
Abstract
Analogous to Toll-like receptors, NOD-like receptors represent a class of pattern recognition receptors, which are cytosolic and constitute part of different inflammasomes. These large protein complexes are activated not only by different pathogens, but also by sterile inflammation or by specific metabolic conditions. Mutations can cause hereditary autoinflammatory systemic diseases, and inflammasome activation has been linked to many multifactorial diseases, such as diabetes or cardiovascular diseases. Increasing data also support an important role in different central nervous diseases such as stroke. Thus, the current knowledge of the functional role of this intracellular ‘master switch’ of inflammation is discussed with a focus on its role in ischemic stroke, neurodegeneration, and also with regard to the recent data which argues for a relevant role in other organs or biologic systems which influence stroke incidence or prognosis.
INTRODUCTION
The intracellular multiprotein complex inflammasome was shown to be involved in various systemic diseases since its initial description more than 10 years ago, including a group of hereditary autoinflammatory diseases. 1 Caspase-1 activation and interleukin-1 (IL-1) release have a central role in many disorders with an inflammatory component, 2 which comprises different diseases of the central nervous system (CNS) such as multiple sclerosis, Alzheimer disease, meningitis, or stroke.3–8
The potential molecular signals, which activate the inflammasomes in the CNS, have already been delineated in detail before.8,9 Thus, this overview aims at a more global, system biologic view of the potential role of the inflammasomes in CNS diseases and accounts for recent discoveries in related research fields. A pure isolated molecular view probably does not match with the complex resulting holistic effects of the activation of the inflammasomes during brain injury. These effects are sustained by complex interactions of multiple systems, multiple pathways, and also multiple diverse inflammasome types, influencing each other in a specific pathogenic situation and also over time. Nevertheless, there are examples where a single dysfunctional inflammasome component can cause complex inflammatory disorders or is able to modulate the incidence or the severity of autoimmune-mediated disorders.10,11
The same is true for another family of pattern recognition receptors of the innate immune system: the membrane-linked Toll-like receptors (TLRs). These receptors were initially characterized as controllers of development. 12 TLRs were later shown to also be responsible for the activation of the innate immune system, 13 the initialization and fine tuning of the adaptive immunity,14,15 the modulation of the microbiome composition in the gut, 16 and the regulation of the metabolism and the autonomic nervous system. 17
Both, TLRs as well as the cytosolic NOD-like receptors (NLRs), which are thought to represent the receptor subunits of the inflammasomes, mediate the activation of the immune system. TLRs survey the endosomes and extracellular milieu, whereas NLRs are mainly cytosolic. However, it remained unresolved up until now whether the inflammasomes are activated by direct binding of danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns or by an indirect mechanism involving a second messenger. Recent reports have shown a functional relevant attachment of NLRs to intracellular organelles such as mitochondria or endoplasmic reticulum. 18 Inflammasome activation is not only influenced by the localization of specific inflammasome subunits, their spatial vicinity, the composition of expressed subunits (e.g., NALP1-inflammasome versus NALP3 inflammasome), or their cell-specific expression, but is also tightly regulated by kinases and inhibitory molecules.8,9,19
The following chapters will discuss the different roles of the inflammasome in central nervous disorders in more detail. The focus will be on stroke, which is the second leading cause of death worldwide. 20
STROKE: INTERPLAY WITH THE IMMUNE SYSTEM
The significance of the immune system in stroke pathophysiology is well recognized. 21 The incidence of stroke is affected by systemic proinflammatory conditions as shown in several clinical studies, but brain injury also by itself initiates a local inflammatory response and systemic inflammation determines stroke prognosis.21,22 However, severe strokes also cause an immune depression, thereby increasing the frequency of systemic infections and the mortality of stroke. 23 Thus, it is highly relevant to understand the interplay between brain injury, activation of the immune system, and the resulting consequences for the long-term outcome.
Stroke represents an almost prototypical paradigm for sterile inflammation. The damage of a substantial mass of endogenous tissue caused by insufficient blood supply, starting at a specific time, initiates a local immune response which targets at different aims: limiting the damage, protecting the still vital adjacent tissue and cells, removing the cell debris and initiating the repair mechanisms to regain tissue homeostasis, as well as protecting against following challenges (preconditioning).21,24
The time point of the initiation of the following waves of immune response were precisely defined in experimental models of stroke, as well as the intensity of the brain injury (as determined by the duration of the occlusion time). Thus, the following events were easily monitored.21,24,25
The acute reduction of blood supply below the necessary threshold in ischemic stroke is thought to cause a plethora of different pathophysiologic consequences, which contribute to ischemic injury.21,24 The resulting energy depletion with loss of adenosine triphosphate (ATP) causes anoxic depolarization resulting in Ca++ influx and the release of excitatory neurotransmitters.
Production of reactive oxygen species (ROS) is increased by mitochondrial dysfunction and lack of energy metabolites but potentially also as a consequence of regulatory changes. Depending on the time and the degree of insufficient blood supply, there is also a more or less intense response of the innate immune system with activation of the complement system, the innate immune receptors, and invasion of immune cells.21,24
Short ischemic periods lead to an ischemic tolerance (preconditioning), which is thought to be a consequence of a reprogramming of cerebral tissue and the immune system. 26 Severe ischemic insults mediate a systemic immune suppression, which is coupled to the activation of the sympathetic nervous system and which increases the risk of following systemic infections. 23 Local release of proinflammatory cytokines in the damaged tissue, activation of immune active cells, and induction of adhesion molecules cause a rapid infiltration of immune cells, particularly neutrophils and monocytes/macrophages. 25 This inflammatory cascade is thought to contribute to tissue injury, but is also hypothesized to be necessary for plasticity and reestablishing tissue homeostasis or wound healing.
Since the process of neuronal death after cerebral ischemia is only partially reversed by rapid reperfusion of the occluded or stenotic vessel in ischemic stroke, the major aim of current neuroprotective treatment concepts is to delay the ischemic brain injury or protect from ischemia-induced cell death.
However, most—if not all—proven concepts of successful neuroprotection in experimental stroke research failed in the clinic. 27 This discrepancy may be because of wrong models, differences of the immune systems in different species, publication bias, or selection of the wrong outcome parameters. It is increasingly recognized, for example, that several processes, detrimental short time (e.g., TLR signaling), may be beneficial long term.28,29 Thus, a more profound knowledge of immune mechanisms in stroke is required, which takes into account opposing effects in different cell types, different organs, at different doses, and at different time points.
It is well known that systemic inflammation can increase the rate of cardiovascular events, but also correlates with a worse prognosis after acute injury.21,30 Inflammation mediates the development of atherosclerotic lesions, the genesis of the metabolic syndrome, as well as the development of ischemic brain damage.
The central contribution of the inflammasome, with regard to regulation of the immune response after ischemic injury, is increasingly recognized and is based on the well-known role of IL-1-mediated effects in stroke.2–5,8,9,21 However, besides its contribution in acute brain injury, the NLRP3 inflammasome was also shown to be involved in atherogenesis, which represents a major risk factor for ischemic stroke.31,32 Moreover, inflammasome-mediated mechanisms were also shown to instigate obesity-induced inflammation and insulin resistance. 33
A ‘crosstalk’ between different organs, such as the central nervous system, the thymus, or the gut has been identified: e.g., rapid apoptotic thymocyte cell death after stroke leads to severe depression of the immune system. 23 Recent studies note that the immunologic barrier in body surfaces not only protects against invasion of foreign pathogens, but is also able to modulate the composition of commensal microbiota.10,16 Alterations of the gut microbiota are able to influence the severity and frequency of various autoimmune diseases such as colitis, immune hepatitis,10,34 or experimental autoimmune encephalomyelitis. 35
Thus, it is increasingly recognized that not only local molecular mechanisms of stroke pathophysiology in the ischemic brain territory have to be taken into account when analyzing the influence of the immune system, but also systemic factors, such as the ‘bacterial burden’ on diverse body surfaces, the basic inflammatory level, as well as the reactivity of the immune system, the state of the autonomous nervous system, and corresponding changes of each system over time, weighted by its relative significance.
INFLAMMASOME: INTRACELLULAR SENSOR FOR DANGER SIGNALS AND ISCHEMIC INJURY
The former view of the central purpose of the immune system to discriminate between infectious nonself from non-infectious self 36 has been modified towards a stress-, or danger-related view.37,38 The danger model allows an explanation for the neuroinflammatory response in sterile injury such as cerebral ischemia, bleeding, or traumatic brain injury. Receptors for danger molecules released by cells under pathologic circumstances have been identified as parts of the molecular mechanisms underlying these abilities of the innate immune system: the TLRs, the RIG-like receptors, the C-lectin like receptors, and the NLRs. Other parts of the innate immune system such as the complement system, adhesion molecules, different immune cells, and cytokines were also shown to contribute to ischemia-reperfusion injury (e.g., reviewed by Iadecola and Anrather, 2011). 21 Moreover, these different factors influence each other and signaling pathways have been identified between the TLRs, the NLRs, and the complement system.39–42
Clinical studies have identified a time window of 4.5 hours for systemic thrombolysis and early recanalization and reperfusion in ischemic stroke as mandatory. 43 Reperfusion after cerebral ischemia restores the necessary blood flow after insufficient supply with energy metabolites and oxygen, and it also contributes to an inflammatory response with invading immune cells, activated endothelial cells, and thereby leads to a further progress of ischemic injury by inflammatory mechanisms. However, a strong neuroinflammatory response could also be observed in stroke models without reperfusion (permanent cerebral ischemia). The signals that mediate this immune activation are only poorly defined. Endogeneous DAMPs released during stroke or other conditions of sterile inflammation are thought to activate pattern recognition receptors such as TLRs and NLRs. These were expressed in both leukocytes as well as activated endothelium, but also in selected neurons and even in platelets. 44
A large amount of reports define the role of TLRs in ischemic brain injury and neurodegenerative diseases, whereas the consequences of NLR-activation during brain injury are only partially understood up until now.
The classic two-step model of inflammasome activation (Figure 1) comprises an initial ‘priming step’, which is normally derived by TLR-stimulation and NFkB-mediated induction of pro-IL-1β mRNA and potential transcripts of inflammasome subunits, such as ASC (apoptosis-speckle like protein containing a CARD domain) or NLRP3. The second stimulus then activates the inflammasome either via direct binding of DAMPs to NLRs (e.g., as it was shown for the bacterial ligand flagellin, which directly binds IPAF/NLRC4, or DNA which binds to AIM2), or via a potential interconnected signal (e.g., mitochondrial dysfunction, release of ROS, cathepsin, K+ efflux, or a yet unknown signal). 45 Most NLRs possess a N-terminal pyrin domain (PYD) that interacts with the PYD of ASC to bridge the complex to pro-caspase-1, which is then activated by proximity-based autocatalytic cleavage. TLRs were initially identified in drosophila, whereas the inflammasome was initially described in mammalian cells. 1 Characterization of inflammasome-activation in simple model systems such as yeast are now revealing fascinating basic mechanisms of innate immune defense by prion-like protein polymerization of inflammasome subunits such as ASC. 46 Together with the work of Lu et al (2014), 47 a unified model of ASC-dependent inflammasome activation evolved: after an insult reaches a threshold, NLRP3 is released from autoinhibition, allowing the interaction of its PYD domain with the PYD domain of ASC, which results in ASC-prionlike nucleation. This perpetuates independently to form large stable ASC filaments that are required and sufficient for inflammasome activation.46–48 Cell-free in vitro experiments confirmed the important role of the expression level of the inflammasome subunits and the potassium concentration, activating factors which have been already realized before.49–51 However, the consequences of this prion-like polymerization for perpetuation of autoinflammatory diseases, or atherosclerosis, and the significance of inflammasome degradation by autophagy 52 still remain to be elucidated.
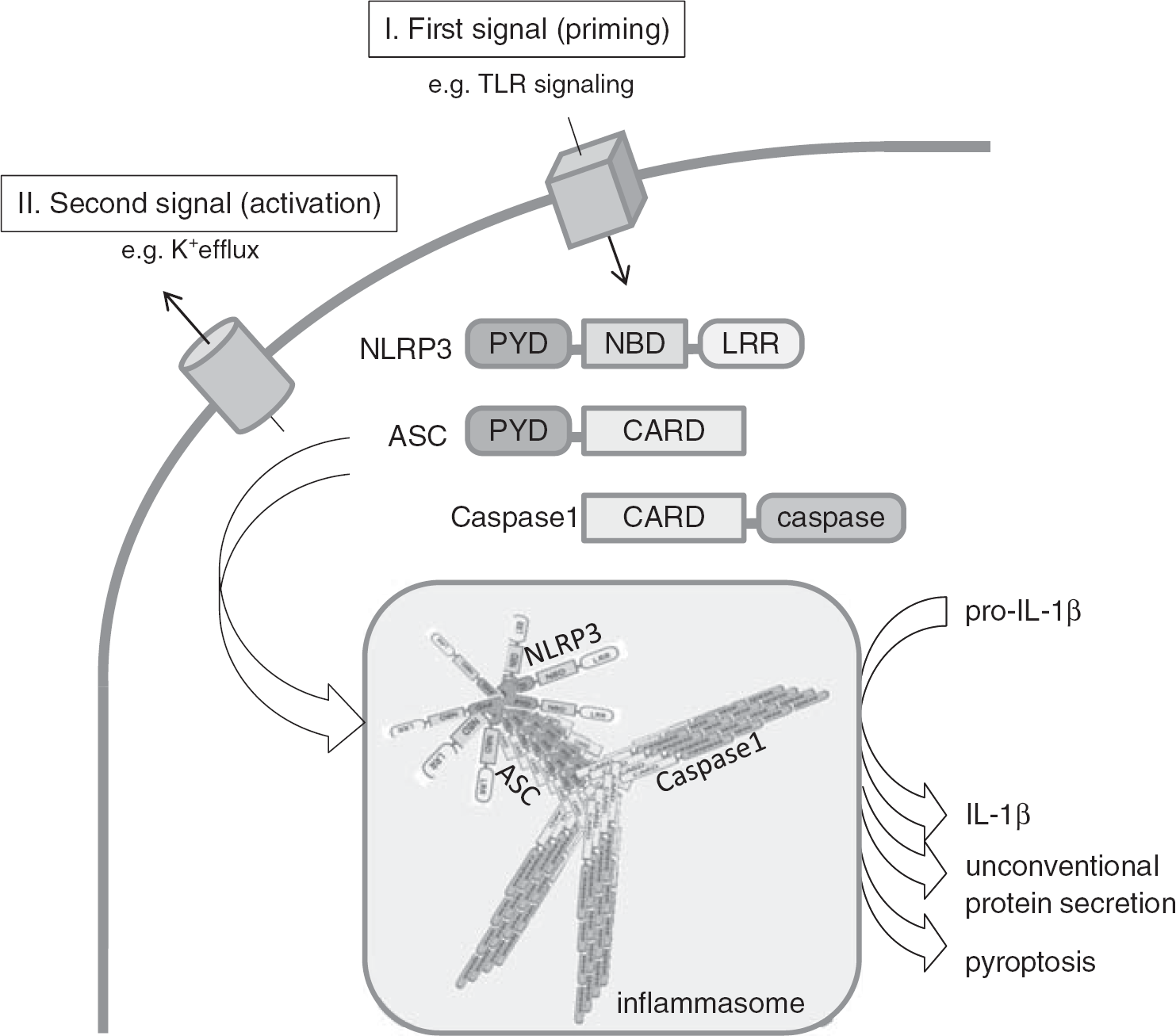
Inflammasome activation by prion-like polymerization. Inflammasomes (e.g., the NLRP3 inflammasome) are activated by a wide range of pathogens or danger signals, including ATP (adenosine triphosphate) and uric acid crystals. A fist signal (‘priming step’), which could be mediated via Toll-like receptors, leads to NFkB-mediated induction of pro-IL-1β mRNA and potentially other inflammasome components. A second signal deactivates the autoinhibition of the NOD-like receptor (NLR; e.g., NLRP3)—most probably by a conformational change—and initiates a self-perpetuating polymerization process of ASC (apoptosis-speckle like protein containing a CARD domain) and caspase-1 proteins, which were recruited via homodimeric binding of their domains (ASC binds NLRP3 via PYD (pyrin domain), and pro-caspase-1 binds ASC via CARD domains). The resulting prion-like polymerization activates caspase-1 and allows a highly sensitive and robust ‘digital’ response to harmful insults. 46
Interestingly, inflammasome activation is also influenced by another part of the innate immune system beyond the well-known interplay with the TLRs: the complement system. The complement subunit C3a has also been demonstrated to regulate NLRP3-mediated IL-1β release in human monocytes by binding to C3a-receptor and via ATP efflux. 53 Moreover, C5a and TNF (tumor necrosis factor) were shown to act as potent primers for cholesterol crystal-induced IL-1β release. Thus, cholesterol crystals use the complement system to induce cytokines and activate the inflammasome. 41
The activation of the inflammasome is also triggered by dysfunction of intracellular organelles, such as the mitochondria 19 or the lysosome. Accordingly, lysosomal membrane rupture after exposure to cell-toxic aggregates such as monosodium urate, or amyloid-beta in Alzheimer's disease activate the inflammasome.6,54 Moreover, a direct link between the intracellular metabolic homeostasis and degree of inflammation has been described involving the inflammasome: inflammasome activation depends on the degree of tubulin-acetylation, NAD+-level, and is triggered in conditions of stress or hyperglycemia. 19 Translocalization of NLPR3 to mitochondria upon activation was shown to require mitochondrial antiviral signaling protein. 55 Stress of the endoplasmic reticulum has also been added recently to the list of conditions, which have been shown to regulate the inflammasome: 56 improperly folded proteins within the endoplasmic reticulum can activate caspase-1 and maturation of IL-1β in the absence of the ‘classic’ inflammasome subunit ASC and without the need of MyD88, but dependent on TRIF and caspase-8. 56 The proapoptotic caspase-8 also mediates upregulation of NLRP3, inflammasome assembly and caspase-1 activation during wound repair. 57 In addition to intersections with the inflammasome activation, caspase-8 has recently been shown to be involved in the regulation of necroptosis 58 (Figure 2). A noncanonical caspase-8 inflammasome is involved in antifungal defense and requires Dectin-1, a C-type lectin receptor, 59 thereby representing a further example for a crosstalk and similarities between different pattern recognition receptor families. Members of the C-type lectin receptor family also sense DAMPs and pathogen-associated molecular patterns, and a subset of these C-lectin like receptors activate the tyrosine kinase Syk (e.g., Clec9a, which recognizes the spliceosome-associated protein SAP-130 upon cell damage), 60 but some of them also negatively regulate inflammation (e.g., Clec12a, which binds monosodium urate crystals and inhibits inflammation in response to cell death). 61
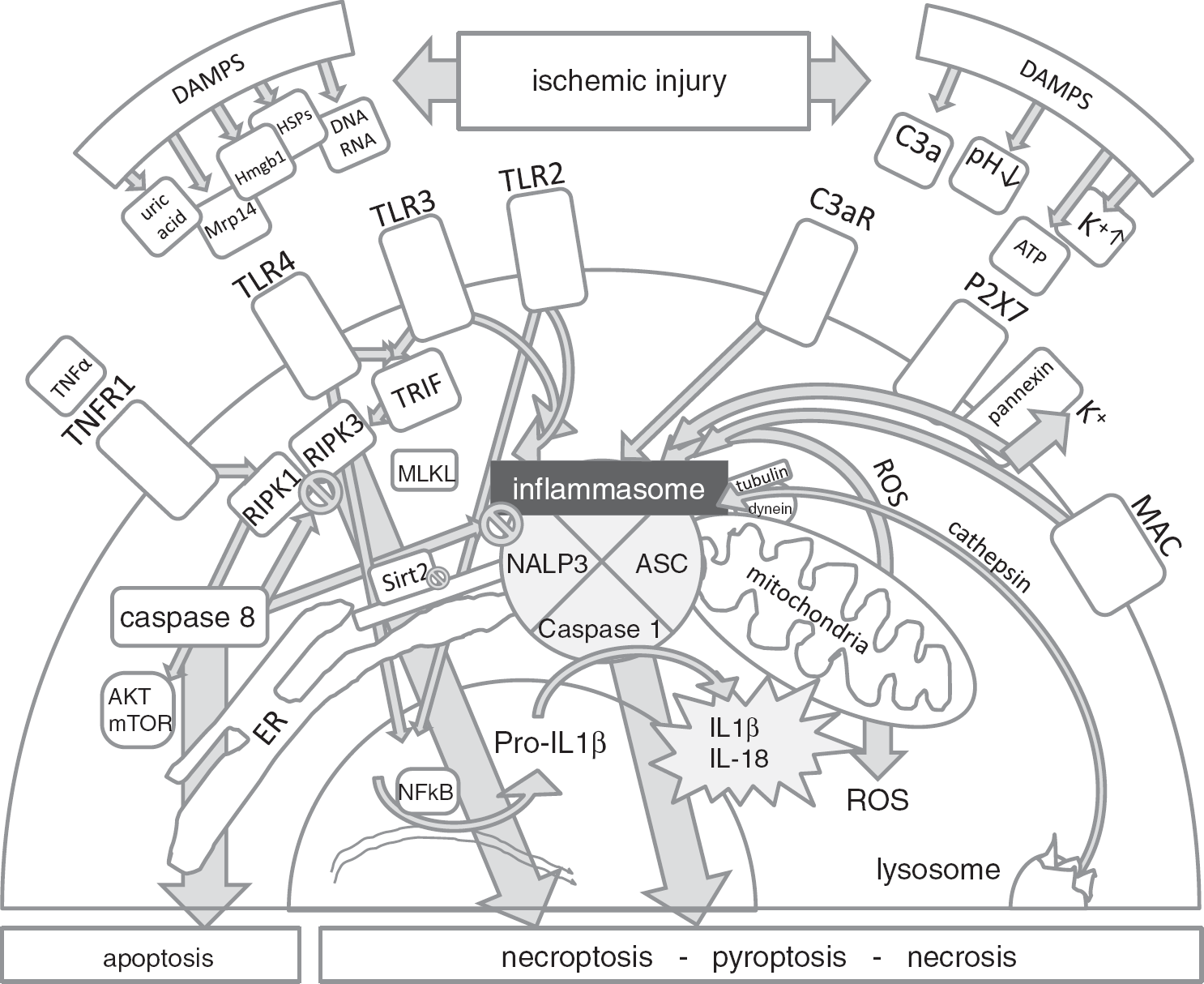
Molecular crosstalk leading to programmed cell death and/or activation of the inflammasome in cerebral ischemia. Several danger signals (DAMPS) could potentially mediate the activation of the inflammasome in cerebral ischemia: e.g. release of Toll-like receptor agonists such as Hmgb1 or heat-shock proteins (HSPs), MRP14, mitochondrial DNA, uric acid, or changes of pH or potassium. But intracellular stress in the endoplasmic reticulum (ER) could also regulate inflammasome activation by the degree of acetylation of intracellular proteins. Other parts of the innate immune system, such as the complement system can activate the inflammasome by sublytic concentrations of terminal membrane attack complex (MAC) or via activation of C3aR. Caspase-8 determines if cells die by necrosis or by apoptosis. 58 cAkt/mTOR being involved downstream of RIPK1 in necroptotic signaling in neurons was shown by Liu et al (2014). 89 The regulatory role of capase 8 was demonstrated by Kang et al (2013). 86 TRIF links TLR3/4 with RIPK3, whereas RIPK1 links TNFR1 (tumor necrosis factor receptor 1) with RIPK3. 58
The subunits of the inflammasome, which are necessary to constitute a full functional active inflammasome, are differentially expressed in different cell types. For example, the NLRP3 inflammasome is mainly expressed in immune cells in the CNS, whereas the NLRP1 inflammasome is mainly expressed in neurons.62–65 However, induction of both, NLRP1 and NLRP3 inflammasomes in ischemic neurons during stroke has been shown. 4 Making it more complex, certain human and murine inflammasome subunits differ substantially in their structure: in contrast to human NLRP1 inflammasome, murine NLRP1 is devoid of a PYD domain, which is thought to be essential to bind the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) to activate caspase-1. 66 Other subunits of inflammasomes, such as capase-5 or CARDINAL only exist in humans.11,45,66 Furthermore, the role of IL-1β and caspase-1 as central parts of the neuroinflammatory response after cerebral ischemia needs to be revised in light of the fact that ‘caspase-1 knockout’ mice are actually caspase-1 and caspase-11 double knockout mice because they harbor a mutation in the caspase-11 locus, as mice from strain sv129 do. 67 This is relevant also for the translation of experimental data to humans, as human caspase-4 and caspase-5 arise from mouse caspase-11 by duplication. 66
As expected, fine-tuning of the activation of the inflammasome (as well as its inhibition) is modulated by various pathways and proteins. Inflammasome activity is not only regulated by transcriptional mechanisms but also posttranscriptionally by the degree of phosphorylation, ubiquitination, acetylation, or nitrosylation.19,52,68–70 TLR-mediated transcriptional regulation of inflammasome subunits contributes to the degree of inflammasome activation, but the amount of aggregated terminal membrane attack complexes of the complement system also determine if a cell is condemned to die or if inflammasome-mediated inflammation is triggered. 42
Similar dose-dependent mechanisms may represent the molecular correlate of the in vivo observation. The strength of a stimulus (e.g., the duration of the vessel occlusion in stroke) could lead to either protection (ischemic preconditioning) or severe brain injury, but may involve the same signaling pathways. 71 The significance of TLR signaling for both possibilities (protective versus detrimental) has already been demonstrated.72–76 It still remains to be shown if NLR-mediated signaling owns similar properties.
CELL FATE UNDER ISCHEMIC STRESS: APOPTOSIS, REGULATED NECROSIS, NECROPTOSIS, AND PYROPTOSIS
Necrotic cell death is a central event in a wide variety of pathologic conditions, including stroke.24,77 Necrosis is thought to occur in the infarct core of ischemic stroke, whereas apoptotic cell death is proposed to be dominating where local energy resources allow a more controlled death of cells without the need of a massive inflammatory reaction. Necrosis activates the inflammasome: 7-Bromoindirubin-3'-oxime, an indirubin oxime derivative induces necrosis and it is a potent inducer of inflammasome activation in a model of sterile inflammation in airway epithelia. 78 Until recently, necrosis was thought to be an uncoordinated form of cell death in situations where programmed cell death (apoptosis) is not feasible. In contrast to that, recent studies revealed that even necrosis could be induced in a regulated fashion by specific pathways and could be triggered by mitochondrial permeability transition, or by receptor-induced pathways, involving receptor-interacting protein kinase (RIPK)1- or RIPK3-mediated signaling.58,77–81 A regulated form of cell death in macrophages called pyroptosis was shown to depend on ASC polymerization and inflammasome activation, 82 but was shown to depend on caspase-11 rather than on caspase-1. 67
Programmed necrosis, or necroptosis (a form of receptor-mediated programmed cell death which morphologically resembles necrosis), shares some features with all three major established mechanisms of cell death: apoptosis, necrosis or type 2 autophagic death.77,83 Teleologically, necroptosis may function as a cellular ‘backup’ mechanism to ensure the elimination of damaged cells under stress conditions when apoptosis is inhibited and DAMPs have to be released to initiate a host immune response.77,83 It is characterized by a rise in cytosolic Ca2+, increased ROS, intracellular acidification, a depletion in ATP, and ultimately plasma membrane rupture, which involves proteins such as RIPK3, mixed lineage kinase domain-like protein, or transient receptor potential melastatin related 7 (TRPM7). 84
Interestingly, mice without RIPK3, which is the downstream partner of RIPK1 in necroptosis, are protected against ischemic kidney injury, and mice double deficient for RIPK3 and Cyclophilin (Cyp)D (acting in the mitochondrial permeability transition pathway) are even more protected. 79 The RIPK1-dependent necroptosis pathway also regulates ischemic injury in the brain. 77 The central role of necroptosis has been demonstrated by the reversal of the lethal phenotype of caspase-8-deficient mice on a RIPK3 knockout background.79,85 Caspase-8 has been shown to be a multivalent controller of innate immune signaling by the fact that it promotes extrinsic apoptosis, blocks the formation of the complex including RIPK1 and RIPK3 (also called the ripoptosome), which initiates programmed necrosis, and now has been further shown to also inhibit the NLRP3 inflammasome.86,87 The ripoptosome has been shown to induce programmed necrosis, but was also shown to activate the inflammasome. 88 One of the regulators is the AKT/mTOR (mechanistic target of rapamycin) pathway, which was shown not only to inhibit apoptotic cell death, but also to be downstream of RIPK1 activation in neuronal cell death. 89 Accordingly, necrostatin-1 (Nec-1), an inhibitor of a key regulator of programmed necrosis (RIPK1), was able to protect against cerebral and cardiac ischemic injury.77,90,91
It was shown by the use of RIPK3-deficient mice and transgenic RIPK3 mice, which lack a catalytic activity, that RIPK3 determines whether cells die by necrosis or apoptosis. 58 Tumor necrosis factor receptor 1, TLRs, and antigen receptors could trigger necroptotic cell death when caspase-8 is inhibited and cells express RIPK3.58,92 The kinase activity of RIPK3, but not RIPK1 was shown to be essential for caspase-8 regulation, and RIPK1 mutant mice without kinase activity are viable in contrast to the RIP1 - / - mice or the mutant RIPK3 mice without kinase activity. 58 Thus it would be interesting to see, if RIPK1 mutant mice without kinase activity or RIPK3 - / - mice are protected against ischemic stroke.79,81
Moreover, there are various examples for intersection points between inflammasome signaling and apoptosis-related pathways: e.g., it has been demonstrated that B cell lymphoma-2 and Bcl-XL could bind and suppress NLPR1 inflammasome, 93 and X-linked inhibitor of apoptosis protein was shown to directly interact with NLRP1 in neurons.94,95
However, to what degree each signaling pathway contributes to each stage of ischemic brain injury or each specific cell type still needs to be determined.
IN VIVO EVIDENCE FOR THE SIGNIFICANCE OF THE INFLAMMASOME IN ISCHEMIC INJURY
NLPR3 is expressed and induced after cerebral ischemia mainly in microglia and endothelial cells, 5 the same cells in which the NLRC4 inflammasome is also expressed. 96 In contrast to that, NLPR1 (de Rivero Vaccari et al 94 ) and AIM2 inflammasomes 97 are mainly expressed in neurons, whereas NLRP2 is expressed in astrocytes. 98 Sterile inflammation caused by necrotic cells is mediated by the NLRP3 inflammasome. 40 Accordingly, NLRP3 deficiency protects against neutrophil influx and tissue injury in a model of renal ischemia. 40 This is in agreement with the observation that inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury: 99 inhibition, silencing, or genetic deletion of NLRP3 in the mouse limited the infarct size in experimental acute myocardial infarction.100–102
Intracerebroventricular injection of different pro-caspase-1 inhibitors was shown to protect against ischemic injury up to 7 days after induction of cerebral ischemia in rodents31,103,104 and NLPR1 inhibition by the use of a blocking antibody demonstrated protective effects in a thromboembolic model 24 hours after induction of cerebral ischemia. 3 Accordingly, NLRP3 deficiency protects against experimental stroke: NLPR3-knockout mice have reduced infarct volumes, and there was reduced neuronal death in vitro, when co-incubated with OGD (oxygen glucose deprivation)-treated microglial cells with reduced NLRP3 expression. 5 Thus, inflammasome-dependent microglial neurotoxicity was shown to mediate neuronal apoptosis in vitro. 5
in vitro experiments with embryonic cortical neurons stimulated with cerebrospinal fluid of patients with traumatic brain injury demonstrated neuronal pyroptosis, which is mediated via AIM2 inflammasome and pannexin-1. 97 The significance of pannexin-1 channel-mediated inflammasome activation in neurons and astrocytes—which express a NLPR2 inflammasome—was shown before.95,98,105
Interestingly, there is also data on NLRP3 inflammasome activation in platelets during viral disease, 44 and platelets stimulated by TLR4 agonists release IL-1β in microparticles. 106 These microparticles have been shown to promote endothelial cell activation and endothelium permeability, thus contributing to the inflammatory response.44,106 Thus, it would be interesting to see if this cellular compartment also substantially contributes to proinflammatory mechanisms in acute stroke patients, possibly in the early phase, as platelets release IL-1β before monocytes. 106
DIFFERENT ORGANS MODULATE AUTOIMMUNE MECHANISMS IN THE CENTRAL NERVOUS SYSTEM
The local activation of the immune system is responsible to limit the harm of local injured tissue for the whole organism, but also to protect the body from pathogenic intruders. However, brain injury leads to a systemic immunodepression, and the resulting breakdown of the immune barrier increases the susceptibility to infections with a high mortality such as pneumonia. 107 CNS injury-induced immunodepression is mediated by the sympathetic nervous system and is characterized by a shift from T helper cell (Th)1 to Th2 cytokine production and a defective interferon gamma response. 23 IL-1β produced during cerebral injury may also contribute to immune depression by its effects on the hypothalamic-pituitary-adrenal axis, as corticotropin-releasing factor producing neurons were activated by IL-1,108 and IL-1-mediated effects on the immune system in rodents were shown to depend on both activation of the sympathetic nervous system and release of corticotrophin-releasing hormone. 109 Moreover, IL-1 receptor antagonist reverses stroke-induced peripheral immunosuppression in patients, demonstrating that IL-1 activity is—at least partially—responsible for stroke-induced immunosuppression.109,110 As Youm et al (2012) demonstrated, 111 age-related increase in ‘lipotoxic danger signals’ such as free cholesterol and ceramides, leads to thymic caspase-1 activation via the Nlrp3 inflammasome. Elimination of Nlrp3 and Asc, a critical adaptor required for inflammasome assembly, reduces age-related thymic atrophy. 111 Thus, it could be speculated that this mechanism also contributes to the massive apoptosis of thymic cells observed after severe stroke. 23
Interestingly, there are arguments for a bi-directional communication between the mammalian immune system and the bacteria on body surfaces. Composition of microbiota in the gut influences the severity of autoimmune encephalitis in experimental in vivo models. 35 Vice versa, the innate immune system is able to modulate the composition of the gut microbiota, as demonstrated by aberrant dysbiosis in TLR-deficient or NLRP-deficient animals.10,31,34 TLR4 also modifies the adaptive immune system by regulating the T-cell activation and survival during autoimmune inflammation: e.g., it has been shown that loss of TLR4 on CD4+ T cells abrogates autoimmune inflammation in models of multiple sclerosis. 112 A similar significance of IL-1β and inflammasome signaling for regulation of adaptive immunity has been demonstrated.113,114 There is emerging evidence that the inflammasome facilitates adaptive immune responses: ASC-deficient CD4+ T cells suppress the T-cell response upon CD3/CD28 stimulation, and ASC-deficient regulatory T cells produce an increased amount of IL-10 when compared with wild-type cells. 115
Thus, the receptors of the innate immune system have been shown to be essential in primary host defense against pathogens or dangerous conditions, but these receptors have also been shown to be essential in shaping the resident bacterial composition, in fine tuning the innate and adaptive immune response, and in orchestrating tissue repair (Figure 3).8,28,116
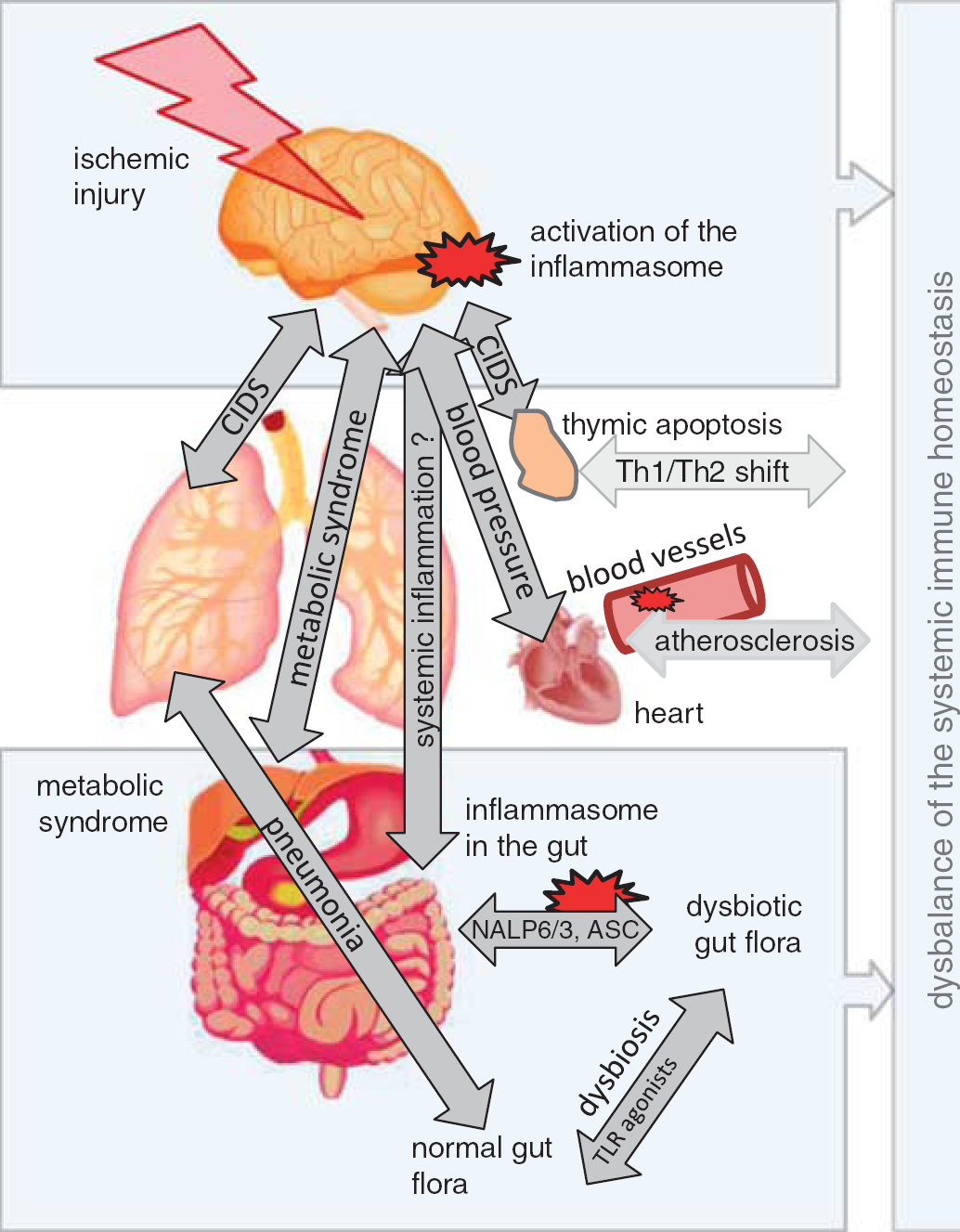
Systemic crosstalk between different organs in cerebral ischemia and influence of the immune system. The activity of the innate and adaptive immune system is modulated by brain injury, but also modifies the course of tissue injury and repair on various levels. Severe brain injury leads to a severe immunodepression (CNS injury-induced immunodepression (CIDS)), which alleviates the development of bacterial infections such as pneumonia. Activation of the sympathetic nervous system is thought to cause apoptotic thymocyte cell death and a T helper cell 1 (Th1)/Th2 shift of adaptive immune response. 23 Local inflammation in atherosclerotic lesions is sustained by cholesterol crystal-induced activation of inflammasomes and platelets could increase the endothelium permeability through NLRP3 inflammasome activation. 44 Inflammasome deficiency reduces the immune barrier in the gut, modifies the microbiom and increases incidence of autoimmune diseases such as autoimmune colitis or hepatitis. NLR, NOD-like receptor.10,34
The metabolic state of cells and the whole organism also determines the inflammatory level of the whole organism, thereby potentially influencing stroke incidence, e.g. by promoting atherogenesis (Figure 4). Interesting molecular links exist between the regulation of inflammation, the metabolic state of cells, and ischemic injury involving regulatory roles of hypoxia-induced factor 1ɑ, AMP-activated protein kinase,117,118 and sirtuin family members. Sirtuins represent a family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, which couple sirtuin-dependent posttranslational modifications with cellular energy levels. 119 Caloric restriction was shown to protect against ischemic injury in the heart and brain, and increased sirtuin-1 (Sirt1) expression after prolonged caloric restriction has been linked to improved recovery after myocardial ischemia. 119 Accordingly, intermittent fasting was very recently shown to suppress ischemia-induced activation of the inflammasome in a murine model of focal transient cerebral ischemia.120,121 Whereas the cardioprotective action of the red wine substance resveratrol and mechanisms of ischemic preconditioning have been ascribed to the action of sirtuins, and whereas resveratrol was shown to inhibit NLRP3 inflammasome, 122 there is still debate on the exact role in ischemic injury. 81 Sirt1 overexpressing mice are not protected in a mitochondrial permeability transition pathway model of neurodegeneration, 123 but Sirt1-KOs are protected against permanent focal cerebral ischemia. 124 This discrepancy may be because of NAD+-depleting effect of sirtuins, potentially contributing to neuronal death in models of excitotoxicity. 119 Sirt2 has been demonstrated to be a repressor of microglial activation and brain inflammation. 125 Aberrant mitochondrial homeostasis diminishes the coenzyme NAD+ levels, leads to reduced Sirt2 activity, increases the level of acetylation of microtubules, and thereby causes NLRP3-inflammasome activation by a dynein-mediated transport of ASC on mitochondria to NLRP3 on the endoplasmic reticulum. 19 Dietary fatty acids have been demonstrated to mediate systemic inflammation and insulin resistance via innate immune receptors, and palmitic acid, the major saturated fatty acid, was shown to induce NLRP3 expression via TLR2-activation and induces inflammasome-mediated IL-1β production in human monocytes. 126
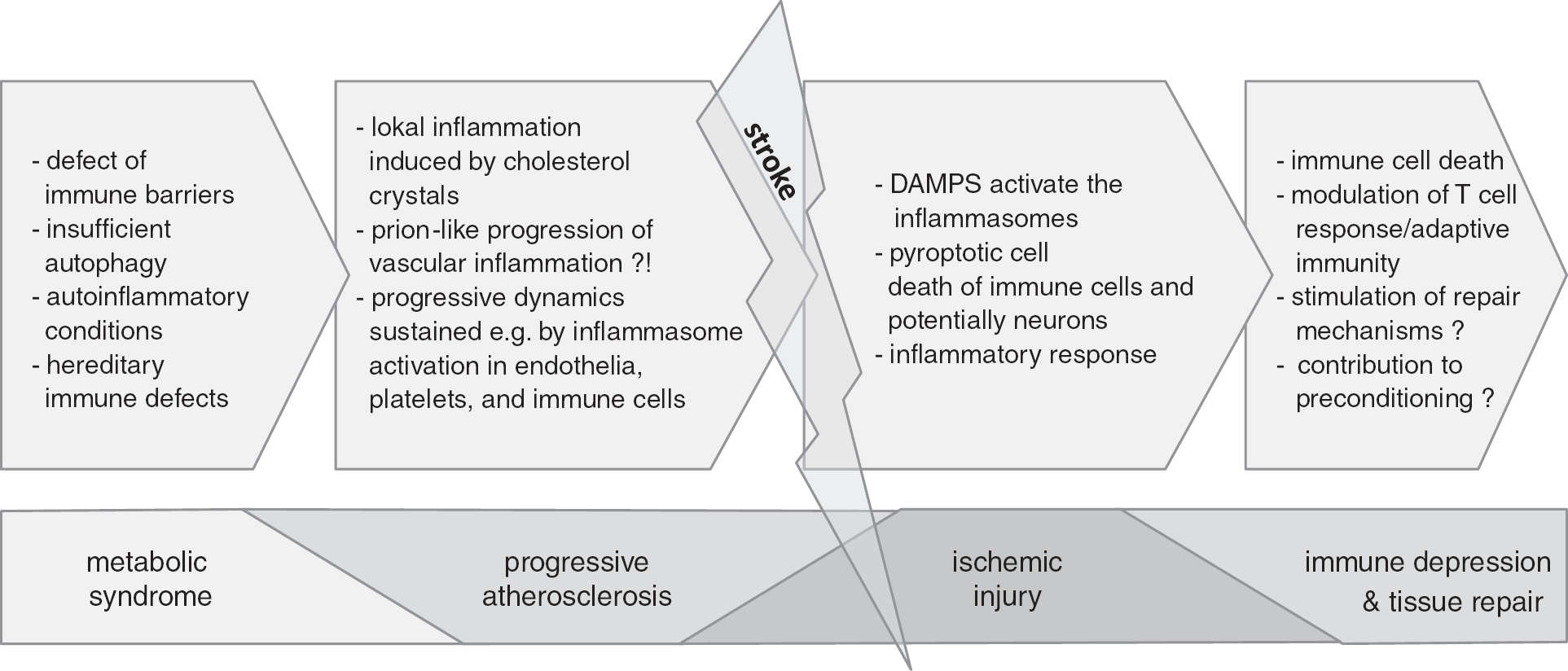
Influence of inflammasomes on cardiovascular events and ischemic injury. The effects of the innate immune receptors, such as TLRs (Toll-like receptors) and the inflammasomes, potentially contribute to the incidence and course of cardiovascular events by increasing the systemic inflammatory level, modifying the immune barriers at the body surfaces (gut, lung, etc.), promoting local inflammatory processes of atherosclerotic lesions, mediating the acute inflammatory response upon sterile inflammation (e.g., ischemic-reperfusion injury), orchestrating mechanisms leading to tissue homeostasis or repair, modifying the adaptive immune system, and triggering preconditioning effects.
INFLAMMASOME: ACTIVATORS IN STROKE
There is a longstanding interest in mitochondria with regard to ischemia-mediated tissue injury, and ROS is hypothesized to be a central signaling pathway for inflammasome activation and stroke-induced inflammation. 5 Oxygen glucose deprivation was shown to activate the NLRP1b inflammasome in fibroblasts in vitro. 127 Various molecular mechanisms identified in vitro potentially activate the inflammasome during ischemic brain injury in vivo. ROS-mediated effects on NLRP3 inflammasome activation were postulated to be upstream of NLPR3 induction, because ROS-inhibitors block priming, but not activation of NALP3 inflammasome. 128 Accordingly, deficiency of one of the most important NADPH oxidase, NOX2, results in attenuated NLRP3 expression in microglial and endothelial cells under ischemic conditions. 5 However, not only ROS may represent a major inflammasome-activating signal in ischemic brain injury, but ATP may also activate inflammasomes in cerebral ischemia. Extracellular ATP was shown to drive systemic inflammation, tissue damage, and mortality not only by inflammasome-mediated cytokines, but also via inflammasome-independent mechanism in a model of systemic inflammatory response syndrome. 129
Energy depletion with loss of ATP potentially activates inflammasomes during stroke, but drop of tissue pH during ischemia may also contribute to inflammasome activation, as extra- and intracellular acidosis was recently demonstrated to activate NLRP3 inflammasome in vitro. 130
Lysosomal damage or increased permeability of phagosomal membrane lead to the release of cathepsin and activation of the inflammasome, a mechanism that may also contribute to inflammasome activation in cerebral ischemia. 131 Lysosomal damage and release of cathepsin B has already been shown to be responsible for immune response to amyloid-beta, 6 and phagolysosomal damage-mediated NLRP3 inflammasome activation is required for cholesterol-crystal-induced atherogenesis. 32
NLRP1 and NLRP3 inflammasomes are activated in vitro by a decrease in intracellular K+ levels or K+ efflux.49,50,95 Potassium efflux in cerebral ischemia is potentially a consequence of energy depletion leading to reduced activity of the Na+/K+ ATPase pump and passive release of potassium. Release of potassium could also be because of P2×4 and P2×7 receptor opening, which is mediated by ATP. Since ATP is released by pannexin channels, this loop may further increase the activation of the inflammasomes during ischemic tissue injury.95,132 The central role of subphysiologic potassium concentrations and its temperature dependency for the formation of large supramolecular ASC aggregates (termed pyroptosome), which were able to activate caspase-1, was demonstrated by elegant in vitro experiments by Fernandes-Alnemri et al (2007). 82
There is data on the activation of the inflammasomes by pannexin channels, and pharmacological inhibition of pannexin1 was shown to protect against cerebral ischemia in vivo and in vitro. 9 However, there is still debate on whether these effects are directly caused by inflammasome inhibition. 133
Additional molecules exist besides the better-known potential danger signals that are supposed to mediate the inflammatory response in cerebral ischemia (e.g., Hmgb1, heat-shock proteins, MRP14, ATP, K+ efflux, ROS, etc.):9,21,83,134 e.g., nucleic acids. Release of oxidized mitochondrial DNA into the cytosol was shown to directly activate the NLPR3 inflammasome, a process which is regulated by autophagy.135,136 Extracellularly delivered viral RNA was shown to cause neurodegeneration upon activation of TLR7, 137 and there are data on significant induction of retrotransponson mRNA in experimental stroke,138–141 which is localized in neurons, and which activates a caspase-independent and p53-dependent death pathway via mitochondrial and lysosomal damage. 142 The fact that retrotransposons account for around 45% of the mammalian genome supports the significance of these findings, 140 but it remains yet unknown to what degree they contribute to neuroinflammation after stroke.
However, when discussing the specific contribution of single danger molecules, one has to be aware of the caveat, that not only the degree of stimulation of receptors of the innate immune system (e.g., preconditioning effect versus cell death), 26 but also the specific receptor of a family which is expressed and activated, is of relevance. To this end, it is reasonable to assume that different endogenous molecules orchestrate a complex activation pattern of immune receptors in cerebral ischemia. Accordingly, cerebral ischemia in TLR-knockout animals revealed different effects of each TLR: e.g., TLR2- and TLR4-deficiency, but not TLR3- or TLR9-deficiency, have neuroprotective effects against focal cerebral ischemia.73,143
Moreover, the kinetic of the stimulation could also be an important factor and may determine the fate of the tissue under danger: e.g., it has been shown recently, that despite a profound and reproducible effect of neuroprotection after TLR2 inhibition in the short term;73,76 in the long-term, TLR2-deficiency could be detrimental. 28
MODULATION OF INFLAMMASOME AND INNATE IMMUNITY IN ISCHEMIC INJURY
It is above the scope of the present review to discuss all the existing substances known to modify inflammasome activation. Some of them are already frequently used in the clinic, thus it seems reasonable to analyze their potential roles in neuroinflammation and stroke. Glyburide, for example, a widely used antidiabetic drug (sulfonylurea) owns NLPR3-inhibitor activity in vitro and its use is correlated with a better clinical outcome in stroke patients; 144 however, the use of glyburide in normoglycemic stroke patients could be associated with detrimental hypoglycemia.45,102 The small molecule 1667 3-34-0 (5-Chloro-2-methoxy-N-[2-(4-sulfamoylphenyl) ethyl]benzamide), an intermediate substrate in the glyburide synthesis, inhibits the NLRP3 inflammasome in cardiomyocytes and protects against myocardial ischemia-reperfusion injury in the mouse without affecting glucose metabolism. 102 Blocking inflammasome activation is neuroprotective in traumatic brain injury: an ASC-neutralizing antibody applied intraperitoneally in a rat traumatic brain injury model was shown to reduce innate immune response and contusion volume. 94 Intracerebroventricular application of NLRP1-blocking antibody reduced levels of proinflammatory cytokines in a thromboembolic stroke model, thus demonstrating its protective potential against the inflammatory response in ischemic brain injury. 3
Treatment of stroke patients with IL-1 receptor antagonist in a phase II placebo-controlled trial reverses peripheral innate immune suppression in the acute phase of stroke, 110 and improved clinical outcome in cortical infarct patients. 145 However, results of the ongoing CANTOS trial of IL-1β inhibition in cardiovascular high risk patients with elevated levels of the inflammatory biomarker C-reactive protein are awaited: in a phase IIb randomized trial, the monoclonal antibody canakinumab was shown to reduce systemic inflammation significantly. 30
There were other IL-1β-pathway inhibiting drugs, which were already used in other inflammatory diseases (e.g., Rilonacept in cryopyrin-associated periodic syndromes), but there is presently no final assessment of their use in stroke patients.8,9
LONG-TERM EFFECTS
Despite exciting new data on the neuroprotective effects of inhibition of the inflammation after acute cerebral injury, e.g., by blocking the receptors of the innate immune system, one has to keep in mind that there may be severe consequences for the long-term outcome, which is influenced by different arms of the immune system. For example, the significance of the TLRs for the T-cell response has been shown in different systems, and albeit modulation of TLR signaling is beneficial short term,73,75,76,146 that does not imply that this is also true for the long-term outcome.28,29 Similar effects with only timely limited protective anti-inflammatory effects of inflammasome inhibition have been observed in models of inflammatory bowel diseases.10,147
Thus, it could be too naive to conclude from short-term neuroprotective observations in experimental stroke models that a given innate immune modulator also improves the long-term outcome in clinical stroke studies.27,28
The knowledge about the molecular pathways and mechanism which connect the ischemic tissue under danger with the immune response is rapidly increasing and reveals different specifically controlled signaling pathways. However, to what extent the modulation of any specific single pathway contributes to the long-term outcome of treated patients needs further exploration. Major aspects of interest are the involved signals (e.g., endogenous danger signals) and compartments/cell types (e.g., neutrophils, T cells, platelets, etc.), which activate the immune system, but also the long-term consequences of an altered immune response, as short-term benefit does not necessarily also mean long-term benefit. Moreover, it seems reasonable to see as to what extent the recently discovered molecular prion-like mechanisms of inflammasome activation contribute to the course or progression of various autoinflammatory diseases and the metabolic syndrome which come along with increased IL-1β processing.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The help of Viktoria Hasselhof is acknowledged for helpful comments and proofreading of the manuscript.