Perlecan and fibronectin have been proposed to participate in Aβ accumulation by accelerating its aggregation by providing a higher stability of Aβ in the basement membrane.10,11 Interestingly, perlecan and fibronectin levels are also elevated during aging in mice, and this could in part explain the impaired drainage of Aβ in the aging brain.9,31 However, whereas perlecan is normally expressed in abundance around healthy cerebral blood vessels, it is absent around amyloid-laden blood vessels in the brains of Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis of the Dutch-type patients, 33 raising the intriguing possibility that diminished perivascular perlecan expression could contribute to cerebral amyloid angiopathy. Our observations suggest that jTBI-induced expression of perlecan and fibronectin participate in the establishment of accelerated brain aging. Other basement membrane proteins like laminin, type IV collagen, and agrin are also known to be involved in the process of Aβ clearance, inducing, e.g., changes in basement membrane composition and thickness, impairing the process of Aβ drainage.10,11 In parallel, several adenosine triphosphate-binding cassette (ABC) transporters other than P-gp have been shown to be implicated in Aβ clearance, like ABCA1 and ABCG233,34 and their expression could be modified long term after a jTBI as it was shown at short term for ABCA1. 34 Therefore, the fact that Aβ accumulation is still increasing between 2 and 6 months, whereas perlecan and fibronectin are not may be consistent with perlecan changes in cerebral amyloid angiopathy and also suggests that other pathways are likely to be involved in the process of Ab increase after a jTBI.
However, besides their possible implication in Aβ clearance, basement membrane proteins also have biologic activities that can be revealed after brain injury-induced proteolysis. They are not merely components of an inert mat supporting brain cells, and growing evidence suggests that those proteins and their proteolytic fragments can be neuroprotective and affect angiogenesis.14–16,18,19 As described previously, the vasoactive DV fragment of perlecan can induce neuroprotection and angiogenesis by increasing the production and release of VEGF by endothelial cells in different rodent stroke models.14,16 It can compete with Aβ for the binding to α2 integrin and thus inhibit neurotoxic pathways, 17 and it can reduce glial scar by inhibiting astrocyte proliferation. 13 Interestingly, DV has also been shown to interact with α5β1 integrin receptor in endothelial cells, inducing Aβ internalization, degradation and clearance, thereby reducing its toxicity. 19 Therefore, a lack of additional DV being generated from total perlecan could contribute to Aβ buildup. The second protein studied, fibronectin, has been shown to exert neuroprotective effects after transient focal cerebral ischemia and spinal cord injury in rats by decreasing apoptosis and promoting axonal regeneration.15,18
Therefore, these augmentations of perlecan and fibronectin staining could also suggest a potential compensatory mechanism aiming at protecting endothelial cells against Aβ toxicity, and maybe in a larger extent protecting the NVU against neurodegenerative processes like Aβ accumulation. In addition to immunostaining, we also performed immunoblotting experiments to detect perlecan DV and fibronectin. No changes in perlecan DV were revealed suggesting that despite an increase in total perlecan staining, there is no concomitant increase in perlecan proteolysis to generate perlecan-cleaved DV. As DV is pro-angiogenic in the brain,14,16 this lack of a change in DV levels is consistent with the lack of increased angiogenesis that we also observed 2 and 6 months after jTBI (discussed further below). Unchanged DV levels in the presence of increased total perlecan staining after jTBI also suggest that the activity of DV-generating proteases are unaltered or perhaps diminished. However, western blots revealed the presence of fibronectin fragments after a jTBI, suggesting that other proteases are activated chronically after jTBI. This activation could be due to Aβ, shown to induce MMPs in atrocytes cultures, leading to the degradation of fibronectin. 35 Although the increase in fibronectin staining in the jTBI group could be related to the ‘degradation’ of fibronectin by providing better access to the epitope, those fragments could also have major implications and consequences in jTBI outcome. Fibronectin fragments have been shown to possess different biologic activities like the inhibition of Schwann cells proliferation or the inhibition of endothelial cells growth.36,37
It is also interesting to note that perlecan DV and fibronectin have been shown to modulate angiogenesis via the same receptor, α5β1 integrin, suggesting its relative importance to brain angiogenesis.
Link between Integrin and Angiogenesis
Perlecan DV binding to α5β1 integrin can induce VEGF release, 16 and whereas α5β1 integrin and fibronectin are both poorly expressed in quiescent endothelium, they are strongly expressed in proliferating vessels. 22 Here, an increase in perlecan and fibronectin could lead to angiogenesis by interacting with α5β1 integrin. However, when we investigated angiogenesis, both t-lectin intensity quantification and microvessel density did not reveal any difference between jTBI and naive animals at 2 or 6 months after TBI. Therefore, there is almost no angiogenesis present at these time points. Perhaps even more surprisingly, although we saw an increase in its ligands, we did not see any increase in the levels of the receptor a5 integrin in brain blood vessels when compared with naive rats. This, and the lack of an increase in perlecan-cleaved DV, may in part explain why we did not observe any angiogenesis in these animals. Indeed, while α5 integrin is expressed in microvessels during angiogenesis, it has been shown to be downregulated after angiogenesis. 23 However, as it was shown for global hypoxia, 24 we can hypothesize that α5 integrin has been upregulated acutely after jTBI, and then returned to baseline after 2 and 6 months. Moreover, α5β1 integrin was previously shown to be involved in the clearance of Aβ, 30 therefore the absence of increase of α5β1 integrin may also contribute to Aβ accumulation observed in our model. To the best of our knowledge, the level of expression of α5β1 integrin after jTBI has never been studied before.
Finally, the absence of angiogenesis after jTBI could also be explained by the presence of Aβ, shown to inhibit angiogenesis by binding to VEGFR-2 and inhibiting VEGF signaling. 38 However, we observed a decrease in microvessel diameters in jTBI animals after 2 and 6 months. A recent study also shows a decrease in microvessel diameters in postmortem brains of patients with Alzheimer's disease and vascular dementia. 39 This observation emphasizes the fact that jTBI is turning in a chronic vascular disease, as the decrease of microvessel diameter is likely to induce a decrease of cerebral blood flow, reducing the brain perfusion and possibly leading to cognitive dysfunctions. 40 In some extend, it looks like that there is an absence of significant angiogenesis in compensation to the possible decrease of cerebral blood flow in contrast with other vascular brain injury like stroke. Absence of compensatory angiogenesis could be due to the absence of induction of α5β1 integrin on blood vessels and increase of DV at long term after jTBI.
Overall Summary
Collectively, our results suggest that an early TBI can induce long-term modifications of endothelial cell phenotype, contributing to acceleration of neurodegenerative processes like accumulation of Aβ in the brain. In particular, transformations of the vascular environment seem to induce a decreased rate of Aβ clearance: the decrease in P-gp levels and the increase in perlecan and fibronectin seen at 2 and 6 months are, among other, potential mechanisms involved in accumulation of Aβ after a jTBI in a vicious circle (Figure 6). Cognitive dysfunctions can be explained by the accumulation of Aβ in the brain and by decreased cerebral blood flow suggested by a decrease of microvessel diameter after jTBI combined with a lack of angiogenesis. Perlecan and fibronectin are interesting components of this mechanism as they participate in Aβ5 accumulation, but at the same time they have the ability to protect the CNS. They could represent a potential therapeutic target if we could promote their beneficial effects, e.g., by treatment with perlecan DV or fibronectin, or by increasing the expression of α5β1 integrin receptor.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Germaine Paris for the technical help, Jacqueline S Coats for surgeries and Monica Romero for a portion of imaging performed at the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core (LLUSM AIM) Facility supported by NSF grant, MRI-DBI 0923559 (to SM Wilson).