
Abstract
Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a hypoxia-inducible, neuroprotective protein that also stimulates proliferation of neuronal precursor cells. Accordingly, HB-EGF may contribute to recovery from cerebral injury through direct neuroprotective effects, by enhancing neurogenesis, or both. When administered by the intracerebroventricular route 1–3 days after focal cerebral ischemia in adult rats, HB-EGF decreased the volume of the resulting infarcts and reduced post-ischemic neurological deficits. HB-EGF also increased the incorporation of bromodeoxyuridine into cells expressing the immature neuronal marker protein TUC-4 in the dentate subgranular and rostral subventricular zones, consistent with increased proliferation of neuronal precursors. However, HB-EGF decreased the number of newborn neurons that migrated into the ischemic striatum, perhaps partly because reduction of infarct size by HB-EGF also reduced the stimulus to migration. To determine if HB-EGF might also directly inhibit migration of neuronal precursors, we co-cultured subventricular zone (SVZ) explants treated with HB-EGF or vehicle together with hypoxic cerebral cortical explants, and measured cell migration from the former toward the latter. HB-EGF reduced directed migration of SVZ cells toward the cortical explants, possibly due to a local chemoattractant effect on neuronal precursor cells, which may be mediated through the HB-EGF-specific receptor, N-arginine dibasic convertase. The delayed neuroprotective effect of HB-EGF may have implications for efforts to prolong the therapeutic window for intervention in stroke.
The rapidity of neuronal death from ischemia and the brain's limited regenerative capacity make treatment of stroke and related disorders difficult. One approach to this problem involves the administration of growth factors or other agents that target downstream mediators of pathways involved in delayed neuronal death. Another possible approach is the use of neuronal precursor cells, which are found in the adult central nervous system, and might be capable of replacing dead cells in the ischemic brain through the process of neurogenesis.
Several growth factors can protect neurons from hypoxia, ischemia, or other froms of injury, and some of these have been found to promote neurogenesis, including fibroblast growth factor-2 (FGF-2), epidermal growth factor (EGF), stem cell factor and vascular endothelial growth factor (VEGF). Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is expressed in proliferating and post-mitotic cerebral neurons and astroglia (Nakagawa et al., 1998). HB-EGF regulates intercellular adhesion (Raab and Klagsbrun, 1997), cell death (Iwamoto et al., 1999;Lin et al., 2001;Opanashuk et al., 1999), and cardiac development (Iwamoto et al., 2003), by activating the EGF receptor EGFR/ErbB1, the related tyrosine kinase ErbB4, and the metalloendopeptidase nardilysin, or N-arginine dibasic convertase (NRDc) (Raab and Klagsbrun, 1997;Nishi et al., 2001). HB-EGF also stimulates proliferation of neuronal precursor cells in vitro (Kornblum et al., 1999) and in vivo (Jin et al., 2002). Whether HB-EGF can elicit a postischemic increase in the number of newborn neurons in neuroproliferative regions of the brain – the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) and the subventricular zone (SVZ) – and whether these cells can migrate to replace ischemically damaged neurons is unknown.
We infused HB-EGF into rats 1–3 days after focal cerebral ischemia, and found reductions in infarct volume and post-ischemic neurological deficits. HB-EGF also increased neurogenesis, measured by incorporation of bromodeoxyuridine (BrdU) into cells that co-expressed the immature neuronal marker TUC-4, but decreased the number of newborn neurons migrating to the ischemic striatum. This suggests that reducing the severity of ischemia may have the unintended effect of diminishing the neurogenic response to ischemia, perhaps by reducing motogenic stimuli emanating from the ischemic lesion. In addition, delivering neurogenic factors with chemotactic properties to neuroproliferative zones, rather than sites of injury, may inhibit migration of neuronal precursors toward the latter.
MATERIAL AND METHODS
Focal cerebral ischemia
Animal experiments were approved by local committee review and carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Ischemia was induced using the suture occlusion technique, as previously described (Jin et al., 2001). Male, 280–310 g Sprague-Dawley rats were anesthetized with 4% isoflurane in 70% N2O/30% O2 using a mask. The neck was incised in the midline, the right external carotid artery (ECA) was carefully exposed and dissected, and a 19-mm long 3–0 monofilament nylon suture was inserted from the ECA into the right internal carotid artery to occlude right MCA at its origin. After 90 minutes, the suture was removed to allow reperfusion, the ECA was ligated and the wound was closed. Sham-operated rats underwent an identical procedure except that the suture was not inserted. Rectal temperature was maintained at 37.0 ± 0.5°C using a heating pad and heating lamp. Mean arterial blood pressure (MABP), arterial blood gases, and blood hemoglobin and glucose concentrations were monitored. Regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry (Moor Instruments, UK) with the probe positioned over the left hemisphere, 1.5 mm posterior and 3.5 mm lateral to the bregma. After reperfusion for various periods, rats were anesthetized and perfused through the heart with 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4). Brains were removed, sectioned and stained.
HB-EGF administration
Rats were re-anesthetized 24 hours after MCA occlusion (MCAO) with 4% isoflurane in 70% N2O/30% O2 and implanted with an Alzet 1003D osmotic minipump (Alza Scientific Products, Mountain View, CA, USA). The cannula was placed in the right lateral ventricle 4.0 mm beneath the pial surface, +0.8 mm anteroposterior relative to bregma, and 1.3 mm lateral to the midline. Each rat was treated for 3 consecutive days with 1 μl/hour of either artificial CSF (aCSF) containing (in mM): 128 NaCl, 2.5 KCl, 0.95 CaCl2 and 1.99 MgC12, or 1 μg/ml of recombinant human HB-EGF (R&D Systems, Minneapolis, MN, USA) in aCSF. Rats were killed 4, 7, 14, or 28 days later.
BrdU administration
BrdU (50 mg/kg; Sigma, St. Louis, MO, USA) was dissolved in saline and given intraperitoneally twice daily at 8-h intervals for the same 3 days days as HB-EGF infusion, and rats were killed at intervals thereafter.
Quantification of BrdU-labeled cells
BrdU-labeled cells in SGZ and SVZ were counted blindly in five to seven DAB-stained, 50-μm coronal sections per animal, spaced 200 μm apart. Cells were counted under high-power (200×) on a Nikon E300 microscope with Magnifire digital camera, and the image was displayed on a computer monitor. Results were expressed as the average number of BrdU-labeled cells per section.
Immunohistochemistry
Rat brains (n = 4 per group) were perfused with saline and 4% paraformaldehyde in PBS. Adjacent 50-μm sections corresponding to coronal coordinates interaural 8.7–10.2 mm, bregma −0.30 to bregma −1.2 mm (SVZ), and interaural 4.48–6.2 mm, bregma −4.3 to bregma −2.8 (DG), were cut with a cryostat. For BrdU immunocytochemistry, sections were pre-treated with 50% formamide, 280 mM NaCl and 30 mM sodium citrate at 65°C for 2 hours, incubated in 2 M HCl at 37°C for 30 minutes, and rinsed in 0.1 M boric acid, pH 8.5, at room temperature for 10 minutes. Sections were incubated in 1% in PBS for 15 minutes, in blocking solution (2% goat H2O2 serum, 0.3% Triton X-100, and 0.1% bovine serum albumin in PBS) for 2 hours at room temperature, and with mouse monoclonal anti-BrdU (Roche, Indianapolis, IN, USA; 2 μg/ml), rabbit monoclonal anti-cleaved caspase-3 (Cell Signaling, Beverly MA, USA; 1:200), or affinity-purified goat polyclonal anti-doublecortin (DCX) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; 1:200), at 4°C overnight. Sections were washed with PBS, incubated with biotinylated goat anti-mouse IgG (Vector, Burlingame, CA, USA; 1:200) for 2 hours at 25°C, washed, and placed in avidinperoxidase conjugate solution (Vector, Burlingame, CA, USA) for 1 hour. The horse-radish peroxidase reaction was detected with 0.05% diaminobenzidine (DAB) and 0.03% H2O2. Sections were examined with a Nikon E800 epifluorescence microscope.
Double-label studies to define the phenotype of BrdU-labeled cells were performed using anti-BrdU and rabbit polyclonal anti-TUC-4 (Chemicon, Temecula, CA, USA; 1:10,000). Double-labeled cells were observed using a Nikon PCM-2000 laser-scanning confocal microscope, and Simple PCI imaging software (Compix, Cranberry Township, PA, USA) was employed to record results and to confirm co-localization.
Quantification of infarct volume
At intervals after reperfusion, rats were anesthetized with isoflurane, decapitated, and transcardially perfused with normal saline followed by 4% phosphate-buffered formaldehyde. Brains were removed and fixed in the latter. After dehydration in phosphate-buffered 25% sucrose, 25-μm cryopreserved coronal sections were stained with hematoxylin, and the hema-toxylin-stained area was used to define uninfarcted tissue (Rodrigues et al., 2002;Demougeot et al., 2003). The area of the nonischemic right hemisphere (A), uninfarcted area of the left hemisphere (B), and total brain was measured in each 20th section by a blinded observer using the NIH Image program. Infarct area was expressed as a percentage of the area of the uninfarcted hemisphere ((A -B)/A*100%), and multiplied by the distance between sections to obtain infarct volumes (Swanson et al., 1990).
Neurological assessment
Neurological evaluations were carried out on days 4, 7, 14 and 28 post-ischemia using a 14-point behavioral rating scale, as described (Li et al., 2001).
DNA damage assay
Single- and double-strand DNA breaks were detected in fresh-frozen 20- μm sections using the Klenow fragment of DNA polymerase I, as described (Jin et al., 1999). To determine nonspecific labeling, some sections were incubated without the Klenow fragment.
Cell migration assay
Brains from postnatal day 3–7 Sprague-Dawley rats were placed in ice-cold PBS and sectioned coronally at 300-μm intervals with a tissue chopper (Stoelting Inc., Kiel, WI). Tissue within the borders of the rostral SVZ was dissected out from the appropriate coronal sections to make SVZ explants 200–300 mm in diameter. These were plated approximately 7 mm away from cortical explants, maintained for the previous 14 hours in 5%N2/5% CO2 as described (Jin et al., 2002), on 24-well tissue culture plates coated with Matrigel (Becton Dickinson, Cockeysville, MD, USA) diluted 1:1 in Neurobasal medium containing 2% B27 supplement, 2 mM glutamine, 1% penicillin, and 1% streptomycin (Life Technology, Rockville, MD, USA). Matrigel containing 20 μl of HB-EGF (1μg/ml) or vehicle was overlaid on the top of SVZ explants, and the SVZ/cortical co-cultures were maintained at 37ºC in humidified 95% air/5% CO2 for 1–5 d. Cultures were then viewed under phase-contrast microscopy, and the distance of cell migration from the margins of the SVZ explant was measured in two directions: along the shortest line that could be drawn connecting the proximal margins of the two explants (distance toward, or t), and along a line 180° therefrom (distance away, or a). The ratio t/ a was then calculated for HB-EGF- and vehicle-treated cultures, as an index of directed migration that corrects for possible nonvectorial effects of HB-EGF on cyto-kinesis (Hamasaki et al., 2001).
Statistical analysis
Values were expressed as mean ± SD. Differences between means were analyzed by t-tests for single comparisons, and by one-way analysis of variance (ANOVA) and post-hoc Scheffe tests for multiple comparisons. A level of P < 0.05 was considered statistically significant.
RESULTS
Our previous studies showed that both focal cerebral ischemia (Jin et al., 2001) and HB-EGF (Jin et al., 2002) increased neurogenesis in the normal adult rat DG and SVZ. To investigate its effect after ischemia, HB-EGF was infused into the right lateral ventricle beginning 24 hours after transient focal ischemia produced by MCA occlusion, and BrdU was injected intraperitoneally. Both treatments were continued for 3 days, and animals were killed 4, 7, 14 or 28 days later (Fig. 1).
Schedule for administration of BrdU and HB-EGF after induction of ischemia by transient MCAO. HB-EGF or vehicle was infused into the ipsilateral lateral ventricle (A) for 3 consecutive days, beginning 1 day after MCAO (B). BrdU was given intraperitoneally twice daily at 8-hour intervals for same 3 days and rats were killed after 4–28 days of reperfusion (B).
Ischemia and HB-EGF stimulate cell proliferation in DG
Fig. 2 shows that ischemia increased BrdU labeling in DG of the ischemic, and to a lesser extent the non-ischemic, hemisphere. On day 4 post-ischemia, the number of BrdU-labeled cells was 4-fold higher in DG of the ischemic hemisphere than at baseline, whereas increased BrdU labeling in the nonischemic hemisphere was not seen until 1 week. The time-course of this effect, its bilaterality and its asymmetry are consistent with our previous findings (Jin et al., 2001). HB-EGF also increased BrdU labeling in DG of both the nonischemic and ischemic hemispheres, and these effects were evident within the first week after treatment, in line with prior results (Jin et al., 2002). Finally, at 4 days post-ischemia, the combination of ischemia and HB-EGF increased BrdU labeling to a greater extent than either ischemia or HB-EGF alone, but their combined effects were less than additive.
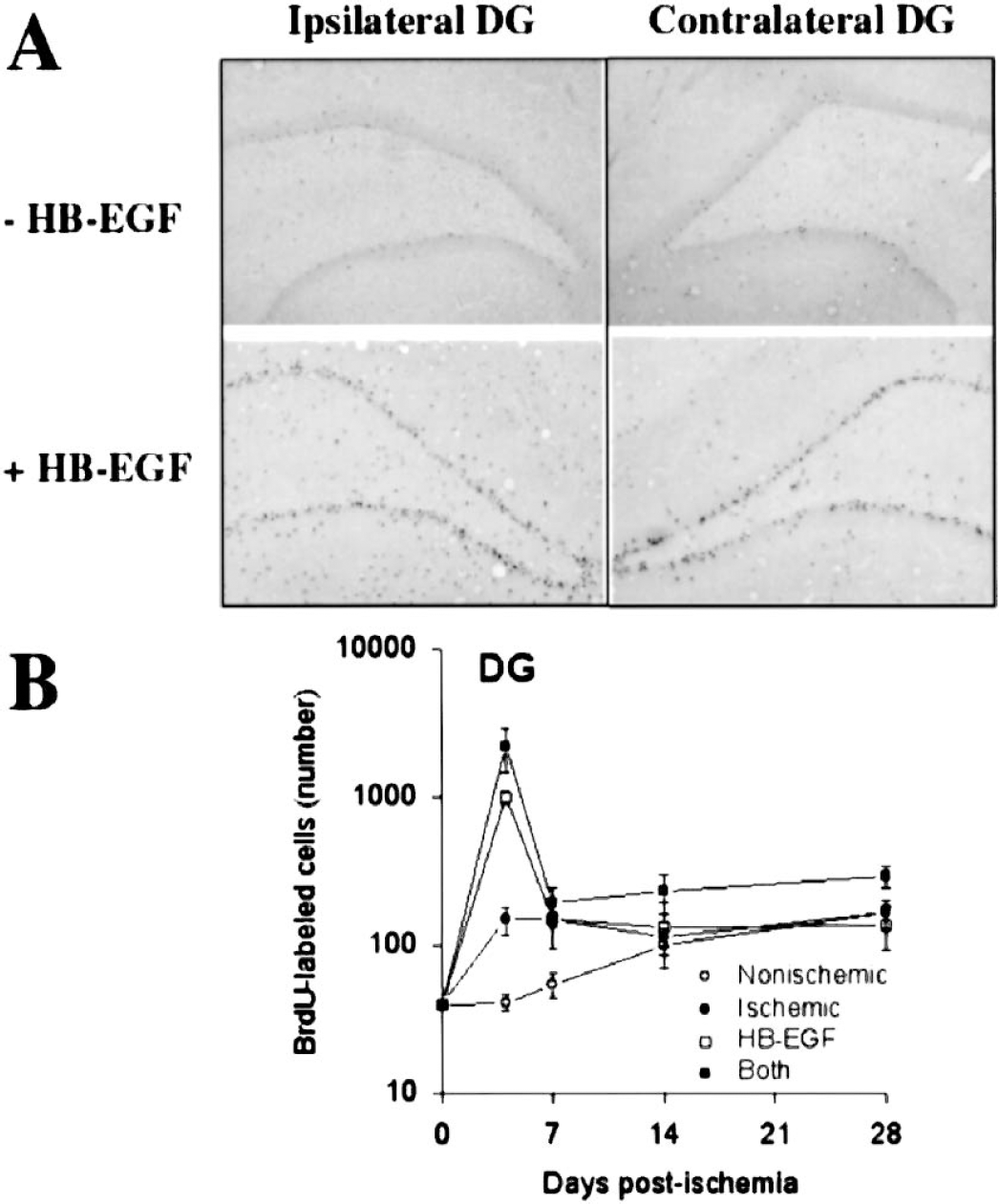
Effects of ischemia, HB-EGF and both on BrdU labeling of cells in SGZ of hippocampal DG. (A) Animals underwent MCAO and were treated with BrdU and infused with HB-EGF or vehicle on the ischemic side, according to the schedule shown in Fig. 1. At intervals thereafter, BrdU incorporation ipsilateral and contralateral to MCAO, HB-EGF infusion, or both was detected by immunohistochemistry. (B) The number of BrdU-immuno-positive cells in each condition was counted and expressed as mean ± SD (n = 3 per group). Nonischemic, DG contralateral to MCAO without HB-EGF; ischemic, DG ipsilateral to MCAO without HB-EGF; HB-EGF, DG contralateral to MCAO with HB-EGF; Both, DG ipsilateral to MCAO with HB-EGF; *P < 0.05 relative to day 0.
Ischemia and HB-EGF stimulate cell proliferation in SVZ
Fig. 3 illustrates that findings in SVZ were similar, but not identical. In previous studies, we found that in SVZ compared to DG, the effect of ischemia on BrdU labeling was similar in magnitude but more bilaterally uniform at 1 week (Jin et al., 2001), and the magnitude of the response to HB-EGF was larger (Jin et al., 2002). These findings were recapitulated in the present study, except that both HB-EGF and ischemia plus HB-EGF increased labeling in DG more so than in SVZ at 4d. This was not detected in our previous studies because the 1-week time point was the earliest one examined. We also found that ischemia and HB-EGF produced greater effects together than separately, although this became apparent only after 2 weeks, and as in DG, the combined effects were less than additive.
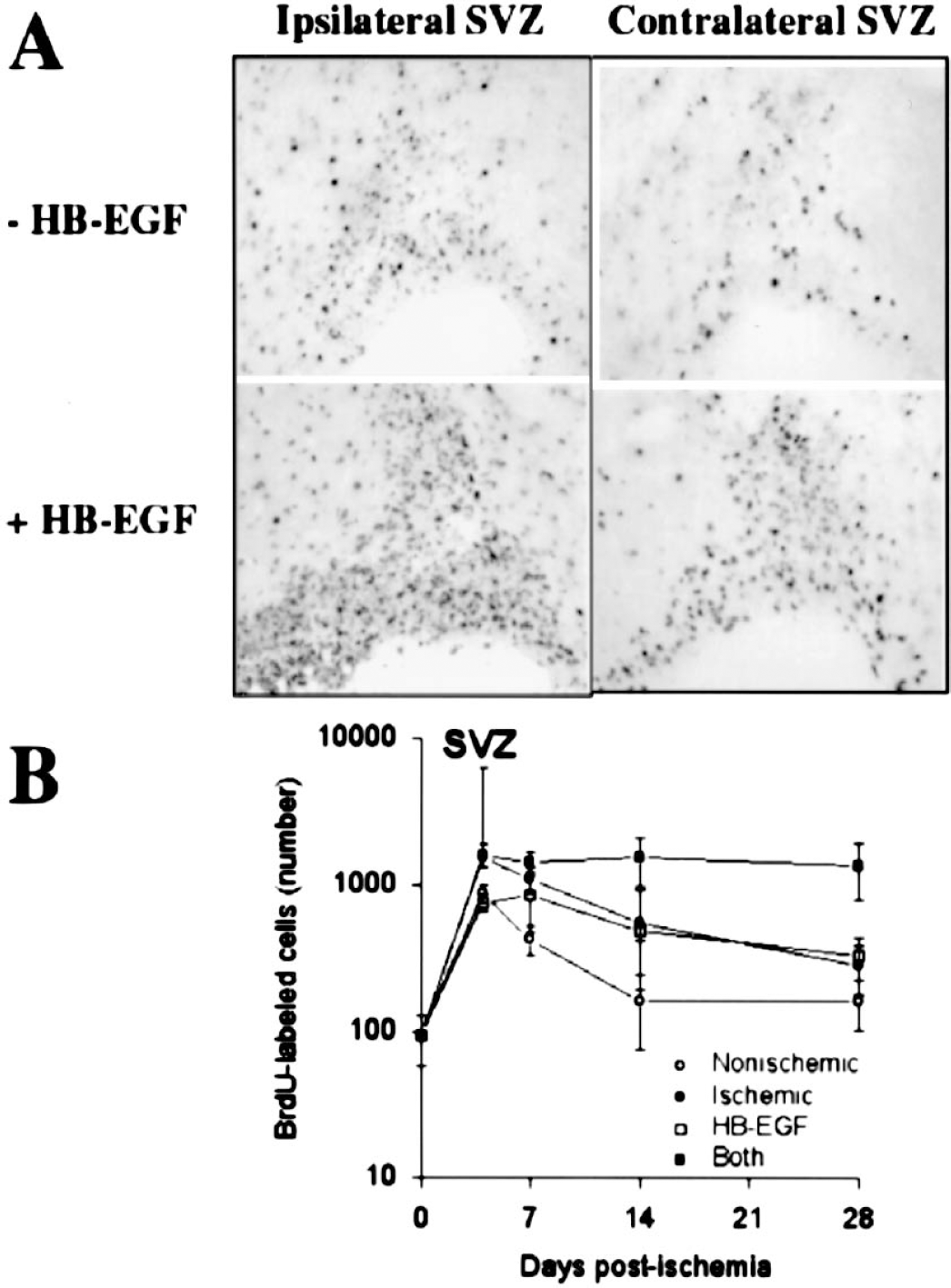
Effects of ischemia, HB-EGF and both on BrdU labeling of cells in rostral SVZ. (A) Animals underwent MCAO and were treated with BrdU and infused with HB-EGF or vehicle on the ischemic side, according to the schedule shown in Fig. 1. At intervals thereafter, BrdU incorporation ipsilateral and contralateral to MCAO, HB-EGF infusion, or both was detected by immunohistochemistry. (B) The number of BrdU-immunopositive cells in each condition was counted and expressed as mean ± SD (n = 3 per group). Nonischemic, SVZ contralateral to MCAO without HB-EGF; ischemic, SVZ ipsilateral to MCAO without HB-EGF; HB-EGF, SVZ contralateral to MCAO with HB-EGF; Both, SVZ ipsilateral to MCAO with HB-EGF; *P < 0.05 relative to day 0.
Cell proliferation after ischemia and HB-EGF involves cells of neuronal lineage
Cells in DG and SVZ that are stimulated to incorporate BrdU after ischemia (Jin et al., 2001) or treatment with HB-EGF (Jin et al., 2002) are of neuronal lineage, because they express neuronal marker proteins. To assess if this is true of cells labeled with BrdU in the setting of combined ischemia and HB-EGF treatment, we stained brain sections from rats that received both treatments with antibodies against BrdU and against the immature neuronal marker TUC-4 (TOAD-64/Ulip/CRMP), a developmentally regulated phosphoprotein involved in axonal growth and guidance (Minturn et al., 1995). Double-label immunohistochemistry in DG and SVZ showed that BrdU was localized to nuclei of cells that expressed TUC-4 (Fig. 4) as well as DCX (not shown), consistent with incorporation of BrdU into cells of neuronal lineage.

Co-localization of BrdU and the immature neuronal marker TUC-4 in dentate SGZ and SVZ of rat following MCAO and HB-EGF treatment. Sections were cut through SGZ and SVZ and double-labeled with antibodies against TUC-4 (green) and BrdU (red). The three-dimensional image (bottom right panel), constructed using Imars software, shows TUC-4 immunoreactiv-ity in the cytoplasm of a cell with BrdU-immunoreactive nucleus.
HB-EGF reduces infarct size after MCAO
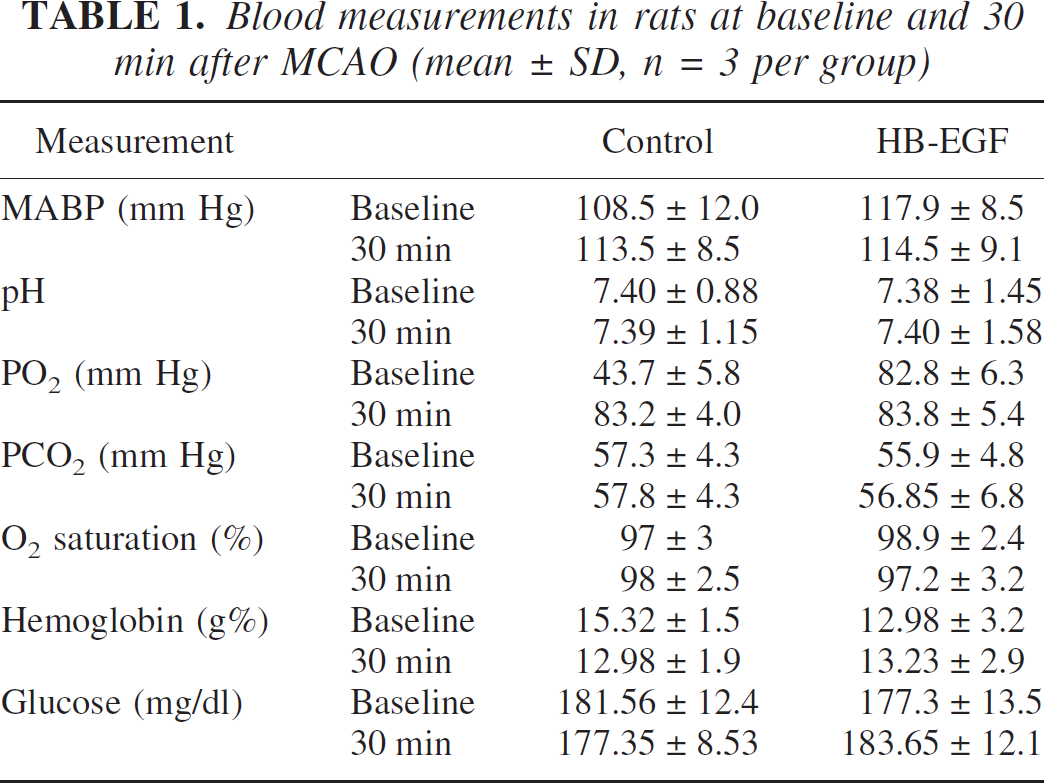
Some growth factors that stimulate neurogenesis, such as FGF-2 (Nozaki et al., 1993) and VEGF (Sun et al., 2003), are also neuroprotective in ischemia, in that they reduce ischemic neuronal loss and infarct size. Although HB-EGF expression in rat brain is induced by neonatal hypoxic-ischemic injury (Tanaka et al., 1999) and global cerebral ischemia (Kawahara et al., 1999), the functional consequences of induction are unknown. To investigate whether HB-EGF affects outcome after focal cerebral ischemia, MCAO was performed, rats were given intra-cerebroventricular infusions of HB-EGF or aCSF vehicle, and infarct volume and neurological deficits were measured. HB-EGF-treated and vehicle-treated rats showed similar MABP, arterial blood gases and blood hemoglobin and glucose concentrations at baseline, as well as 30 minutes after the onset of ischemia (Table 1). In addition, there was no difference in pre-, intra- or post-ischemic rCBF between the HB-EGF-treated and vehicle-treated groups (Fig. 5). However, infarct volume, measured in hematoxylin-stained brain sections, was reduced by 45% at 1wk, 40% at 2 weeks and 35% at 3 weeks in HB-EGF-treated compared to vehicle-treated rats (Fig. 6A—B). This decrease in infarct size resulted primarily from a decrease in the medial extent of cortical involvement, consistent with an effect on the ischemic penumbra. The observation that infarct size was reduced at post-ischemic intervals as long as 3 weeks indicates that HB-EGF decreased, and did not merely delay, ischemic damage.
Blood measurements in rats at baseline and 30 min after MCAO (mean ± SD, n = 3 per group)
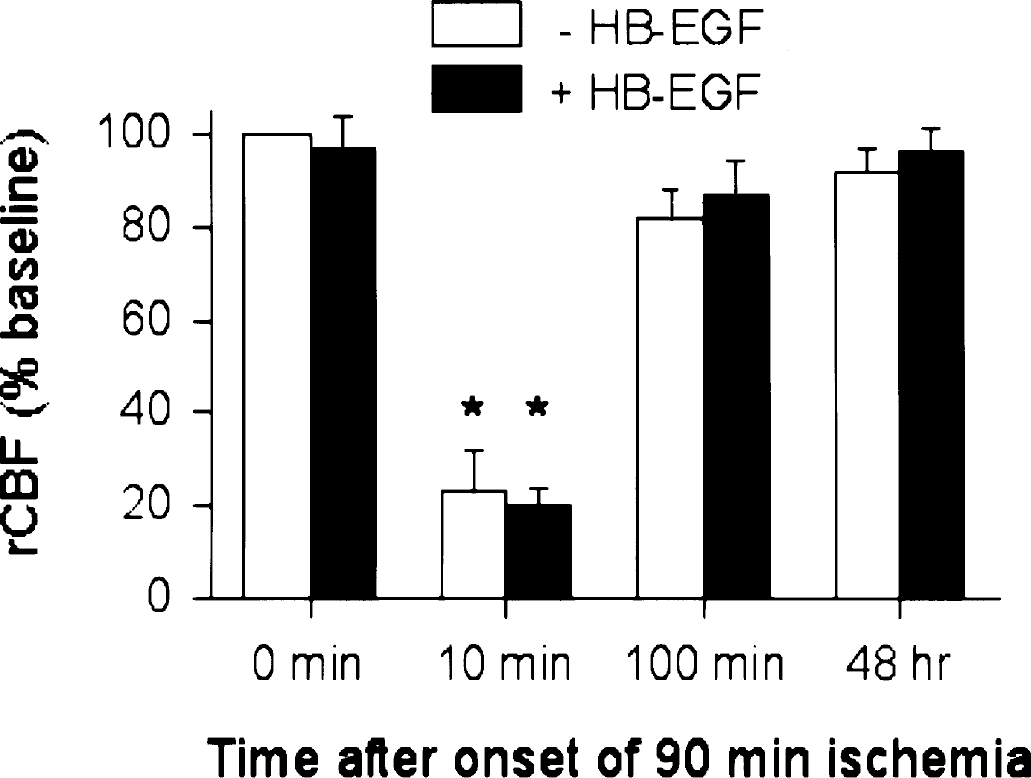
rCBF in vehicle-treated and HB-EGF-treated rats before, during and after 90 minutes of MCAO. rCBF was measured by laser-Doppler flowmetry, as described in Materials and Methods. Data are mean values ± SD (n = 3 per group) from measurements taken prior to the onset of ischemia (0 minutes), during ischemia (10 minutes), and after reperfusion (100 minutes, 48 hours). *, P < 0.05 compared to baseline (−HB−EGF, 0 minutes). There were no significant differences in rCBF between −HB-EGF and +HB-EGF groups at any time.
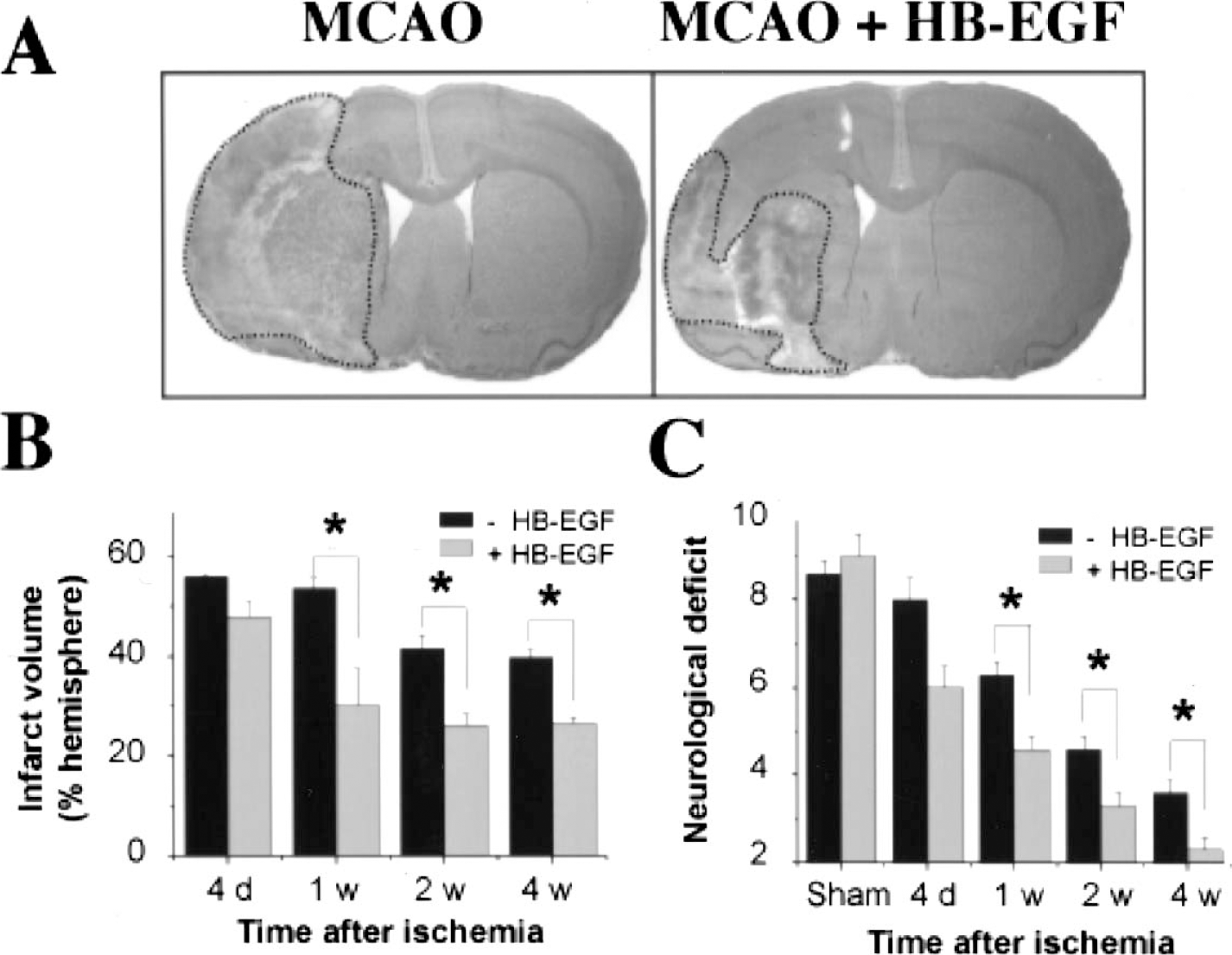
Effects of HB-EGF on infarct size and neurological deficit after MCAO. Rats were given aCSF vehicle or HB-EGF by the intracerebroventricular route for 3 d, beginning 24 hours after MCAO. (A) Brains from aCSF (left) and HB-EGF (right) treated rats were sectioned and stained with hematoxylin to delineate infarct area (dotted lines). Infarct volume (B) and neurological deficit (C) were determined at the post-ischemic intervals shown. Data are mean values ± SD (n = 3 per group). *, P < 0.05 compared to –HB-EGF.
HB-EGF protects cerebral cortex and striatum from ischemic cell death
At the microscopic level, in penumbral regions of ischemic cerebral cortex and striatum, HB-EGF treatment increased the number of cresyl violet-stained neurons with normal morphology, and decreased both DNA damage revealed by Klenow staining and caspase activation demonstrated by immunoreactivity for the 17–20 kDa cleavage product of caspase-3 (Fig. 7). Maximal Klenow staining occurred at 24 hours, followed by a gradual decline at 3 and 7 days post-ischemia (not shown), which is similar to the time course of ischemia-induced DNA damage described in previous reports (Sasaki et al., 2001;Ren et al., 2003). Caspase-3 cleavage also peaked at 24 hours and subsequently decreased, but remained detectable for at least 1 week. These findings indicate that the reduction in infarct size and improved functional outcome produced by HB-EGF after MCAO is likely due, at least in part, to increased neuronal survival.
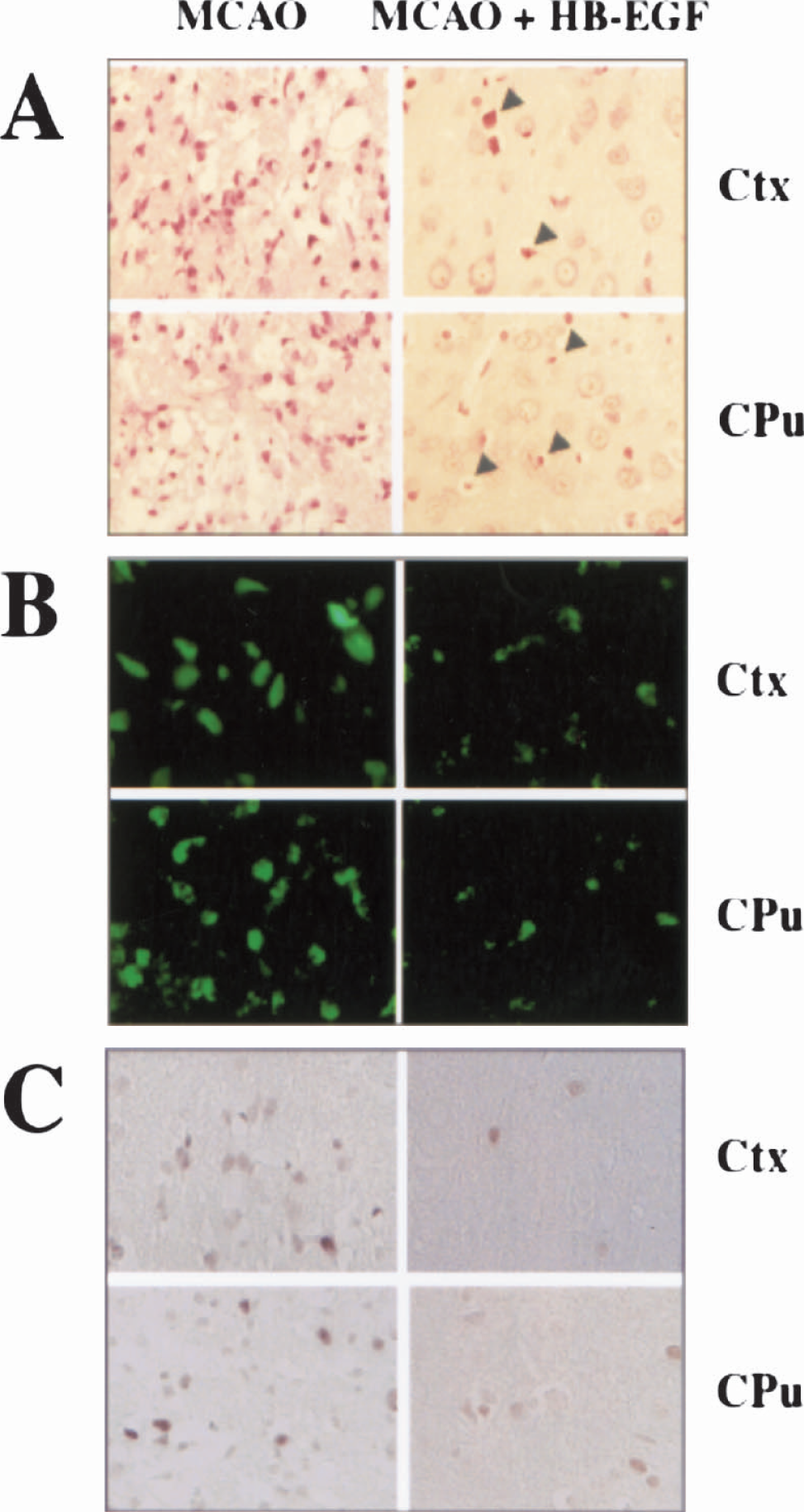
Histological evidence for protection from cell death in cerebral cortex (Ctx) and striatum (caudate-putamen, CPu) after MCAO by HB-EGF. (A) Cresyl violet staining (red) shows an increase in the number cells with preserved morphology after HB-EGF treatment. Arrowheads indicate shrinkage associated with cell death. (B) Cells with DNA damage detected by Klenow assay (green) are decreased in number after HB-EGF. (C) Caspase-dependent cell death, demonstrated by labeling with an antibiody against the 17–20 kDa cleavage product of caspase-3 (brown), is also decreased at 1 week post-ischemia by HB-EGF.
HB-EGF reduces post-ischemic neurological deficits
The amelioration of ischemic injury by HB-EGF was accompanied by improvement in functional outcome, as measured by neurological testing (Fig. 6C). Neurological scores (corresponding to the severity of neurological deficits) were reduced by 30% at 1 week post-ischemia in HB-EGF-treated compared to untreated rats and, although the severity of deficits declined as the post-ischemic interval increased in both treated and untreated rats, outcome remained superior in treated rats for at least 3 weeks.
HB-EGF modifies migration of immature neurons from SVZ into ischemic striatum
Newborn neurons can migrate into ischemic regions of the adult rat brain, where they may participate in tissue repair and help restore function (Arvidsson et al., 2002). After MCAO without HB-EGF treatment, we found newborn neurons, identified by immunostaining for the neuronal migration marker DCX (Gleeson et al., 1999), extending beyond the SVZ and into the adjacent striatum, where they surrounded the infarct core (Fig. 8). This is consistent with our previous results showing directed migration of new neurons from SVZ into the ischemic striatum and cerebral cortex (Jin et al., 2003a). However, in rats treated with HB-EGF after MCAO, the number of DCX-expressing cells in the ischemic striatum was reduced by 60%. Therefore, contrary to expectations, HB-EGF appeared to inhibit the migratory phase of ischemia-induced neurogenesis.
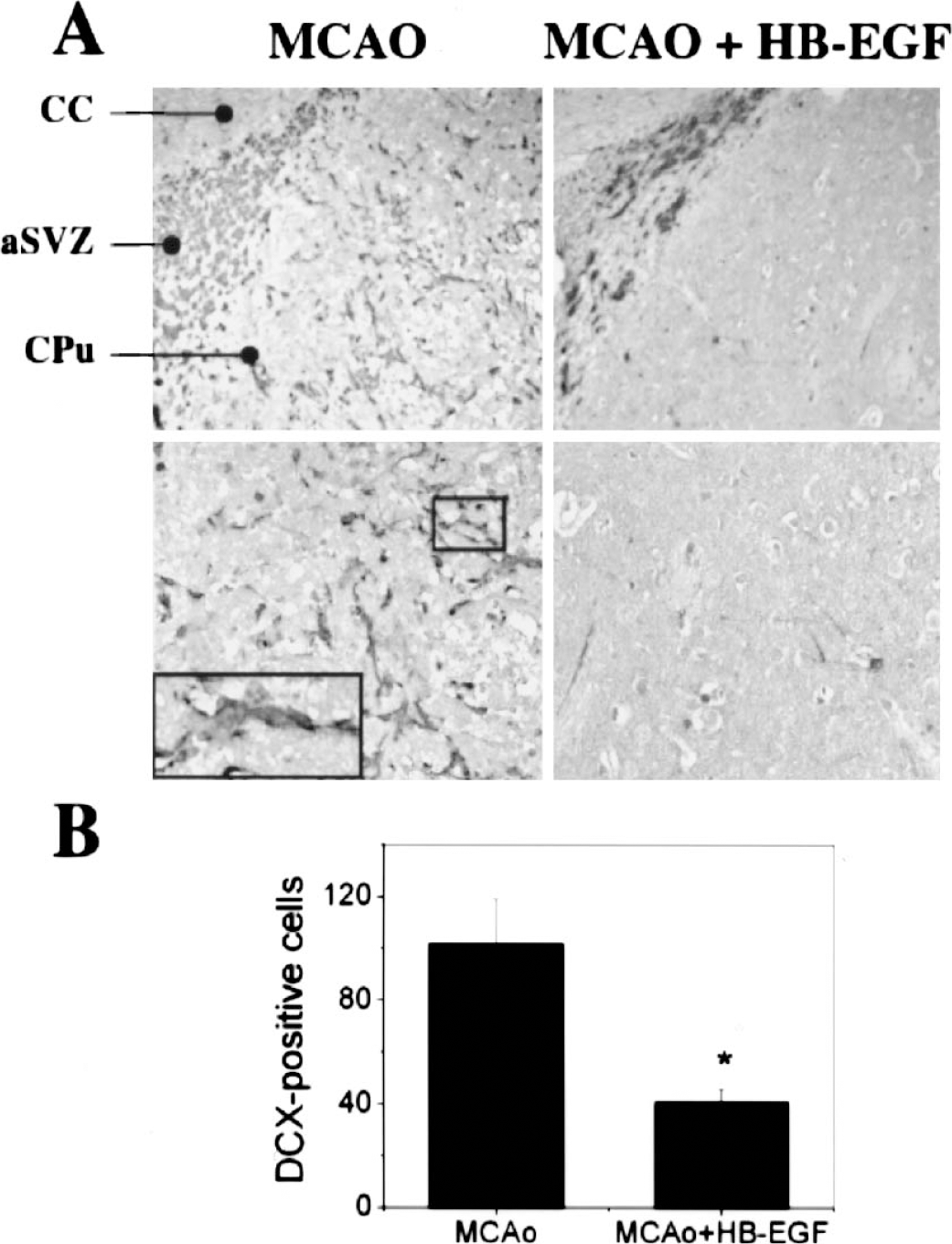
Effect of HB-EGF on migration of DCX-immunoreactive immature neurons from SVZ into ischemic striatum 1 week after MCAO. (A) DCX-immunopositive cells (black) are seen in rostral SVZ near corpus callosum (CC) and in adjacent striatum (caudate-putamen, CPu) after MCAO at low (upper left panel) and high (lower left panel) magnification. The box and magnified inset at lower left show chain-like orientation of DCX-expressing cells. HB-EGF treatment reduced post-ischemic staining for DCX in the same regions, shown at low (upper right) and high (lower right) magnification. Coordinates for the sections shown were bregma –0.3 mm, interaural 8.7 mm. (B) DCX-positive cell counts per field show a decrease in the number of cells with HB-EGF. Data are representative fields from 3 independent experiments (A) or mean ± SD (n = 3; *, P < 0.05 compared to MCAO alone) (B).
HB-EGF modifies migration of SVZ cells toward hypoxic cortical explants in vitro
HB-EGF could inhibit neuronal migration after MCAO indirectly, by reducing the severity of ischemia and thereby the chemotactic stimulus to migration. This is not necessarily inconsistent with the observed increase in neuroproliferation produced by HB-EGF, because cell proliferation and cell migration could be triggered by different factors released from ischemic brain tissue or by interaction of a single factor with different receptors (Nishi et al., 2001). Alternatively, HB-EGF might inhibit cell migration after MCAO by a direct effect on proliferating neurons. HB-EGF exerts not only mitogenic, but also chemoattractant effects on a variety of non-neuronal cell types (Faber-Elman et al., 1996), and HB-EGF-induced cell migration appears to depend on two HB-EGF receptors: EGFR/ErbB1 and NRDc (Nishi et al., 2001). We found previously that both of these receptors (but not another HB-EGF receptor, ErbB4) are expressed on cells in the SVZ (Jin et al., 2002), where they would be in a position to mediate these responses. In addition, a neutralizing antibody against EGFR/ErbB1 blocked HB-EGF-induced proliferation of neuronal precursors in vitro (Jin et al., 2002). Therefore, we hypothesized that intracerebroventricular administration of HB-EGF leading to increased concentrations in periventricular regions might enhance neuroproliferation in SVZ, while chemo-attractant effects resulting from the same elevation of local HB-EGF levels could retain proliferating cells within the SVZ. To test this latter hypothesis, we adapted an in vitro neuronal migration assay (Wichterle et al., 1997;Chazal et al., 2000;Hack et al., 2002) in which an explant from rat SVZ was cultured in Matrigel for up to 5 days with or without HB-EGF, together with an explant from rat cerebral cortex maintained for 14 hours previously in a hypoxic environment. As illustrated in Fig. 9, hypoxic cortical explants induced preferential migration of cells from SVZ explants toward the cortical tissue. However, when HB-EGF was applied to the SVZ explants, no such preferential migration was observed. This appeared to be due to specific inhibition of directional migration, rather than a nonspecific reduction in cell motility, because the absolute distance of migration at 180° to (away from) the cortical explant was unaffected by HB-EGF (0.41 ± 0.08 mm, n = 18 versus 0.45 ± 0.06 mm, n = 15, P = 0.702).
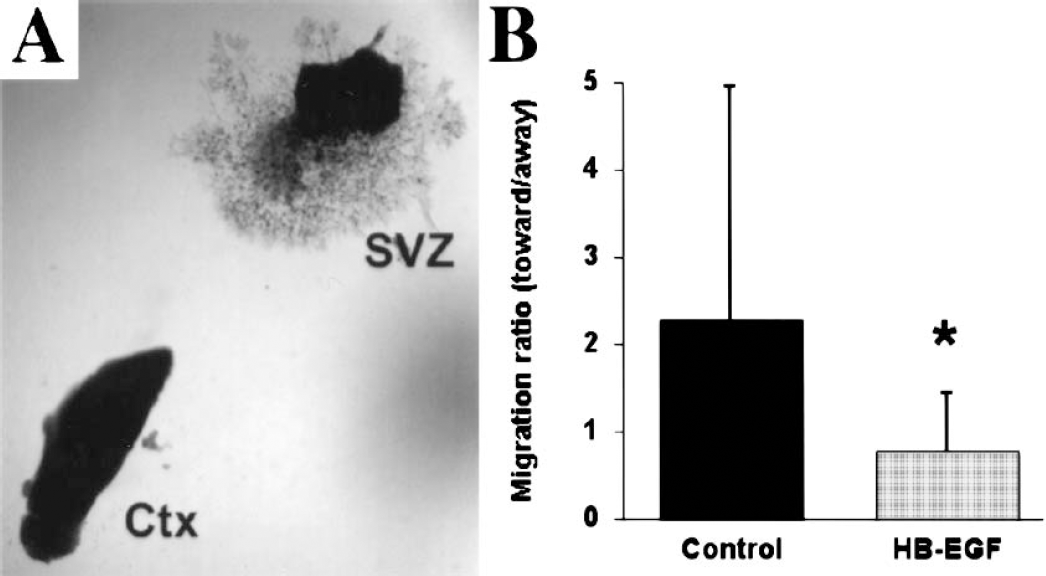
Effect of HB-EGF on in vitro migration of SVZ cells. SVZ explants to which either aCSF vehicle (control) or HB-EGF was applied were cultured together with hypoxic explants from cerebral cortex (Ctx), and migration of SVZ cells toward and away from Ctx was measured. (A) Bright-field photomicrograph shows outgrowth of SVZ cells from explants treated with aCSF. (B) Migration ratio, defined as the maximum distance of migration toward, divided by the maximum distance of migration 180° away from, Ctx was reduced by HB-EGF treatment, indicating that HBEGF inhibited migration toward Ctx. Data are mean values ± SD n = 15−18). *, P < 0.05 compared to aCSF Control.
DISCUSSION
The major findings of this study are that HB-EGF (a) reduces infarct volume and improves neurological outcome when given by the intracerebroventricular route beginning 1 day after focal cerebral ischemia and (b) increases the proliferation of neuronal precursors in SVZ and SGZ of ischemic brain, while decreasing the number of these new neurons that migrate into the ischemic striatum. The ability of HB-EGF to reduce infarct size and post-ischemic deficits when its administration is delayed until 1 day after ischemia is noteworthy; although we (Sun et al., 2003) and others (Zhang et al., 2000) have observed a similar phenomenon in the case of VEGF, such delayed administration of neuroprotective agents following ischemia is usually ineffective. As hypothesized previously (Sun et al., 2003), growth factors may be better able to confer delayed neuroprotection because they act at downstream steps of cell-death pathways. Alternatively or in addition, transient MCAO of 90 minutes duration, as used here, may lead to more slowly evolving cell death than is observed in some other systems, providing a more prolonged time window for neuroprotection.
Growth factors have been shown previously to exert acute neuroprotective effects in the ischemic brain. Shigeno and colleagues showed that administration of nerve growth factor up to 5 minutes after the onset of transient global ischemia reduced delayed neuronal death in the CA1 sector of gerbil hippocampus (Shigeno et al.,1991). FGF-2 decreased neuronal damage after hypoxicischemic brain injury in neonatal rats (Nozaki et al., 1993) and produced a persistent, 25% reduction in infarct size when infused from 30–210 minutes after MCAO in adult rats (Sugimori et al., 2001). VEGF also attenuates cerebral ischemic injury in rats, independent of its angiogenic effect. Topical application of VEGF to the brain surface during reperfusion following MCAO for 90 minutes decreased infarct size at 24 hours by 45% (Hayashi et al., 1998). Intravenous administration of VEGF as much as 48 hours after MCAO improved neurological function (Zhang et al., 2000), and intraventricular infusion of VEGF from 1–3 days after MCAO improved neurological function and reduced infarct size by 35%, when measured 3 days to 4 weeks post-ischemia (Sun et al., 2003).
We are not aware of previous studies on the effect of HB-EGF in cerebral ischemia, but studies in other systems are consistent with the protective effect that we observed. HB-EGF reduces hypoxic death of intestinal epithelial cells in vitro (Pillai et al., 1998), and this is associated with down-regulation of inducible nitric oxide synthase (Xia et al., 2001) and diminished production of reactive oxygen species (Kuhn et al., 2002). HB-EGF also exhibits neuroprotective effects in vitro. For example, HB-EGF enhances the survival of cultured mesencephalic dopamine neurons (Farkas and Krieglstein, 2002) and inhibits the kainate-induced death of cultured hippocampal neurons (Opanashuk et al., 1999).
What signaling mechanisms might be involved in the neuroprotective action of HB-EGF that we observed? The effects of HB-EGF are mediated primarily through the receptor tyrosine kinases EGFR/ErbB1 and ErbB4, although HB-EGF also interacts with heparan sulfate proteoglycan and N-arginine dibasic convertase (NRDc), and can activate ErbB2 and ErbB3 indirectly (Raab and Klagsbrun, 1997;Iwamoto et al., 2003;Nishi et al., 2001). Two principal cell-survival pathways – mediated through mitogen activated protein kinase (MAPK) and phosphatidyl inositol 3-kinase (PI3K)/Akt – are mobilized following ErbB activation (Olayioye et al., 2000;Grant et al., 2002). PI3K/Akt has also been implicated in the neuroprotective effects of FGF-2 (Miho et al., 1999) and VEGF (Miho et al., 1999;Wick et al., 2002;Li et al., 2003;Jin et al., 2000), as well as in reduction of ischemic and related forms of neuronal damage by a range of protective treatments (Luo et al., 2003). It is therefore plausible, although unproven, that HB-EGF protects cerebral neurons from ischemia via activation of the PI3K/Akt pathway.
In addition to direct neuroprotective effects, another way in which growth factors might improve recovery from ischemia is by stimulating the proliferation of new neurons (Cameron et al., 1998). We found previously that HB-EGF stimulates neurogenesis in vitro and in SVZ and SGZ in adult (Jin et al., 2002) and aged (Jin et al., 2003b) rat brain in vivo. However, ischemia also increases neurogenesis in these regions (Jin et al., 2001), so it was unclear if HB-EGF could evoke an incremental response beyond that induced by ischemia itself and, if so, how this response might be modified in the ischemic brain. Both ischemia and HB-EGF increased BrdU incorporation in SVZ and SGZ, but the response to a combination of ischemia and HB-EGF was no greater than that to HB-EGF alone until about 1 week. This delayed effect is consistent with enhanced survival rather than enhanced proliferation of BrdU-labeled cells. However, we observed another interaction between ischemia and HB-EGF related to the migration of new neurons from SVZ into the adjacent striatum following ischemia: a reduction in the number of newborn (DCX-immunoreactive) cells migrating into the striatum. At least two factors may explain this finding. First, because HB-EGF reduces infarct size, it may also reduce the ischemia-induced stimulus to neuronal migration, although it did not reduce the stimulus to neuronal proliferation. Second, because HB-EGF is a chemoattractant, its application at the ventricular surface may induce proliferating cells to remain in the SVZ, and inhibit their migration to ischemic brain regions (Arvidsson et al., 2002;Jin et al., 2003a;Parent et al., 2002). Our finding that HB-EGF inhibits cell migration from SVZ explants toward hypoxic cortical explants in vitro supports this possibility, but does not address whether an additional influence on migration may be exerted by infarct size.
A curious feature of the present study is that it contrasts with a recent report showing that EGF stimulates both neuronal proliferation and neuronal migration into ischemic regions of the mouse brain (Teramoto et al.,2003). Both EGF and HB-EGF interact with the EGFR/ErbB1 receptor, but HB-EGF is also a ligand for ErbB4 and NRDc. NRDc is prominently expressed in the SVZ of adult rat brain (Jin et al., 2002), and binding of HB-EGF to NRDc promotes cell migration (Nishi et al., 2001). Taken together, these observations may provide an explanation for the disparate effects of EGF and HB-EGF on the migration of neuronal precursors into ischemic brain: EGF stimulates neuronal proliferation through its interaction with EGFR/ErbB1; HB-EGF acts similarly, but in addition, it acts on NRDc to elicit a chemo-attractant effect. When the principal source of HB-EGF is ischemic tissue (Jin et al., 2002), the site of ischemia becomes a preferred destination for proliferating neurons. However, if HB-EGF is administered into the ventricles of the ischemic brain, its high concentrations in the adjacent SVZ counterbalance those tactic influences arising from the area of ischemia, and SVZ-derived cells fail to migrate toward the ischemic tissue.
Current therapeutic approaches to stroke are limited by the fact that existing treatments are effective only when given before or shortly after the onset of ischemia, whereas relatively few patients are seen and evaluated within this time span. The finding that growth factors like VEGF (Sun et al., 2003;Zhang et al., 2000) and HB-EGF can reduce cerebral ischemic injury and its functional sequelae in animals when given at more prolonged postischemic intervals provides encouragement that drugs targeting the protective pathways involved might be effective.