
Abstract
Clinical studies suggest that traumatic brain injury (TBI) hastens cognitive decline and development of neuropathology resembling brain aging. Blood–brain barrier (BBB) disruption following TBI may contribute to the aging process by deregulating substance exchange between the brain and blood. We evaluated the effect of juvenile TBI (jTBI) on these processes by examining long-term alterations of BBB proteins, β-amyloid (Aβ) neuropathology, and cognitive changes. A controlled cortical impact was delivered to the parietal cortex of male rats at postnatal day 17, with behavioral studies and brain tissue evaluation at 60 days post-injury (dpi). Immunoglobulin G extravasation was unchanged, and jTBI animals had higher levels of tight-junction protein claudin 5 versus shams, suggesting the absence of BBB disruption. However, decreased P-glycoprotein (P-gp) on cortical blood vessels indicates modifications of BBB properties. In parallel, we observed higher levels of endogenous rodent Aβ in several brain regions of the jTBI group versus shams. In addition at 60 dpi, jTBI animals displayed systematic search strategies rather than relying on spatial memory during the water maze. Together, these alterations to the BBB phenotype after jTBI may contribute to the accumulation of toxic products, which in turn may induce cognitive differences and ultimately accelerate brain aging.
INTRODUCTION
Traumatic brain injury (TBI) is defined as damage resulting from a direct or indirect mechanical force on the brain, and pathophysiological outcomes emphasize damage to neurons and glia. However, the blood–brain barrier (BBB) and vascular integrity are also compromised following TBI. Primary injury occurs at the moment of TBI impact, with disruption of blood vessels and the BBB, contributing to vasogenic edema formation. 1 Blood–brain barrier disruption precedes several downstream events contributing to secondary injuries, such as changes in cerebral blood-flow and hypometabolism, brain swelling and increased intracranial pressure, hypoxia/ischemia, and related molecular events such as cell death, inflammation, oxidative stress, and pathology. 1 Blood–brain barrier disruption normalizes within 1 week in several injury models, yet recent studies show BBB permeability up to 30 days after ischemic insult. 2 Collectively, these observations suggest that BBB integrity represents a complex and dynamic sequelae meriting attention during acute and delayed stages post-TBI.
Vascular dysfunction is a critical element of brain aging and neurodegeneration, especially in Alzheimer disease (AD).
3
Age-related brain Aβ accumulation may depend on progressively impaired clearance mechanisms, as measured by
Clinically, several lines of evidence demonstrate long-term pathological and behavioral modifications after TBI. These long-term changes may lead to premature aging and neurodegenerative processes like AD with higher risk for aberrant Aβ protein accumulation. 8 In support of this, brain Aβ immunolabeling was detected within hours after clinical TBI 9 and in long-term survivors (1 to 47 years) of a single injury. 10 As for patient outcome, many TBI survivors endure lifelong consequences, with 3.2 to 5.3 million US residents currently suffering physical and/or mental disability, which can result in long-term complications. 11 Young children, followed by adolescents and older adults, are at greatest risk for incurring TBI. 12 Therefore, long-term studies on cellular and molecular changes after TBI are needed in juvenile experimental models, especially regarding collective changes in the BBB phenotype, neuropathology, and behavior.
We hypothesized that an early life juvenile TBI (jTBI) may result in several brain changes. We evaluated BBB components (tight junctions, influx/efflux transporters) in parallel with neuropathology and aberrant protein accumulation (Aβ) and cognitive outcomes (learning and memory). A controlled cortical impact (CCI) was delivered to the parietal cortex of juvenile (17-day-old) rats and outcomes are described in adults at 60 days postinjury (dpi).
MATERIALS AND METHODS
Animals
All protocols and procedures were approved by the Institutional Animal Care and Use Committee of Loma Linda University. Juvenile (Literature descriptions for ‘juvenile’ include a broad range of postnatal ages.
13
Based on previous publications and our recently published data,
14
we used postnatal day 17 as ‘juvenile’ which is identical to methods in the present manuscript.) (17-day-old) male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) were housed with their dams on a 12-hour light–dark cycle at constant temperature and humidity. Only male rats were selected, as prior studies suggest the existence of gender differences following TBI.
15
Pups were weaned 7 days after the surgery, housed two rats per cage, and fed with standard lab chow and water
Juvenile Traumatic Brain Injury Model
Controlled cortical impact was induced in rats as previously described. 14 Briefly, rats were anesthetized with isoflurane (Webster Veterinary Supply, Sterling, MA, USA) and given a 5-mm diameter craniotomy over the right fronto-parietal cortex (1 mm posterior, 2 mm lateral from Bregma) and CCI was delivered to jTBI animals using a 3-mm impactor at a 20° angle to cortex, 1.5 mm depth, 200 milliseconds impact duration, 6 m/s velocity. Body temperature was maintained at 37°C during surgery. A subcutaneous buprenorphine injection was administered for pain relief (0.01 mg per mL per kg at 1 hour and 24 hours after surgery) and animals were returned to their dams. All sham animals underwent the same procedures as jTBI animals, except for the CCI.
Water Maze Design and Testing
Water maze testing has been previously described for this model. 14 Briefly, testing occurred at 30 and 60 dpi over a 3-day paradigm, with 5 blocks of 2 trials each (10 total trials) including cued testing (visible platform), spatial learning (hidden platform), and probe trials for spatial memory (no platform). Swim strategy analysis 16 revealed eight different swim patterns grouped into three general learning strategies (Table 1; Figure 5A): Spatial strategy (spatial direct, spatial indirect, focal correct), Systematic strategy (scanning, random, focal incorrect), or Looping strategy (chaining, peripheral looping).
Description of MWM swim strategies observed after jTBI
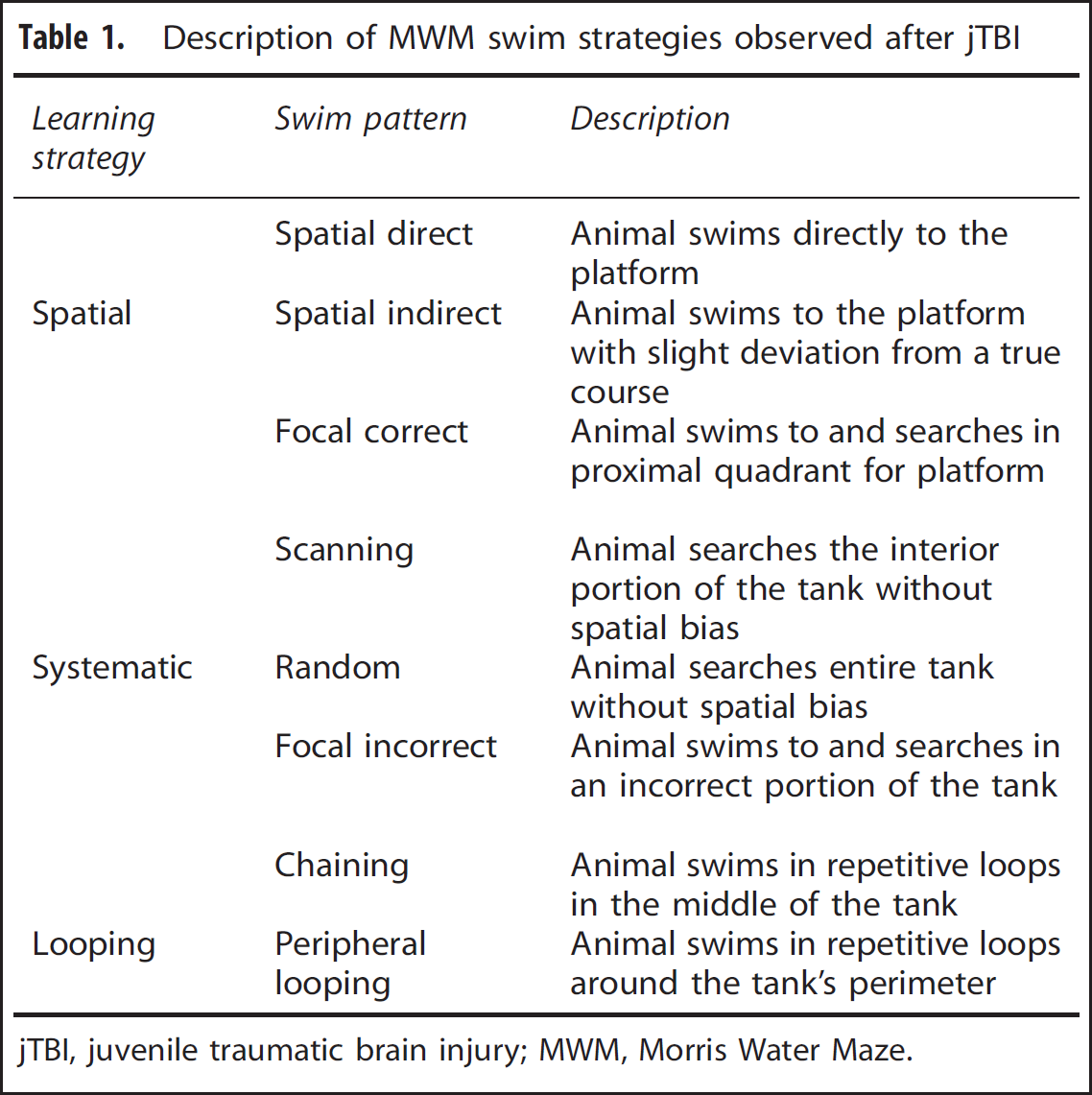
jTBI, juvenile traumatic brain injury; MWM, Morris Water Maze.
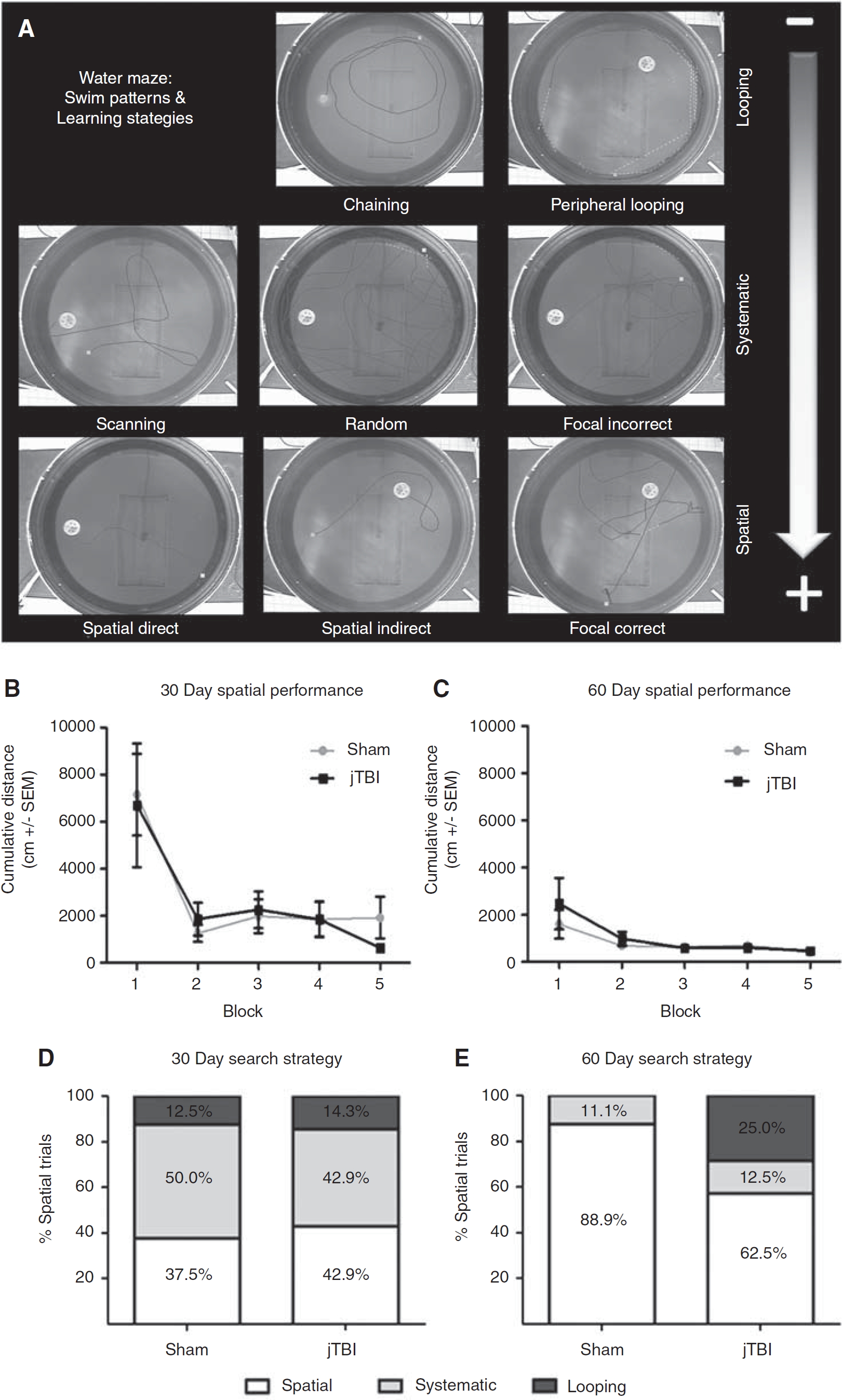
Differences in water maze strategies in adulthood. (
Brain Tissue Processing and Immunohistochemistry
At 60 dpi, rats were anesthetized with a combination of Ketamine (Ketaject 100 mg/mL, Phoenix, St Joseph, MO, USA) and Xylazine (AnaSed 100 mg/mL, Lloyd Laboratories, Shenandoah, IA, USA) at the appropriate dose/body weight, and then transcardially perfused with 4% paraformaldehyde. The brains were excised and cryoprotected in 30% sucrose solution for 48 hours, and frozen on dry ice. Coronal cryostat free floating sections (50
Immunoglobulin G Extravasation Staining for Blood–Brain Barrier
Sections were rinsed in phosphate-buffered saline (PBS) and blocked for 1 hour in 1% bovine serum albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA) made in PBS, pH = 7.4 (Fisher Scientific, Pittsburg, PA, USA) before incubation for 2 hours at room temperature with Alexa-Fluor-800 biotinconjugated affinity purified goat anti-rat immunoglobulin G (IgG) (1:500, Rockland Immunochemicals, Gilbertsville, PA, USA) in PBS containing 0.25% Triton X-100 and 0.25% BSA (Sigma-Aldrich) made in PBS, pH = 7.4. After washing, sections were scanned on an Odyssey infrared scanner to quantify fluorescence in the cortex and striatum.
Immunolabeling of Blood–Brain Barrier Proteins
For P-gp staining, sections were pretreated for antigen retrieval using 33% acetic acid + 66% EtOH solution for 10 minutes at 20°C (Fisher Scientific). For claudin 5, glial fibrillary acidic protein (GFAP), P-gp, LRP1, and RAGE, sections were blocked for 1 hour in 1% BSA in PBS before overnight primary antibody incubation at 4°C. All antibody incubations were in 0.25% BSA with 0.25% Triton X-100 made in PBS, pH = 7.4. For immunolabeling, we used mouse anti-claudin 5 (1:500, Life Technologies: Invitrogen, Grand Island, NY, USA), mouse anti-P-gp (1:100, Abcam, Cambridge, MA, USA and Calbiochem, EMD Chemicals, Merck KGaA, Darmstadt, Germany), mouse anti-LRP1 (1:1,000, Calbiochem, EMD Chemicals, Merck KGaA), rabbit anti-RAGE (1:500, Abcam, Cambridge, MA, USA) and chicken anti-GFAP (1:1,000, Millipore, Temecula, CA, USA). After PBS rinses, we incubated in secondary antibody for 2 hours at room temperature at 1:1,000 as appropriate for each primary antibody: goat anti-mouse secondary antibody coupled with Alexa-Fluor-488 or with Alexa-Fluor-594, goat anti-chicken coupled with Alexa-Fluor-568, and for infrared analysis either goat anti-mouse secondary antibody coupled with Alexa-Fluor-800 or goat anti-rabbit secondary antibody coupled with Alexa-Fluor-680 (all secondaries from Invitrogen, Grand Island, NY, USA). After washes in PBS, sections for classical immunofluorescence were mounted on glass slides and coverslipped with vectashield antifading medium containing DAPI (Vector, Vector Laboratories, Burlingame, CA, USA). Sections for infrared analysis were mounted on glass slides and air-dried. Control sections for all studies in which primary or secondary antibodies were omitted resulted in negative staining (not shown).
Immunolabeling for Aβ
Sections for amyloid analysis were pretreated for 4 minutes in 88% formic acid at room temperature and all other immunostaining procedures were identical to the procedure described above. We used a monoclonal antibody raised in mouse, against rodent Aβ at the N-terminal amino acids 1 to 16 (1:1,000, from M Paul Murphy) and visualized staining with goat anti-mouse secondary antibody Alexa-Fluor-488 (1:1,000; Life Technologies: Invitrogen, Grand Island, NY, USA). Adjacent sections were coincubated with the mouse rodent-Aβ antibody and either the C-terminal antibody against Aβ1-42 (rabbit, 1:1,000, Covance, Emeryville, CA, USA), or the microglial marker ionized calcium binding adaptor molecule 1 (IBA1) (rabbit, 1:1,000, Wako Chemicals, Richmond, VA, USA). To control for nonspecific binding of mouse antibodies on rat tissue (Figure 3F), sections received only the secondary antibody, goat anti-mouse Alexa-Fluor-594 (staining was negative), followed by a full protocol with both the rodent-Aβ antibody and goat anti-mouse Alexa-Fluor-488. Sections were also stained with the 6E10 anti-human-Aβ1-16 antibody (mouse, 1:5,000, Covance) or using protocols excluding primary or secondary antibodies, resulting in negative staining (not shown).
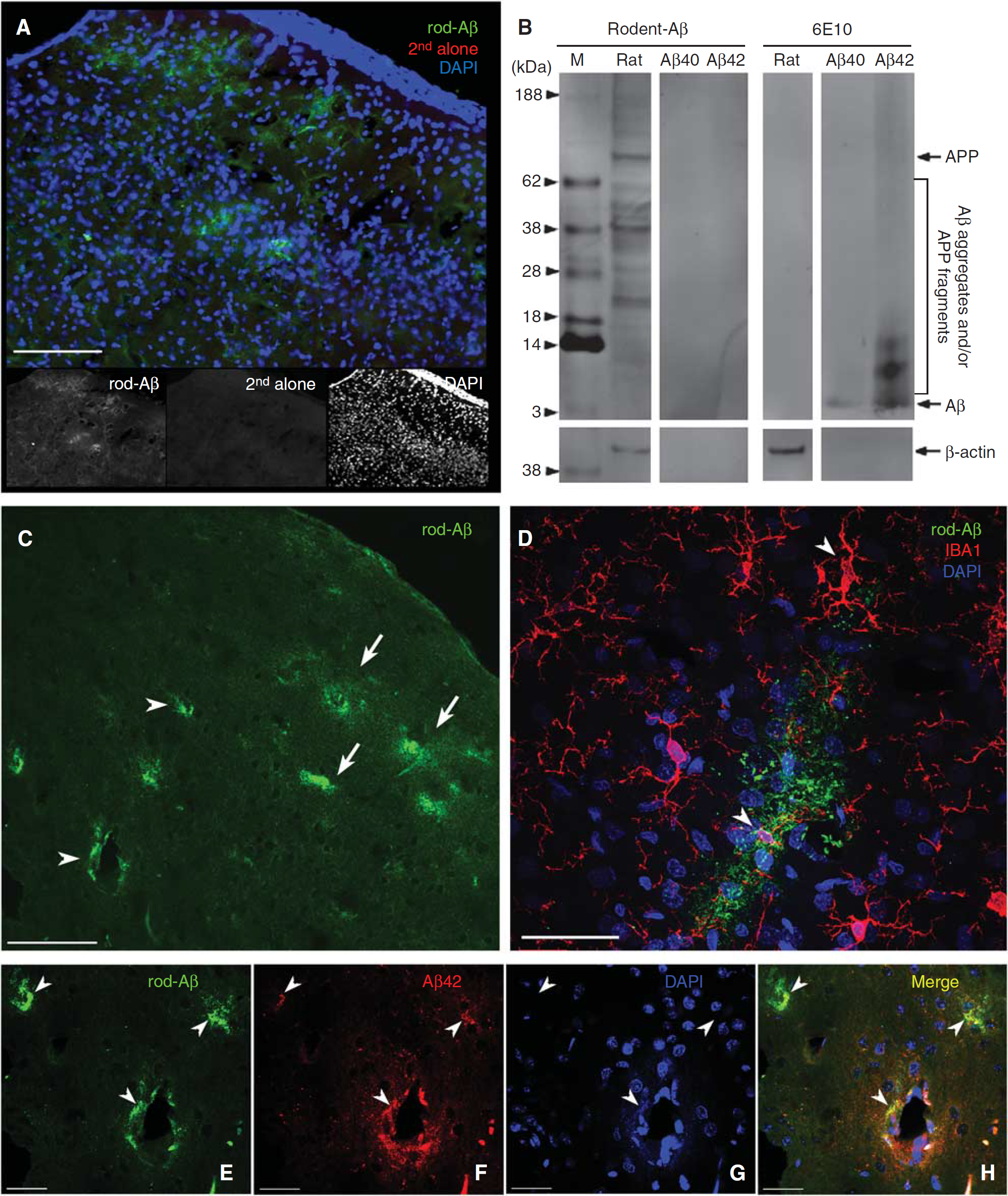
Immunoreactivity patterns with a rodent-Aβ antibody. (
To determine any fibrillar β-pleated sheet structures, sections were double stained with a rodent-Aβ (see above) followed by Thioflavin S staining. Briefly, tissue was immersed for 5 minutes in double-distilled (dd) H20, followed by a 5-minute incubation in 1% Thioflavin S (Sigma-Aldrich) made in ddH20, and subsequent differentiation using consecutive 5 minutes washes in 70% EtOH, 95% EtOH, and ddH20 (Fisher Scientific). Slides were then coverslipped as described above.
Quantification of Immunohistochemistry
Coronal sections immunostained with infrared secondary antibodies (IgG and RAGE) were scanned using the same parameters for sham and jTBI at a 21-
For quantification of classical immunolabeling of claudin 5, P-gp, LRP1, and Aβ, images were evaluated and collected using an epifluorescent microscope (Olympus, BX41, Center Valley, PA, USA). The threshold and morphological user-defined parameters were selected to maximize visualization of positive staining in the ROIs for each protein staining pattern. These parameters were kept consistent for all animals during image acquisition. For claudin 5, P-gp, and LRP1 a series of images were taken in the parietal and temporal cortex, both above and below the rhinal fissure on each hemisphere ipsilateral and contralateral to the lesion or craniotomy using a −20 objective (422
For quantification of rodent-Aβ immunoreactivity, we used eight whole serial coronal slices per animal spaced 1.2 mm apart, from bregma levels +3.2 to −5.2 mm as shown in Figure 4 (
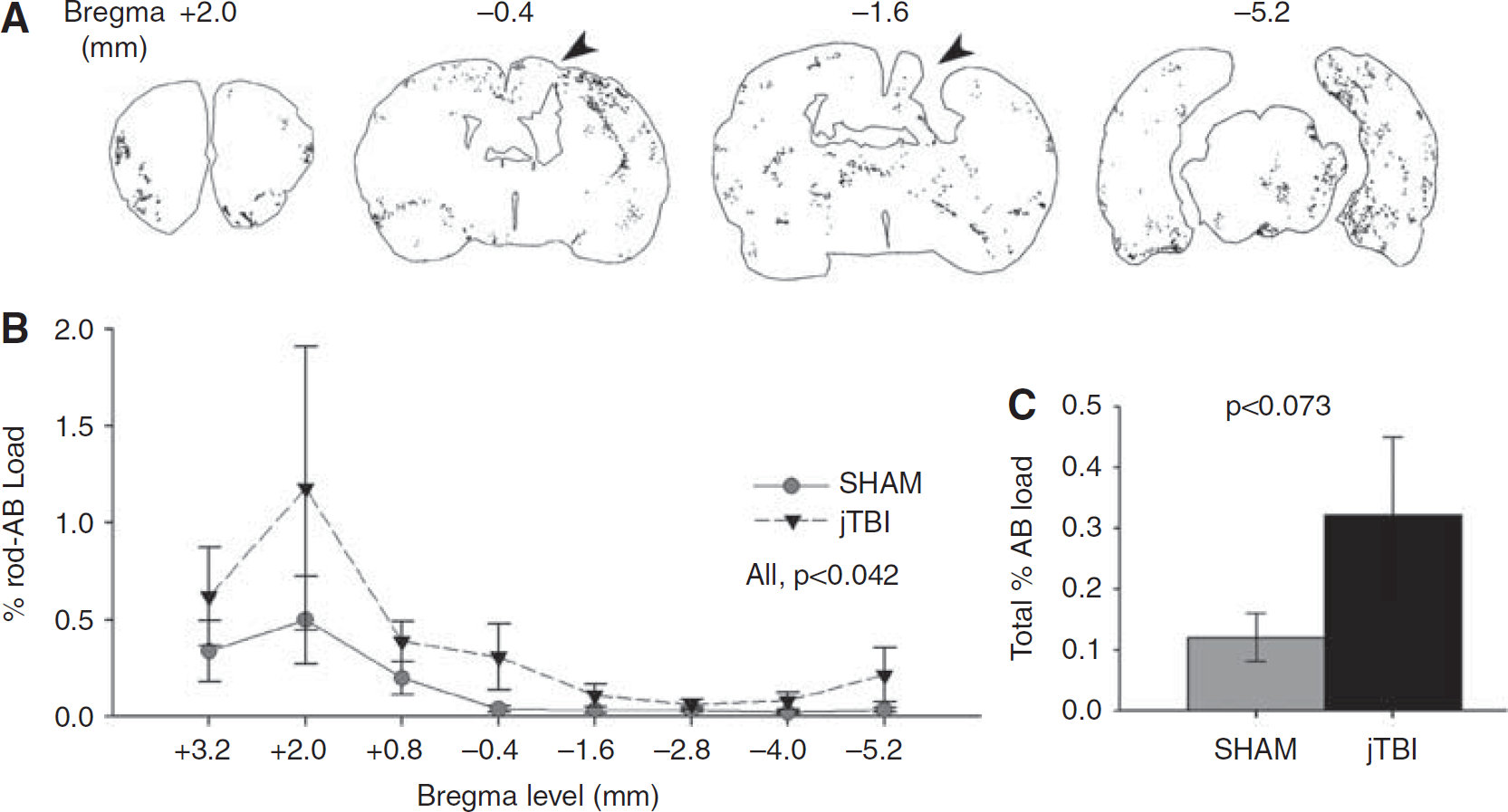
Anterior to posterior patterns of rodent-Aβ distribution. (
Brain Tissue Processing for Western Blotting
A Protein FFPE extraction kit (Qiagen, Hilden, Germany) was used to process perfused brain slices for Western blotting. Parietal and temporal cortical tissue above the rhinal fissure was excised from three coronal sections at bregma levels −1.6, 2.8, and −4.0 mm, that were adjacent to slices of interest used for immunohistochemistry. Briefly, tissue was homogenized and processed according to the kit instructions, and then samples were assayed for total protein concentration by bicinchoninic assay (Pierce Biotechnology, Rockford, IL, USA). The human Aβ1-40 and Aβ1-42 peptides (Biopeptide, San Diego, CA, USA) were prepared using a 1-mg sample that was reconstituted to obtain 231
Statistical Analyses
All data are presented as mean ± s.e.m., statistical analyses were done using SPSS (New York, NY, USA), and graphs obtained using SigmaPlot (San Jose, CA, USA). For the Aβ analyses, we utilized a repeated measures ANOVA (analysis of variance) with group (jTBI, sham) × bregma level (eight serial coronal sections) and a conservative Huyhn-Feldt adjustment to the degrees of freedom was used to protect against any violations of the sphericity and compound symmetry assumptions underlying this ANOVA model. For the behavioral strategy analysis, data were analyzed using independent samples
RESULTS
Blood–Brain Barrier Phenotype
While disruption of the BBB after jTBI lasts only for a short period of about 7 days, 1 little is known about its recovery and structural properties at a delayed post-jTBI time point. We evaluated several BBB markers at anatomical sites near the lesion, as well as on coronal slices at a distance from the injury site at 60 dpi.
Immunoglobulin G extravasation staining (Figures 1A and 1B) and quantification (Figure 1C) did not show any difference between sham and jTBI groups in perilesional parietal cortex and striatum, suggesting that the BBB is no longer disrupted. Immunoglobulin G extravasation was observed as expected in regions without a BBB, such as the median eminence (Figures 1A and 1B). In accordance with this result, claudin 5 immunolabeling was positive and outlined the elongated endothelial membrane structure of large vessels (Figures 1D and 1E) and microvessels (Supplementary Figures 1A and 1B) of both sham and jTBI animals. Claudin 5 staining was significantly increased in the large intracortical blood vessels of jTBI animals compared with sham (Figure 1F,
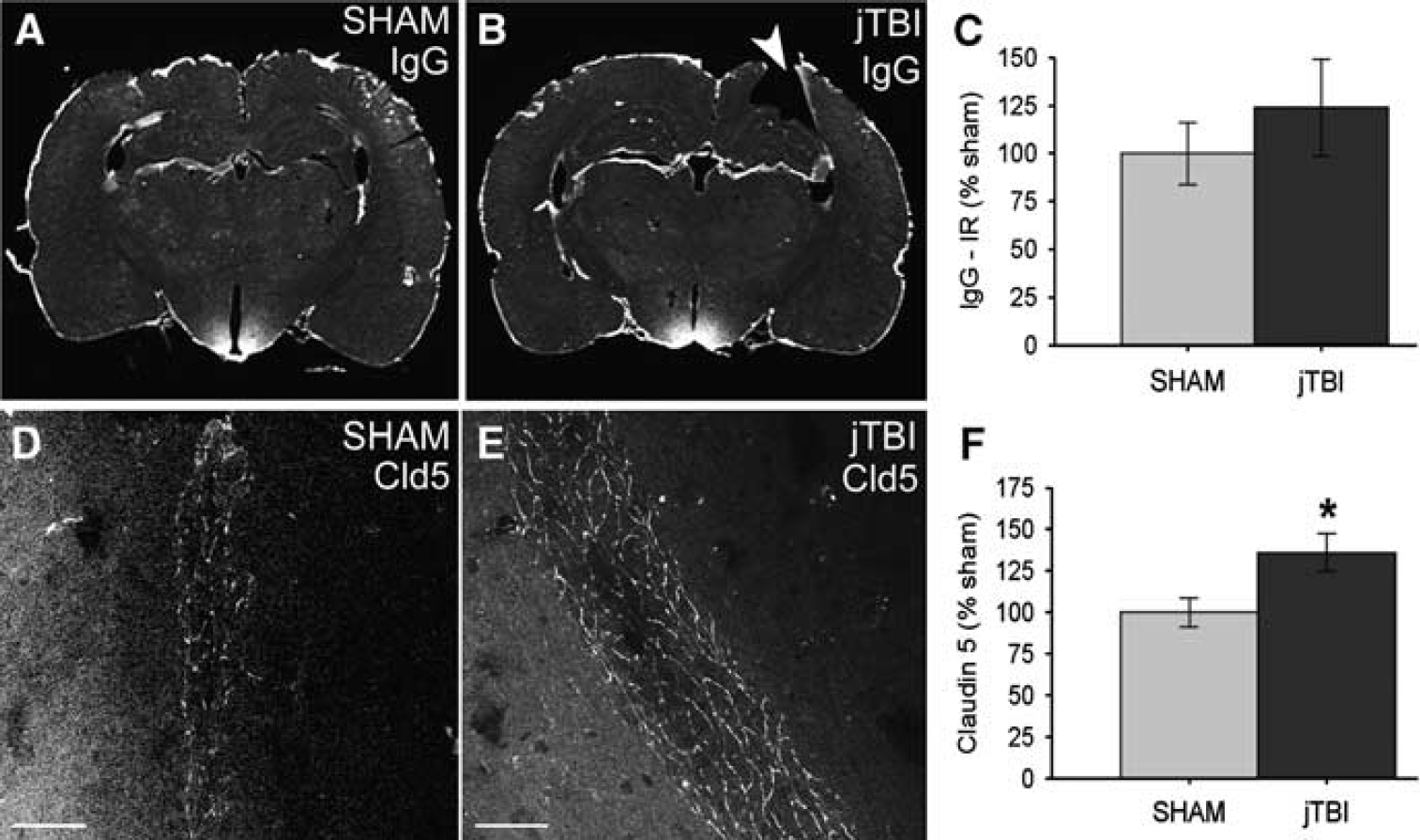
Changes in endothelial tight junctions 2 months after juvenile traumatic brain injury (jTBI). (
To further address phenotypic changes at the BBB, we characterized the distribution of P-gp, LRP1, and RAGE proteins known to be involved in BBB trafficking. P-glycoprotein immunostaining was observed primarily on endothelial cells, in both jTBI and sham animals colabeled with the astrocytic marker, GFAP (Figures 2D and 2F). Sham animals exhibited more intense and widespread staining patterns on microvessels (Figure 2D), whereas jTBI had more diffuse vascular staining of P-gp (Figure 2E). Quantification of images from the parietal and temporal cortices revealed significantly decreased levels of P-gp immunoreactivity in jTBI cortical vessels compared with sham (Figure 2F,
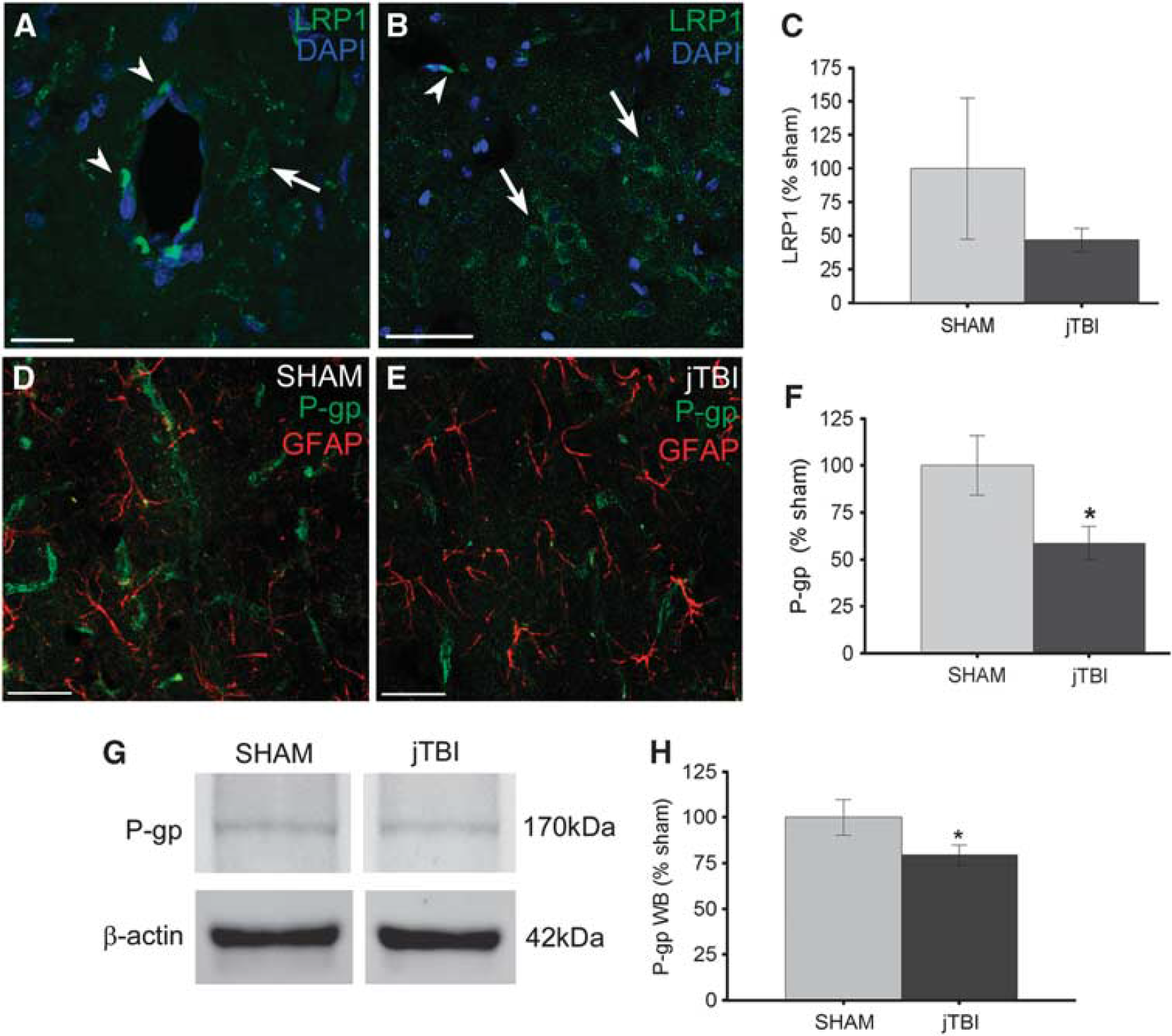
Juvenile traumatic brain injury (jTBI) changes proteins involved in cellular trafficking at the blood–brain barrier (BBB). (
Widespread Aβ Accumulation
Using a rodent-Aβ antibody specific to the N-terminus and a formic acid pretreatment, we detected a diffuse pattern of immunoreactivity that is both extracellular (Figures 3A and 3C–3H) and perivascular (arrowheads in Figures 3C and 3E–3H). Examples of positive rodent-Aβ immunolabeling are shown at a distance from the injury, such as the frontal (Figures 3C and 3H), temporal (Figure 3A), and parietal cortex (Figure 3D). Positive staining was also detected in the entorhinal cortex, striatum, and thalamus (Figure 4A), with no staining using the secondary antibody alone (Figure 3A).
Western blot showed that the rodent-Aβ antibody was specific for rat tissue but not for human Aβ1-40 and Aβ1-42 peptides, while the anti-human 6E10 antibody detected bands for human Aβ peptides, but no signal for rat tissue (Figure 3B). The 6E10 antibody detected monomeric Aβ species (4 kDa) in both human Aβ1-40 and Aβ1-42 peptide preparations, and several Aβ aggregates that are more abundant in the Aβ1-42 preparation (Figure 3B). In the rat tissue, the rodent-Aβ antibody reveals an APP (β-amyloid precursor protein) signal near the expected region ~110 kDa and several additional bands, possibly APP fragments or Aβ aggregates of various sizes (Figure 3B).
Rodent-Aβ immunoreactivity was associated with microglial cells, stained with IBA1. Microglia exhibited an elongated soma, little somatic cytoplasm, and multiple thinner processes, as shown in an example from the jTBI parietal cortex (Figure 3D). No activated microglia were detected at this time point (data not shown). Rodent-Aβ immunoreactivity colocalized with C-terminal-Aβ1-42 staining in several brain regions, such as the frontal cortex (Figures 3E–3H). Thioflavin S staining (data not shown) is negative despite positive rodent-Aβ immunoreactivity, suggesting a lack of fibrillar β-pleated sheet structures. Overall, these data are consistent with an early Aβ accumulation pattern having a diffuse morphology and containing Aβ1-42 species.
Total rodent-Aβ immunoreactivity was quantified using the Mercator program and expressed as % Aβ load relative to total coronal brain area at each bregma level (Figures 4A–4C). Representative jTBI animals are shown with the lesion location (black arrowheads in Figure 4A) and outlines of positive staining at individual bregma levels (Figure 4A). By 60 dpi, the cortical jTBI lesion cavity is apparent from bregma −1.0 mm up to −4.0 mm (not shown) and higher rodent-Aβ immunoreactivity was observed in more anterior and posterior levels from the lesion site (Figure 3B). Repeated measures ANOVA showed significant changes across all coronal sections regardless of group (Figure 4B,
Changes in Strategy During Water Maze
As previously reported, sham and jTBI animals showed no overall performance differences across trials on swim speed, cued learning, or spatial learning/memory at 30 or 60 dpi.
14
The cumulative distance traveled for both groups showed no differences in spatial learning at 30 dpi (Figure 5B) and 60 dpi (Figure 5C). As expected due to repeated testing, performance during block 1 of day 60 was better than block 1 of day 30 (Figures 5B versus 5C,
DISCUSSION
We evaluated long-term changes following TBI in a juvenile rat model to address the concordance with clinical observations of delayed behavioral modifications, and emerging data on changing BBB properties during brain aging and neurodegenerative disease. Our results at a delayed 2-month time point show parallel changes in structural BBB phenotypes of claudin 5, altered P-gp expression, rodent-Aβ immunoreactivity, and altered water maze performance. Together, our data suggest that an early brain injury may commence with vascular damage that promotes long-term phenotypic changes to the BBB that can influence cognition.
Physiopathological Changes in the Blood–Brain Barrier
We explored BBB phenotypes at a delayed time point after juvenile injury as a unique platform for addressing whether jTBI disrupts the neurovascular unit and contributes to brain pathophysiology. Within the first days after injury, we observe high IgG staining. 1 Blood–brain barrier disruption is repaired by 2 months in our jTBI model, as evidenced by the absence of IgG extravasation and increases in tight-junction protein claudin 5 (Figure 1). Other studies also report that improved BBB integrity coincides with altered expression of tight-junction proteins at later time points. Similar to our model, claudin 5 levels are upregulated after evans blue and IgG leakage subsides at 1 and 2 weeks after rat cortical injury. 17 Claudin 5 is also increased, along with resolution of evans blue staining, after 4 and 8 weeks in mice exposed to an infectious agent targeting the BBB. 18 Thus, altered levels of claudin 5 long after jTBI suggest delayed modifications in BBB structural properties. Furthermore, our differential observations on large and small vessels (Figure 1; Supplementary Figure 1) indicated a more complex phenotypic response of the endothelium, which may be related to location along the vascular tree.
We investigated the triad of endothelial cell proteins (P-gp, LRP1, and RAGE) previously shown as potential mediators of Aβ trafficking and other proteins across the BBB (Figure 6).5,19 P-glycoprotein is well known as an efficient gatekeeper on the luminal side and often pharmaceutically by-passed to allow efficient drug delivery. Its expression is influenced by several distinct molecular pathways and its putative role in disease and injury is emerging.20,21 We observed a significant decrease in P-gp in cortical vessels following jTBI by immunostaining and protein blot analyses. Similar to our findings, brain P-gp protein level and function decreased after irradiation of normal rats, 22 in aged 3-year-old rats, 6 and in sporadic AD. 21 Additionally, P-gp expression and function were impaired during multiple sclerosis pathology and its dysfunction coincides with lymphocyte infiltration and inflammation in experimental allergic encephalomyelitis. 20
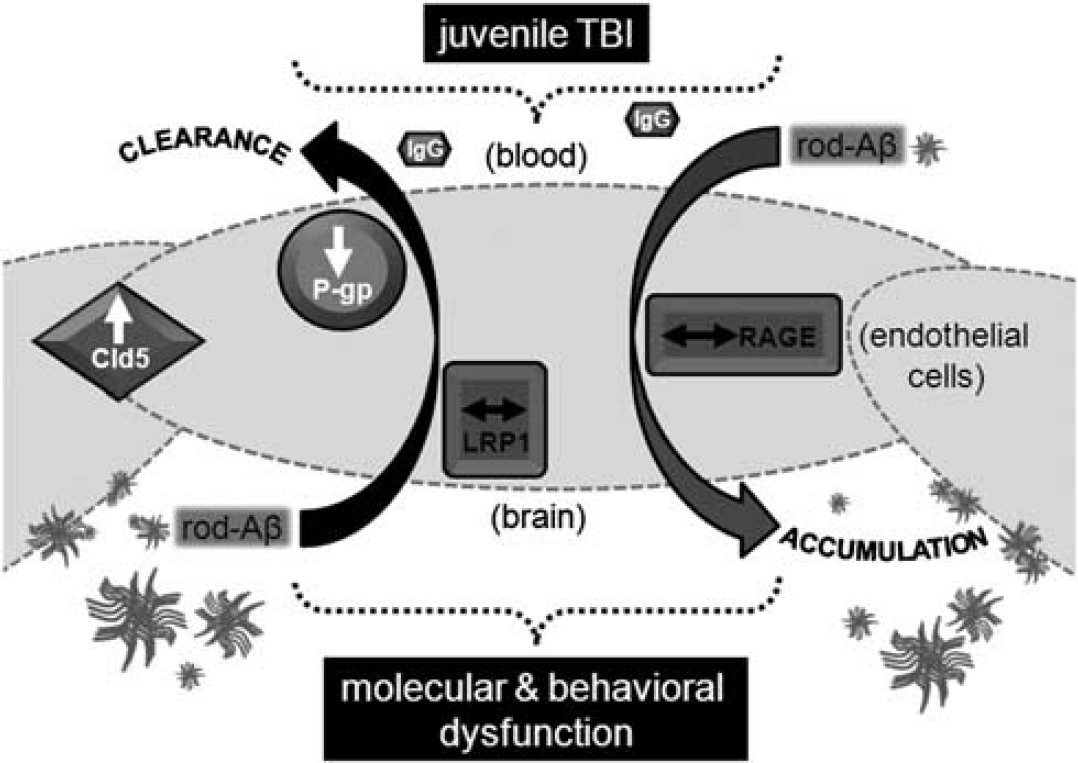
Summary of parallel changes in adulthood after early brain injury. A schematic representation of events 2 months after juvenile traumatic brain injury (jTBI) shows phenotypic alterations to the blood–brain barrier (BBB) changes occurring in parallel with molecular and behavioral dysfunction. Specifically, lack of immunoglobulin G (IgG) extravasation from the blood into the tissue and increased levels of tight-junction marker claudin 5 (cld5) indicate an intact BBB during adulthood. However, in an environment with decreased levels of P-glycoprotein (P-gp), and unchanged levels of lipoprotein-related receptor protein 1 (LRP1) and receptor for advanced glycation end products (RAGE), proper metabolism of toxic proteins such as Aβ can be hindered. As a result, the BBB phenotype impairs normal clearance of rod-Aβ and may promote its abnormal accumulation inside the brain in the parenchyma and vascular walls (adapted from Andras
Our observed P-gp decreases may be one result of long-lasting neuroinflammation, contributing to an increase in rodent-Aβ immunoreactivity. Importantly, P-gp knockout mice exhibited decreased LRP1 in brain capillaries without changes in RAGE proteins 7 and aged 3-year-old rats showed decreased vascular levels of P-gp and LRP1 associated with accumulation of Aβ. 6 We did not detect significant changes in LRP1 between sham and jTBI, but LRP1 was positive in blood vessels and neurons within the temporal cortex of jTBI animals. Our findings are in line with several model systems and age groups, which emphasize the putative role of the BBB in chronic brain pathophysiology after acute injury. Our observed P-gp decrease may have a complex relationship to accumulation of rodent-Aβ in multiple brain areas. Frontal and temporal lobes are vulnerable to neuropathology after TBI. 10 In our model, these same regions exhibited reduced P-gp (Figure 1) and increased rodent-Aβ immunoreactivity in the brain parenchyma and vessel walls (Figures 3 and 4). Clinically, our findings are similar to low endothelial P-gp that correlated with increased parenchymal Aβ1-40 or Aβ1-42 plaques in the medial temporal lobe of aged nondemented humans. 23 Following peripheral Aβ1-42 injections in adult mice, brain endothelial cells show downregulated expression of RAGE, LRP1, and P-gp at the mRNA level; however, P-gp immunostaining remained unchanged. 24 Collectively, these findings highlight complex interactions between Aβ species and the BBB transporters that may be influenced by injury type and study time point.
Aβ Accumulation and Blood–Brain Barrier Changes After Juvenile Traumatic Brain Injury
The important consequences of cerebrovascular dysfunction and the role of the BBB neuropathological protein trafficking are gaining momentum. In normal rodents, classical AD-like pathology with fibrillar amyloidosis is rare. This may be due to Aβ sequence differences of three amino acids at the N-terminal. 25 However, we used an N-terminal specific antibody for rodent Aβ, 26 and endogenous accumulation has been reported long after a brain insult in normal experimental injury models. For example, increased APP and Aβ immunostaining was observed in thalamic nuclei for up to 9 months after stroke in normal rats. 27 In normal adult rats, intranasal nerve growth factor administration resolved increases of Aβ1-42 for up to 2 weeks post-TBI. 28 In a rodent model of aging, senescence accelerated mice (SAMP8) showed increased BBB disruption at 12 months and exhibited vascular Aβ compared with their littermate controls. 29 These data indicate the close relationship of BBB disruption and brain pathology in aging and postinjury.
Blood–brain barrier phenotypic changes at 2 months occurred in parallel with higher rodent-Aβ immunolabeling in the parenchyma and vasculature of jTBI animals (Figures 3 and 4). Notably, we observed diffuse Aβ deposition in regions remote to the original site of impact, especially in the superficial cortical layers. Additionally, diffuse Aβ in superficial cortical layers is the primary conformation present in other models of natural Aβ accumulation, such as aged beagles.
30
Diffuse Aβ is also more common in clinical cases with mild cognitive impairment
31
and is becoming recognized as an important contributor to disease progression by the National Institute of Aging-Alzheimer's Disease.
32
Aβ load, evaluated by immunohistochemical staining similar to our methodology, was the best predictor for dementia in very old individuals.
33
Interestingly, we observed sparse rodent-Aβ immunoreactivity in some shams (
Parallel Changes in Vascular Phenotype and Behavioral Dysfunction
In our injury model, we observed changes in behavioral outcome by 2 months after injury, 14 which indicate that the brain may be undergoing compensatory mechanisms. Here, we observed strategic water maze impairment without overt memory deficits, which may reflect underlying brain-repair mechanisms occurring after injury. Interestingly, clinical defects of initial strategy formulation affected performance on a task of complex visuospatial executive function in individuals with amnestic mild cognitive impairment who are more likely to develop AD. 35 In a CCI study in adult mice, sham and TBI animals exhibited similar water maze outcomes after 2 months, but TBI animals exhibited thigmotaxic strategy impairment with increased percent time swimming along maze walls, similar to our looping category. 36 In comparison with adult models, juvenile rats may be especially vulnerable due to a lack of cognitive reserve. While the neural correlates are unclear, clinically it is thought that having increased cognitive reserve from education and other life experiences may be protective against brain aging and disease. 37 Thus, brain injury at a developmental stage may influence underlying mechanisms and reduce cognitive reserve. Our observations of delayed impairments in jTBI suggest a parallel with acceleration of brain aging and disease due to trauma.
Concluding Remarks
Juvenile traumatic brain injury may induce initial BBB dysfunction that progresses over time to affect protein trafficking at a time point relevant for cognition. Our data are consistent with clinical and experimental findings in studies of TBI and Aβ in transgenic models, as well as studies on BBB dynamics with aging and nontraumatic brain injuries. However, only the use of further target-specific studies may address whether these changes are interconnected, or whether they are independent parallel events in the same trauma-affected brain. In this study, we described lasting effects of jTBI with phenotypic BBB changes after 2 months, including a lack of IgG extravasation and compensatory increases in structural tight-junction protein claudin 5 (Figure 6). 38 However, the reduction in Pgp may be sufficient to impair normal Aβ clearance and promote its accumulation inside the brain. Overall, we show the presence of continued modifications in the BBB phenotype long after injury, and propose the BBB as a meaningful therapeutic target for resolving detrimental posttraumatic dysfunction.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Dr André Obenaus (Loma Linda University) for project assistance and Monica Rubalcava for a portion of imaging performed at the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core (LLUSM AIM) supported by NSF Grant, MRI-DBI 0923559 (to SM Wilson).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.