
Abstract
Following an ischemic stroke, T lymphocytes become activated, infiltrate the brain, and appear to release cytokines and reactive oxygen species to contribute to early inflammation and brain injury. However, some subsets of T lymphocytes may be beneficial even in the early stages after a stroke, and recent evidence suggests that T lymphocytes can also contribute to the repair and regeneration of the brain at later stages. In the hours to days after stroke, T-lymphocyte numbers are then reduced in the blood and in secondary lymphoid organs as part of a ‘stroke-induced immunodeficiency syndrome,’ which is mediated by hyperactivity of the sympathetic nervous system and the hypothalamic—pituitary—adrenal axis, resulting in increased risk of infectious complications. Whether or not poststroke T-lymphocyte activation occurs via an antigen-independent process, as opposed to a classical antigen-dependent process, is still controversial. Although considerable recent progress has been made, a better understanding of the roles of the different T-lymphocyte subpopulations and their temporal profile of damage versus repair will help to clarify whether T-lymphocyte targeting may be a viable poststroke therapy for clinical use.
Introduction
Stroke remains a leading cause of death and disability worldwide. Ischemic stroke is characterized by the disruption of cerebral blood flow, which produces a central core of dead neurons surrounded by a penumbra of damaged but partially functional neurons (Dirnagl et al, 1999). Early restoration of blood flow remains the treatment of choice for limiting brain injury following ischemic stroke. Improved educational efforts that emphasize the early signs and symptoms of stroke, coupled with the widespread application of thrombolytic therapy to patients with acute ischemic stroke have increased the number of patients benefiting from reperfusion (Davis et al, 2007). While reperfusion of the ischemic brain is desirable, tissue damage may result from reperfusion per se. Reperfusion appears to enhance the inflammatory response and causes additional injury to adjacent brain tissue (Schaller and Graf, 2004). Although there is no clear consensus of opinion concerning the ultimate mediator of ischemia—reperfusion (I—R)-induced brain injury, several factors related to inflammation have received considerable attention in recent years (for review see Iadecola and Anrather, 2011; Macrez et al, 2011).
T lymphocytes are central to the development of a sustained inflammatory response and there is now good evidence that these cells accumulate in the postischemic brain within a few hours of reperfusion (Brait et al, 2010; Jander et al, 1995). The purpose of this review is to briefly summarize the emerging evidence of complex roles of T cells in brain injury after stroke. As shall be discussed, T cells are sources of pro-inflammatory cytokines and cytotoxic substances, such as reactive oxygen species, in the brain after stroke, which likely contribute to neuronal death and poor outcomes. However, future therapeutic efforts to treat stroke by targeting T cells must take into account the crucial roles that T cells have in host defense against invading pathogens, which appears to be critically important in the period following a stroke. Furthermore, recent evidence for a novel role of T cells in promoting brain tissue repair and regeneration in the weeks and months after stroke will also be discussed.
T-lymphocyte subtypes and characteristics
T lymphocytes are bone marrow-derived leukocytes that mature in the thymus and have an integral role in the adaptive immune system (i.e., the division of the immune system that recognizes specific pathogens and can generate immune memory). As such, they are vital for clearing pathogens that cannot be cleared by the innate immune system alone (i.e., the division of the immune system that initially defends the body from infection, in a nonspecific manner) (Harrison et al, 2008). Several T-cell subtypes exist, and these can be differentiated by their unique expression profiles of specific coreceptor proteins. CD3 is expressed on the surface of 95% of the T cells that have exited the thymus and which are immunocompetent. The CD3+ subset comprises both CD4+ (or T-helper cells; TH) and CD8+ (or cytotoxic T cells; TC) cells in approximately equal proportions (Harrison et al, 2008; Seder and Ahmed, 2003) (Figure 1).
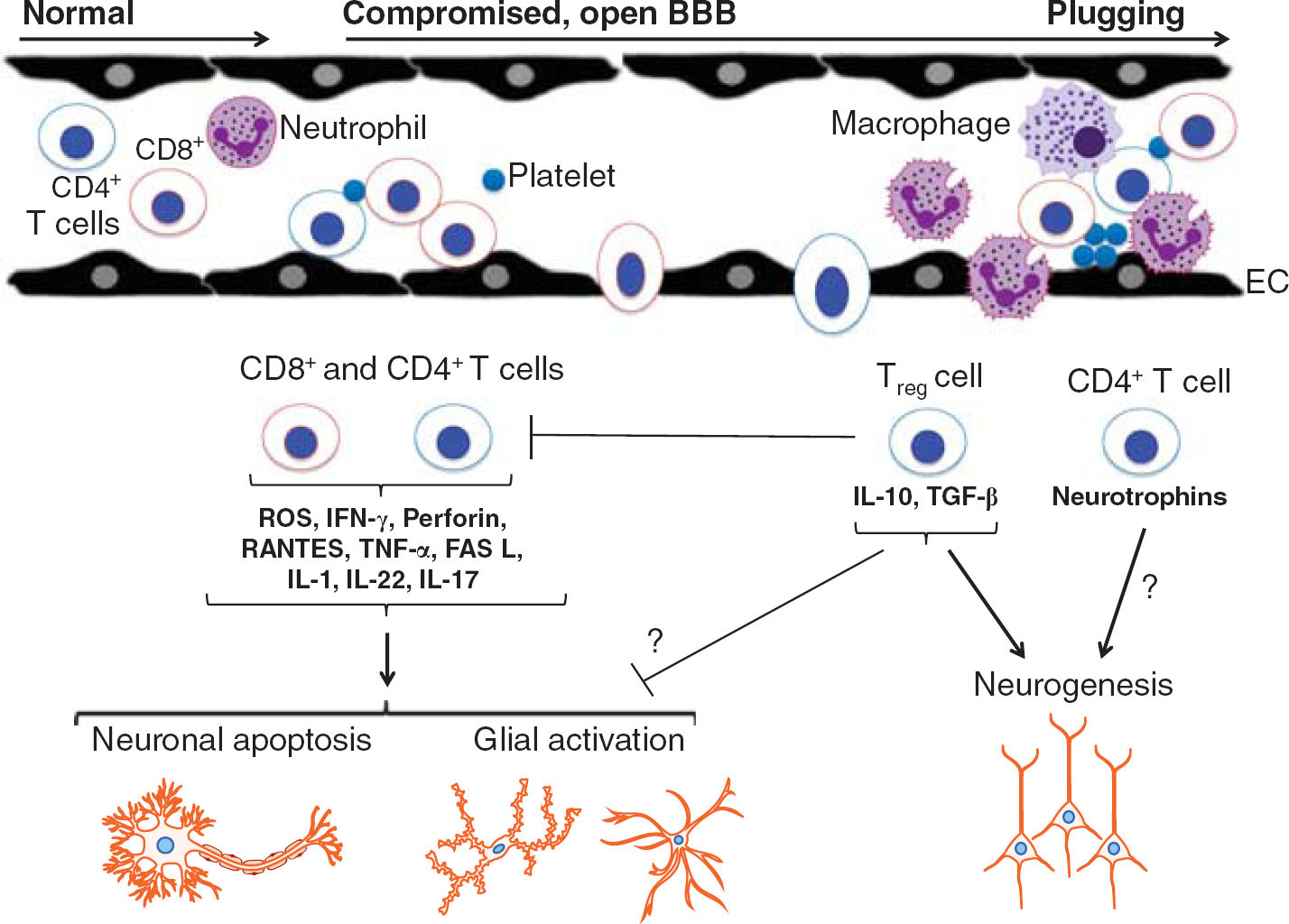
Potential actions of T lymphocytes in the brain following stroke. Following cerebral ischemia and reperfusion, circulating T lymphocytes interact with neutrophils, macrophages, platelets, and endothelial cells (ECs), and may cross the blood—brain barrier (BBB) to infiltrate the injured tissue. The two broad subpopulations of mature circulating T lymphocytes are T-helper cells (i.e., CD4+) and cytotoxic T cells (i.e., CD8+). It appears that both subpopulations contribute significantly to acute poststroke brain injury via several mechanisms. The small subpopulation of CD4+ regulatory T cells (i.e., Tregs) may act to limit some of these damaging effects by other T cells. In addition, CD4+ T cells, perhaps especially Tregs, may contribute to later repair processes, such as neurogenesis, during brain recovery.
TH cells do not kill cells directly, but instead help to activate other immune cells including TC cells, and can be further differentiated into three major types defined by the cytokines they secrete (Korn et al, 2009; Santana and Rosenstein, 2003). Thus, TH cells can promote either: cell-mediated or inflammatory immunity (i.e., TH1 cells); humoral and allergic responses (i.e., TH2 cells) (Abbas et al, 1996; Glimcher and Murphy, 2000; Santana and Rosenstein, 2003); or inflammatory immunity to clear pathogens distinct from those handled by TH1 or TH2 cells, including fungi (i.e., TH17 cells) (Korn et al, 2009). On activation, TH1 cells secrete pro-inflammatory cytokines such as IFN-γ (interferon-γ), TNF (tumor necrosis factor), and LT-α (lymphotoxin), whereas TH2 cells produce antiinflammatory cytokines such as interleukin-4 (IL-4) and IL-10. TH2 cells are thought to suppress pro-inflammatory immune responses, and commonly appear later in an immune response (Abbas et al, 1996). TH17 cells secrete IL-17, IL-21, and IL-22 (Korn et al, 2009). An important CD4+ T-cell subtype that accounts for ~10% of all CD4+ T cells (Zouggari et al, 2009) and which expresses CD25 and the transcription factor Foxp3, is the regulatory T cell (Treg; i.e., CD4 + CD25 + Foxp3+ cells). Treg cells act to limit the immune response and thus prevent autoimmune disorders (Sakaguchi et al, 2006) via their release of transforming growth factor-β and IL-10 (Liesz et al, 2009b).
As their name suggests, the cytotoxic TC cells can directly kill cells that contain intracellular pathogens, such as viruses, through the release of the cytotoxins, perforin, and various granzymes (resulting in necrosis), or by the Fas-FasL pathway (resulting in apoptosis) (Barry and Bleackley, 2002; Russell and Ley, 2002). TC cells also produce pro-inflammatory cytokines, such as IFN-γ and TNF, which serve to block viral replication as well as to promote the activation of other elements of the immune system (Phillips et al, 2010). Finally, ‘unconventional’ T lymphocytes that act to link the innate and adaptive immune systems (and are often called innate-like lymphocytes) include γδ T cells (~5% of T cells; the only T cells that do not express CD3) and natural killer T (NKT) cells (Cerundolo and Kronenberg, 2010; D'Cruz et al, 2010).
Evidence for damaging effects of T lymphocytes after stroke
Although most studies of the effects of immune cells on stroke outcome have focused on cells of the innate immune system, especially neutrophils and monocytes, several recent studies have evaluated cerebral I—R in T-cell-deficient mice and have consistently reported a smaller infarct volume and improved functional outcome than in wild-type controls (Hurn et al, 2007; Kleinschnitz et al, 2010b; Shichita et al, 2009; Yilmaz et al, 2006). Hurn et al (2007) reported that male SCID (severe combined immunodeficient) mice (deficient in both T and B cells), developed an ~40% smaller infarct volume after 90 minutes focal ischemia and 22 hours reperfusion compared with wild-type mice. Two further studies similarly found ~60% to 70% smaller infarct volumes as well as improved functional outcomes at 24 to 72 hours in recombinase activating gene-deficient (Rag1−/−) mice, which also lack T and B lymphocytes (Kleinschnitz et al, 2010b; Yilmaz et al, 2006). Moreover, there is good evidence that this protection of lymphocyte-deficient mice following stroke is due to the lack of T cells, and not B cells, as the reconstitution of B cells does not alter the protection observed in the Rag1−/− mice. By contrast, when wild-type CD3+ T cells are transplanted back into Rag1−/− or Rag2−/− mice (also deficient in T and B lymphocytes), this protection is lost (Kleinschnitz et al, 2010b; Shichita et al, 2009; Yilmaz et al, 2006), confirming that T-cell-mediated damage occurs acutely after experimental stroke. Moreover, mice that lack RANTES (CCL5), a chemokine that recruits T cells (as well as other immune cells) into inflammatory sites (Appay and Rowland-Jones, 2001), have smaller infarct volumes than wild-type littermates. Thus, taken together, existing data suggest that a deficiency in T cells confers protection in the acute period following cerebral I—R. In contrast, when permanent ischemia (i.e., ischemia with no reperfusion) was induced in SCID versus wild-type mice, no difference in infarct volume was observed at 6, 24, 72 hours, or 7 days after the induction of stroke (Saino et al, 2010).
Recent studies have investigated the effects of deficiency in either TH or TC subpopulations of T cells in stroke (Yilmaz et al, 2006; Liesz et al, 2011a, b ). Yilmaz et al (2006) reported that mice deficient in either TH or TC cells had smaller infarct volumes than wild-type mice (TH−/−: ~53% smaller; TC−/−: ~73% smaller) and reduced neurologic deficit, although this latter functional outcome at 24 hours did not reach statistical significance in either group. Liesz et al (2011b) also reported significantly reduced infarct volumes in mice depleted of TH and TC cells following permanent ischemia. Thus, these findings suggest that both TH and TC cells contribute to the development of brain injury following stroke.
Importantly, it is apparent that not all T-cell subtypes are detrimental for acute stroke outcome. It was recently reported that neither γδ T cells nor NKT cells contribute to stroke injury (Kleinschnitz et al, 2010b). Furthermore, the role of Treg cells in stroke outcome is still in question. Infarct volume and sensorimotor dysfunction were significantly increased in mice treated with an anti-CD25 monoclonal antibody to neutralize Treg cells, compared with controls (Liesz et al, 2009b). However, this protection was only observed after 7 days, and only after a relatively modest ischemic insult that produced a small infarct volume (Liesz et al, 2009b; Ren et al, 2011). Moreover, a recent finding by Ren et al (2011) that selective Treg depletion, using Foxp3DTR mice, did not affect stroke infarct volume 96 hours after cerebral I—R, thus failed to implicate this regulatory pathway in limiting poststroke brain damage. In addition, there is now remarkable evidence that the function of NKT cells resident in the liver is profoundly impaired due to augmented sympathetic neurotransmission following stroke (see below for more detail), and that this loss of NKT cell activity substantially contributes to the immunosuppression and susceptibility to infections that occur following stroke (Wong et al, 2011).
Involvement of T lymphocytes and ‘thrombo-inflammation’ at the neurovascular unit after cerebral ischemia
Cerebral I—R is established to cause endothelial cell dysfunction involving production of reactive oxygen metabolites, swelling, or detachment from underlying basement membrane, and compromised barrier function, leading to increased protein extravasation and interstitial edema (Ishikawa et al, 2004; Yilmaz and Granger, 2008). These events generally occur in postcapillary segments of the microvasculature and are often accompanied by the adhesion of leukocytes. (Figure 1) Within capillaries, the swollen endothelial cells can restrict the movement of activated and less deformable leukocytes, leading to nutritive perfusion failure and tissue hypoxia (Yilmaz and Granger, 2008). Activated perivascular cells may also release pro-inflammatory mediators that further promote adhesion molecule expression on endothelial cells and leukocytes. The excessive quantities of reactive oxygen species generated after cerebral ischemia by activated endothelial cells, adherent leukocytes and platelets, and perivascular cells can initiate the peroxidation of membrane lipids, resulting in reduced microvascular integrity manifested as increased vascular permeability and altered basal lamina structure (Yilmaz and Granger, 2010).
During cerebral I—R, leukocytes make contact with the endothelium via a tethering process mediated by selectins on the endothelial surface, resulting in leukocyte rolling (Yilmaz et al, 2006). Rolling enables leukocytes to detect chemokines on the endothelial surface, and the subsequent activation of their chemokine receptors causes rapid conformational changes of leukocyte integrins, resulting in their transition from a low- to high-affinity/avidity state. High-affinity/avidity integrins interact with their endothelial counter receptors of the immunoglobulin superfamily (i.e., VCAM-1 (vascular cell adhesion molecule-1), intercellular adhesion molecule-1, ALCAM (activated leukocyte cell adhesion molecule)). Next, the leukocytes will traverse the endothelial basement membrane into the perivascular space, and migrate to sites of brain injury and inflammation (Yilmaz and Granger, 2010) (Figure 1). Matrix metalloproteinases are zinc-containing endoproteinases involved in the maintenance and remodeling of the extracellular matrix. Activated T cells can produce a variety of matrix metalloproteinases that degrade extracellular matrix and facilitate leukocyte migration through the basement membranes (Korpos et al, 2010).
Recent studies of the postischemic brain microcirculation have revealed that the accumulation of T cells in postcapillary venules is generally accompanied by the recruitment of adherent platelets, suggesting that the venules assume both a pro-inflammatory and pro-thrombogenic phenotype after ischemic stroke (Ishikawa et al, 2003). The net impact of the accumulated platelets in the postischemic microcirculation remains unclear; however, inhibition of platelet tethering and adhesion to vascular endothelial cells (the early phases of platelet activation) has produced reductions in cerebral infarct volume (Elvers et al, 2010; Kleinschnitz et al, 2007, 2009; Zhao et al, 2009). Furthermore, inhibition of early platelet adhesion and activation maintains cerebral blood flow during reperfusion (Pham et al, 2011). A novel concept of ‘thrombo-inflammation’ suggests that there is a strong link between pathways of thrombus formation and inflammation, and recent evidence suggests that specific early platelet adhesion/activation mechanisms may in fact link these pathways to exacerbate infarct development following cerebral I—R (Nieswandt et al, 2011; Stoll et al, 2010). However, it is currently unclear as to whether specific interactions between T lymphocytes and platelets have an important role in stroke outcome, as T-cell deficiency is reported to have no effect on thrombus formation (Kleinschnitz et al, 2010b).
Stroke-induced immunodeficiency syndrome
While it is established that T lymphocytes contribute to early poststroke neuronal injury, the levels of these and other immune cells in the circulation are relatively quickly reduced (Liesz et al, 2009a; Martin et al, 2008; Prass et al, 2003), possibly as an endogenous protective mechanism. Thus, a profound systemic immunodepression—or ‘stroke-induced immunodeficiency syndrome’ —occurs as early as 12 hours after ischemic stroke, and may be maintained for several weeks (Gendron et al, 2002; Liesz et al, 2009a; Prass et al, 2003). This phenomenon also involves reduced numbers of T cells and other immune cells present in the spleen (Gendron et al, 2002; Liesz et al, 2009a; Martin et al, 2008; Offner et al, 2006b; Prass et al, 2003), thymus, and lymph nodes (Liesz et al, 2009a; Martin et al, 2008; Offner et al, 2006b; Prass et al, 2003), and is mediated by hyperactivity of the sympathetic nervous system (SNS) and the hypothalamic—pituitary—adrenal axis (HPA) (Prass et al, 2003). This leads to increased apoptosis of immune cells in the spleen, thymus, and lymph nodes, and as a result, these secondary lymphatic organs undergo atrophy (Liesz et al, 2009a; Offner et al, 2006b; Prass et al, 2003). Furthermore, there is a shift from TH1 to TH2 cytokine production (Prass et al, 2003; Theodorou et al, 2008). It is unclear whether the infiltration of these cells directly into the brain might also partly contribute to lower circulating cell numbers. T cells appear in the ischemic hemisphere within 24 hours of focal cerebral I—R (Brait et al, 2010; Jander et al, 1995). This infiltration is reported to peak at ~ 72 hours after cerebral I—R (Gelderblom et al, 2009; Shichita et al, 2009; Stevens et al, 2002), and greater numbers of TH cells than TC cells are typically seen (Gelderblom et al, 2009; Saino et al, 2010).
Infectious complications, predominantly chest and urinary tract infections, occur in many stroke patients within the first few days after stroke, and development of an infection early after stroke is known to be associated with worse outcomes (Aslanyan et al, 2004; Hilker et al, 2003; Langhorne et al, 2000). The signals and mechanisms that trigger the SNS and the HPA to induce stroke-induced immunodepression remain unclear. One cause appears to be the increased pro-inflammatory cytokine production that occurs early after stroke and central nervous system (CNS) injury-specific neurogenic signaling (for review see Dirnagl et al, 2007; Meisel et al, 2005). An early study by Prass et al (2003) blocked the SNS and HPA by administration of a β-adrenoceptor antagonist (propranolol) and a glucocorticoid receptor inhibitor (RU486), respectively. Interestingly, although both treatments significantly reversed the percentage of apoptotic splenocytes to levels seen in sham-operated mice, and prevented the decrease in circulating lymphocytes, bacterial infections, and mortality following stroke were only reduced with propranolol treatment, and not RU486, suggesting that the inhibition of SNS activation is critical in preventing bacterial infections (Prass et al, 2003). This finding was further supported by a more recent study (Prass et al, 2006).
Consistent with experimental reports, several recent clinical studies have found evidence that SNS-mediated stroke-induced immunodepression and subsequent susceptibility to poststroke infections also occurs in patients. In a prospective study, Chamorro et al (2007) found evidence that acute ischemic stroke is associated with an early activation of the sympathetic adrenomedullar pathway that lowers the threshold of infection and increases the risk of death independent of the blood-borne effects of pro- and antiinflammatory cytokines, and circulating leukocytes. Urra et al (2009) reported increased apoptosis and a reduction in the circulating levels of TH, TC, Treg, and B cells following stroke. They found a correlation between SNS and HPA activation (assessed by elevated metanephrine and cortisol levels, respectively), lower levels of T cells (CD3+ and CD8+) and infection. Furthermore, there was an inverse correlation between lower levels of T cells (CD3+ and CD8+) and infarct volume. However, poor long-term outcome was not associated with infection (Urra et al, 2009). Klehmet et al (2009) confirmed in the PANTHERIS (Preventive Antibacterial Treatment in Acute Stroke) trial on the efficacy of short-term antibacterial therapy to prevent the development of poststroke infections, that a rapid loss and functional deactivation of T cells are common changes in stroke patients consistent with immunodepression after brain ischemia. Furthermore, a stronger decrease in cellular immune responses and an increased sympathetic activity after stroke are associated with a higher risk of infections (Klehmet et al, 2009). A post hoc analysis of the PANTHERIS trial by Harms et al (2011) investigated the impact of distinct lesion patterns on SNS activation, immunodepression, and frequency of poststroke infections. Large stroke volume, lesions affecting distinct regions of the MCA cortex, and SNS activation (assessed by elevated norepinephrine levels) were each associated with an impaired immune function and a higher susceptibility to poststroke infections. Whereas neither stroke severity nor stroke volume was independently associated with poststroke infections, increased levels of norepinephrine and infarction of the anterior MCA cortex were both identified as independent risk factors for poststroke infections (Harms et al, 2011). A recent study by Hug et al (2011) found that reduced costimulatory efficacy of circulating costimulatory cells (i.e., splenic non-T cells) in mice is an important feature of stroke-induced immunodepression, and, if confirmed in humans, points to such cells as potential targets for therapies to prevent secondary inflammatory damage to the brain after stroke.
In addition to the well-established pro-inflammatory cytokine-mediated activation of the SNS and the HPA, another pathway of communication between the nervous and immune systems, known as the vagal ‘cholinergic antiinflammatory pathway’ has been identified. When the vagus nerve is activated by pro-inflammatory cytokines, it releases acetylcholine, which results in inhibition of the release of more pro-inflammatory mediators by macrophages (for review see Pavlov et al, 2003; Tracey, 2002, 2007). Experimental studies have shown that vagal nerve signaling inhibits the release of pro-inflammatory cytokines and improves outcomes following different models of ischemia—reperfusion (Tracey, 2007). Therefore, the vagal ‘cholinergic antiinflammatory pathway' is another potential mediator of this stroke-induced immunodepression.
Potential mediators of damage by T lymphocytes after stroke
Cytokines, Chemokines, and Cytotoxins
The mechanisms of T-cell-mediated brain injury following stroke are currently unclear. Classically, T cells kill bacteria- and virus-infected cells either via the release of cytokines (in the case of TH and TC cells) or cytotoxins (also by TC cells) (Harrison et al, 2008), and so it is plausible that similar actions may exacerbate poststroke brain inflammation. Cytokines and chemokines released by TH and TC cells are likely to increase expression of vascular adhesion molecules and attract other immune cells into the brain, resulting in widespread apoptosis (Arumugam et al, 2005). Alternatively, TC cells may directly cause either cell necrosis or apoptosis via the release of cytotoxins or the activation of the Fas receptor (for review see Barry and Bleackley, 2002). Neutralization of certain T-cell-derived cytokines (e.g., IL-17, IL-12, IL-23) was found to reduce infarct volume and improve neurologic outcome scores over 7 days poststroke (Konoeda et al, 2010; Shichita et al, 2009), consistent with these T-cell-derived cytokines contributing to brain injury early after I—R. Furthermore, Liesz et al (2009b) showed that CD3+ T cells are a major source of the damaging pro-inflammatory cytokine, IFN-γ, in the ischemic hemisphere. When neutralizing antibodies against IFN-γ were administered into the cerebral ventricles, there was a significant reduction in infarct volume (Liesz et al, 2011b). Consistent with these results was the finding that reduced mRNA expression of various pro-inflammatory cytokines, chemokines, and chemokine receptors (TNF-α, IL-6, IL-10, IP-10, CCR1, CCR2, CCR3, and CCR5) occurs after stroke in the brains of SCID mice compared with wild-type mice (Hurn et al, 2007). Taken together, current evidence suggests that T cells (potentially excluding the immunomodulatory subset of Treg cells) represent a major source or stimulus of pro-inflammatory cytokines in the brain following stroke. Moreover, stroke in perforin-deficient mice produce a significantly smaller infarct volume, suggesting that the cytotoxin perforin, released by TC cells, contributes to ischemic damage (Liesz et al, 2011b).
Reactive Oxygen Species
There is recent evidence that T lymphocytes may contribute to oxidative tissue injury following stroke, potentially via the release of NADPH oxidase type 2 (Nox2)-derived superoxide. First, it is quite clear that oxidative stress due to excessive levels of reactive oxygen species is a major mechanism of poststroke brain injury. Studies of stroke in mice either overexpressing antioxidants or deficient in pro-oxidant enzymes have reported smaller infarct volumes than in wild-type controls (Iadecola et al, 1997, 2001; Kinouchi et al, 1991; Sampei et al, 2000; Weisbrot-Lefkowitz et al, 1998; Yang et al, 1994), and conversely, studies of antioxidant-deficient mice have found larger infarcts (Crack et al, 2001; Kondo et al, 1997; Murakami et al, 1998). Moreover, several studies in Nox2-deficient mice have reported substantially smaller infarct and edema volumes, and less blood—brain barrier disruption than wild-type controls, pointing to Nox2 oxidase as a key source of damaging superoxide in the brain after stroke (Chen et al, 2009; Jackman et al, 2009; Kahles et al, 2007; Kunz et al, 2007; Walder et al, 1997). T lymphocytes are now known to contain a functional Nox2 oxidase (Brait et al, 2010; Jackson et al, 2004; Purushothaman and Sarin, 2009) and, at 24 hours following stroke, circulating T cells produce ~ 7-fold greater amounts of Nox2-derived superoxide than do T cells from control mice (Brait et al, 2010). Interestingly, much greater levels of superoxide are generated by circulating T cells compared with spleen-derived T cells after stroke (Brait et al, 2010), consistent with the possibility that exposure to the circulation (and/or postischemic brain tissue) upregulates Nox2 oxidase activity in T lymphocytes. In addition to Nox2, a recent study by Kleinschnitz et al (2010a) has identified NADPH oxidase type 4 (Nox4) as an important source of oxidative stress and an effective therapeutic target in acute stroke. Nox4 was induced in human and mouse brain following ischemic stroke, and mice deficient in Nox4 (Nox4−/−) developed smaller infarct volumes, had improved functional outcomes, and were largely protected from oxidative stress, blood—brain barrier leakage, and neuronal apoptosis, after both I—R and permanent cerebral ischemia.
Mechanism(s) of T-lymphocyte activation after stroke
There is currently a major unanswered and controversial question regarding the mechanism(s) of T-lymphocyte activation following stroke. Based on the information gained from decades of study in the area of immunology, it is apparent that the activation and infiltration of T cells into the brain following stroke is too quick for it to occur via a classical antigen-dependent response (Offner et al, 2006a). Traditionally, it has been believed that for T cells to infiltrate into tissues, cellular activation leading to a change in the expression of surface molecules (e.g., VLA-4, CCR5, and CD44) is required (Harrison et al, 2008; McLachlan and Jenkins, 2007). Classical antigen-dependent activation of naive T cells comprises two main steps and takes 7 to 10 days (Abbas and Lichtman, 2011). The first step involves the binding of the T-cell receptor (TCR) to the antigen presented on the major histocompatability complex on the surface of an APC (antigen-presenting cell). The second step involves the binding of costimulatory molecules on the T cell and the APC, such as CD28 on the T cell and CD80 (B7.1) and CD86 (B7.2) on the APC (Santana and Rosenstein, 2003). An elegant recent study by Kleinschnitz et al (2010b) indeed found evidence that the antigen-dependent activation of T cells is not required for them to contribute substantially to the infarct volume present at 24 hours after cerebral I—R. Through the use of various transgenic mice, including Rag1−/− mice reconstituted with CD3+ T cells, TCR-transgenic mice (bearing a single CD4+ or CD8+ TCR) or mice lacking costimulatory molecules, it was found that neither the first signal (antigen recognition) nor the second signal (costimulation) of classical T-cell activation was required for the T-cell-dependent damage to occur after stroke (Kleinschnitz et al, 2010b). That is not to say that previously activated T lymphocytes, for example, due to preexisting infection or even cardiovascular disease (Andersson et al, 2010; Guzik et al, 2007), could not cause additional damage in the brain following I—R. The mechanism(s) of this antigen-independent T-cell ‘activation' within hours after stroke are currently unknown. On the other hand, antibodies to neuroantigens reportedly increase following stroke (Bornstein et al, 2001; Dambinova et al, 2003), as do myelin basic protein reactive T cells (Wang et al, 1992). Furthermore, another two studies reported that administration of the recombinant TCR ligand, RTL551 (which blocks classical antigen-dependent T-cell activation) linked to a CNS antigen, resulted in a reduced infarct volume following cerebral I—R (Dziennis et al, 2011; Subramanian et al, 2009). Because this protective effect only occurred when RTL551 was linked to a neuroantigen, rather than a nonneuronantigen (Subramanian et al, 2009), it suggests that an adaptive immune response to brain antigens occurred following stroke, and that classical T-cell activation may indeed have contributed to postischemic brain damage. Moreover, tolerance against brain antigens by mucosal administration of the antigen before stroke has been reported to improve outcome after stroke (Becker et al, 1997, 2003; Frenkel et al, 2005; Gee et al, 2008), further suggesting that antigen-dependent lymphocyte activation occurs following stroke, and that it contributes to brain injury. Therefore, the contribution of antigen-dependent T-cell activation in the damaging effects of T cells acutely poststroke remains unclear.
T-lymphocyte-targeted experimental therapies
Anti-α4 Integrin Antibody
Several studies have sought to prevent T-cell infiltration into the brain after stroke, for example, by targeting the α4 integrin with neutralizing antibodies (Becker et al, 2001; Relton et al, 2001). α4 Integrin is part of the VLA-4 protein that is expressed on > 70% of all leukocyte populations, including T cells (Liesz et al, 2011b), and which binds to VCAM-1 expressed on endothelial cells. Vascular cell adhesion molecule-1 mRNA (Jander et al, 1996) and protein (Justicia et al, 2006; Liesz et al, 2011b) are reported to be strongly induced in the microvasculature of the ischemic area following cerebral ischemia, and this expression is important for T-cell infiltration into the brain (Baron et al, 1993; Engelhardt et al, 1995). These studies reported 31% to 58% smaller infarct volumes in animals treated with the anti-α4 integrin antibody (Becker et al, 2001; Relton et al, 2001), with an improved neurologic score at 24 and 48 hours also observed by Becker et al (2001). In both of these studies, there was a higher peripheral blood leukocyte count (with a lymphocyte/monocyte predominance) in the animals that received the anti-α4 integrin antibody compared with those receiving the isotype control antibody (Becker et al, 2001; Relton et al, 2001), consistent with a beneficial effect involving reduced extravasation of immune cells. Furthermore, another study that treated mice with a monoclonal antibody against the α4 integrin reported a reduced infarct volume at 7 days after a 30-minute I—R or permanent cerebral ischemia, as well as an improved sensorimotor activity 3 and 7 days after permanent ischemia (Liesz et al, 2011b). This protection was confirmed to occur via limiting the effects of lymphocytes, as α4 integrin blockade had no effect in Rag2−/− mice. In addition, in both TH and TC cell-depleted mice, α4 integrin blockade had no further effect on infarct volume, providing good evidence that the efficacy of α4 integrin blockade is mediated by targeting T cells (Liesz et al, 2011b). Interestingly, at 5 days after cerebral ischemia, the brain infiltration of all T-cell subsets examined (including TH, TC, Treg, and NKT cells) was significantly reduced, as were the infiltration of B cells and granulocytes. Together, these findings might suggest that even though multiple leukocyte types enter brain lesions after transient I—R, much of the (at least α4 integrin inhibitable) damage is mediated by T cells.
Anti-Vascular Cell Adhesion Molecule-1 Strategies
The means by which VCAM-1 is targeted appears to be critical in the outcome achieved by anti-VCAM-1 therapy after stroke. Intravenous or intraperitoneal administration of anti-VCAM-1 antibodies have been unsuccessful at protecting against ischemic brain damage either in rats or in mice (Justicia et al, 2006; Liesz et al, 2011b). While VCAM-1 blockade reduced the number of monocytes/macrophages/reactive microglia present in the ischemic rat brain (Justicia et al, 2006), it did not reduce the numbers of infiltrating neutrophils and lymphocytes (Justicia et al, 2006; Liesz et al, 2011b). In contrast, when utilizing hydrodynamic in vivo administration of VCAM-1 small interfering RNA, granulocyte and T-cell infiltration into the brain was reduced in association with a reduced infarct volume at 6 days after stroke. Moreover, after this in vivo silencing of VCAM-1, anti-α4 integrin antibody administration produced no further reduction of infarct volume, suggesting that VCAM-1 is the main endothelial receptor for VLA-4 on infiltrating T lymphocytes (Liesz et al, 2011b). Interestingly, a very recent study found that a high plasma level of ALCAM in patients with acute ischemic stroke was predictive of a poor prognosis, raising the possibility that anti-ALCAM strategies might be considered clinically for acute stroke therapy (Smedbakken et al, 2011).
FTY720
Another widely tested therapeutic in experimental stroke is the immunosuppressant FTY720, a stable analog of the lipid signaling mediator sphingosine 1-phosphate. Many experimental stroke studies have found reductions in infarct volume (Czech et al, 2009; Hasegawa et al, 2010; Pfeilschifter et al, 2011a, b ; Shichita et al, 2009; Wei et al, 2011), and improvements in functional outcome (Czech et al, 2009; Hasegawa et al, 2010; Pfeilschifter et al, 2011a, b ; Wei et al, 2011) following administration of FTY720, a drug known to sequester lymphocytes in lymph nodes, preventing them from moving to the CNS for autoimmune responses in multiple sclerosis (Mehling et al, 2011). One study even found improvements in functional outcome up to 15 days after cerebral ischemia (Wei et al, 2011). Shichita et al (2009) found FTY720 to inhibit T-cell infiltration into the infarcted zone, and others have reported fewer infiltrating neutrophils and activated microglia/macrophages in the ischemic lesion (Czech et al, 2009; Pfeilschifter et al, 2011b; Wei et al, 2011). Furthermore, FTY720 was found to reduce apoptotic cell death in the ischemic hemisphere (Czech et al, 2009; Hasegawa et al, 2010; Wei et al, 2011) as well as the expression of intercellular adhesion molecule-1-positive blood vessels in the brain (Wei et al, 2011). However, a recent study could not detect any reduction of infarct volume or behavioral dysfunction following permanent cerebral artery occlusion despite a reduced lymphocyte brain invasion after FTY720 treatment (Liesz et al, 2011a). This lack of neuroprotection despite effective lymphopenia was suggested to be due to a divergent impact of FTY720 on cytokine expression and possible activation of innate immune cells after brain ischemia (Liesz et al, 2011a).
FK506 and Cyclosporin A
Other immunosuppressive drugs, such as FK506 (Tacrolimus) and CsA (Cyclosporin A), have also been tested in experimental stroke studies. The immunosuppressive actions of FK506 and CsA involve the inactivation of NFAT (nuclear factor of activated T cells) via the inhibition of calcineurin. FK506 was first shown by Sharkey and Butcher (1994) to be a powerful neuroprotective agent in an in vivo model of focal cerebral ischemia when administered up to 60 minutes after occlusion. The minimum effective neuroprotective doses of FK506 and CsA are comparable with the immunosuppressant doses in humans, however, suggesting that a broad immunosuppressive effect predisposing to infection may complicate the clinical use of these drugs as a treatment for stroke (Bochelen et al, 1999; Brecht et al, 2003, 2009; Li et al, 2006; Sharkey et al, 1996; Uchino et al, 1998; Vachon et al, 2002; Yoshimoto and Siesjo, 1999).
RTL551
As mentioned above, two recent studies reported that postischemic administration of a recombinant TCR ligand (RTL551, which acts as a partial agonist at the TCR and blocks T-cell activation) linked to a CNS antigen, reduced cerebral infarct volume by up to 33% at 96 hours (Dziennis et al, 2011; Subramanian et al, 2009). These protective effects were associated with less brain infiltration by T cells, B cells, NK cells, and macrophages/microglia, and especially dendritic cells and activated microglia/macrophages (Subramanian et al, 2009), as well as fewer activated TH (CD44+CD4+) cells in the circulation (Dziennis et al, 2011).
Chemokine Receptor Antagonists
Chemokine receptors could be attractive novel targets for modulating T-cell-mediated damage poststroke. Chemokine receptors are expressed on leukocytes and, by binding with a high degree of specificity to chemokines released from damaged tissues, they promote the migration of leukocytes to sites of injury (Gerard and Rollins, 2001; Mackay, 2001). Several chemokines are upregulated in the brain following cerebral I—R in mice, underpinning the attraction of different leukocyte subtypes into the brain (Brait et al, 2011). We have provided proof-of-concept that inhibition of chemokine/chemokine receptor interactions with a small molecule chemokine receptor antagonist (SB225002, a CXCR2 antagonist) can modulate the leukocyte profile in the brain following stroke (Brait et al, 2011). We speculate that more detailed identification of the specific chemokine/chemokine receptor interactions that are involved in the attraction of damaging T-cell subsets (and any other leukocytes responsible for poststroke brain injury) may enable selective inhibition of their infiltration into the brain following stroke, without preventing the entry of those T-cell subsets that are involved in brain repair and regeneration (see below). Moreover, targeting T-lymphocyte migration into the brain following stroke (as opposed to targeting their activation) may have the added advantage of maintaining the levels of these cells in the periphery where they are needed to fight infections.
Evidence for T lymphocyte involvement in brain regenerative processes after stroke
Following the acute pathological events that occur in the hours to days after an ischemic stroke, regenerative processes associated with neurologic recovery take place in the brain, over several weeks or even months. During this process, the brain repairs and reorganizes in a manner similar to that which occurs during the early stages of development (for review see Cramer and Chopp, 2000). The creation of new neural networks through neurogenesis, neuroplasticity, synaptogenesis, angiogenesis, and gliogenesis helps to repair and reorganize the brain. Inflammation has been suggested to have a key role in the promotion of these reparative processes (for review see Kriz and Lalancette-Hebert, 2009; McCombe and Read, 2008; Wieloch and Nikolich, 2006), mainly through the release of growth-related proteins and cytokines, potentially from T lymphocytes as well as other peripheral and resident immune cells. Neurogenesis occurs continually in the hippocampus throughout adult life (Cameron and McKay, 2001), but when T cells are depleted, this neuronal cell proliferation is impaired, suggesting that T cells are required for neurogenesis (Wolf et al, 2009; Ziv et al, 2006). This appears to be specifically due to the actions of CD4+ and not CD8+ T cells (Wolf et al, 2009), and it occurs through classical CNS antigen-dependent T-cell activation (Ziv et al, 2006; Moalem et al, 1999) via the release of neurotrophic factors, such as nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 (Moalem et al, 2000). Thus, CNS-specific CD4 + T cells could be involved in the promotion of neurogenesis after brain injury, such as in stroke, and antigen-activated T cells at the site of injury could have a role in the repair of damaged tissue.
By contrast, a recent study by Saino et al (2010) reported that CD4+ T cells in the brain might inhibit neurogenesis following stroke. In mice depleted of CD4+ (but not CD8 +) T cells, there was a greater proliferation of neurons at 28 days after stroke. However, it is important to note that these studies were performed using a model of permanent ischemia, and it is possible that the effect of T cells is different if reperfusion does not occur. Interestingly, when Treg cells are selectively depleted, neurogenesis is also reduced (Saino et al, 2010), suggesting that Treg cells may be a key CD4+ subpopulation of T cells that is required for neurogenesis. Moreover, Treg cells are also reported to modulate postischemic neovascularization following femoral artery ligation (Zouggari et al, 2009), and so any similar action by Treg cells to promote cerebral neovascularization after stroke would represent a further important role by this subset of T cells in the recovery of brain function following ischemia.
Complications and limitations to be considered in targeting T lymphocytes as poststroke therapy
With tPA (tissue plasminogen activator) still being the only therapy available for acute ischemic stroke patients, novel effective therapies that can be administered beyond 4.5 hours are desperately needed. As acute inflammation appears to occur in the brain for many hours or even days after ischemic stroke (Dirnagl et al, 1999), targeting key circulating immune cells to prevent their activation and/or extravasation would, at face value, seem to be a rational approach. Although therapies that target T cells in this manner relatively soon after stroke may reduce brain injury and improve poststroke outcome, there are several potential complications and likely limitations to the usefulness of T-cell inhibition after stroke. First, in the short term, as the profound systemic immunodepression that rather quickly follows ischemic stroke will already have substantially weakened the immune system and thus increased the risk of infection (Chamorro et al, 2007; Harms et al, 2011; Klehmet et al, 2009; Prass et al, 2003; Urra et al, 2009), further pharmacological approaches that attenuate immune cell function beyond this point (possibly within only a few hours) may do more harm than good by exacerbating the immunodepression. It should be noted that infection is the most common cause of death in the postacute phases of stroke with 23% to 65% of all stroke patients acquiring infections within the first few days (Heuschmann et al, 2004; Vernino et al, 2003). Second, in the longer term (i.e., days to weeks) as mentioned above, inflammation—including via effects of T cells—appears to eventually have a role in promoting brain regeneration in the postacute phase of an ischemic stroke (for review see Kriz, 2006). For example, neurogenesis is reported to begin 5 days after stroke (Wieloch and Nikolich, 2006), and thus any therapy that interfered with these processes would be contraindicated. Third, it is unclear how important T cells are as contributors to acute brain injury after ischemic stroke in the absence of effective reperfusion (either spontaneous or tPA-induced), which still represents the majority of clinical cases. If their role is minimal, then anti-T-cell therapy may only be appropriate to be given in combination with successful reperfusion by tPA. Fourth, the possibility of blocking T-cell subtypes with important antiinflammatory actions (e.g., Treg cells or NKT cells) would also be a limitation to its efficacy. More research is therefore needed to clarify the plausibility of safely and effectively inhibiting T-cell-mediated brain injury in the early stages after stroke, and perhaps even at later times using mild pro-inflammatory treatments (including neurotrophic factors) to promote repair and regeneration (for review see Kriz, 2006; Wieloch and Nikolich, 2006). Additionally, the recent work by Wong et al (2011) has raised the possibility of using selective activators of NKT cells to prevent stroke-associated infections.
Summary and conclusions
In the last 5 years or so, there have been substantial advances in our understanding of the pathogenic role of T lymphocytes in ischemic stroke. These cells, traditionally belonging to the adaptive immune system, are now established to contribute to the infarct volume in the postischemic brain. Current evidence suggests that following an ischemic stroke, a systemic immunodepression occurs, T lymphocytes enter the brain and release cytokines/chemokines and superoxide, and contribute substantially to neuronal injury. Importantly, some types of T cells may be beneficial (e.g., Treg cells and NKT cells), and T cells may also have a longer-term role in general regenerative processes that occur in the later stages following an ischemic stroke. T lymphocytes may produce their damaging acute effects following activation via a nonclassical, antigen-independent process but this point is still controversial. After 7 to 10 days, T lymphocytes may additionally become activated in the classical manner, producing neurotrophins and contributing to neurogenesis. In conclusion, the role of T lymphocytes in ischemic stroke is complex and remains poorly understood. More research is needed to gain a greater understanding of which T-cell subpopulations produce the most damage, how and when this occurs, and when T cells may cease causing damage and begin contributing to regeneration. With this information, it will become apparent whether therapeutic targeting of T lymphocytes is likely to offer benefits for stroke patients.
Footnotes
Acknowledgements
CGS and GRD are Senior Research Fellows of the National Health and Medical Research Council of Australia (350327 and APP1006017). TVA is a Future Fellow of the Australian Research Council (ARC FT100100427).
The authors declare no conflict of interest.