
Abstract
Glibenclamide is neuroprotective against cerebral ischemia in rats. We studied whether glibenclamide enhances long-term brain repair and improves behavioral recovery after stroke. Adult male Wistar rats were subjected to transient middle cerebral artery occlusion (MCAO) for 90 minutes. A low dose of glibenclamide (total 0.6 μg) was administered intravenously 6, 12, and 24 hours after reperfusion. We assessed behavioral outcome during a 30-day follow-up and animals were perfused for histological evaluation.
Keywords
INTRODUCTION
The therapeutic potential of the administration of low doses of glibenclamide has been confirmed in different central nervous system pathologies. 1 In experimental stroke models it reduces cerebral edema and infarct volume, and decreases mortality.2,3 In addition, this has been confirmed in stroke patients, where a retrospective study found that patients with diabetes mellitus taking glibenclamide that suffered an acute ischemic stroke had a better neurological outcome. 4
Glibenclamide blocks the sulfonylurea receptor 1 (SUR1), the regulatory subunit of the KATP
5
and the NCCa-ATP channels,1,6 which under ischemic conditions are expressed in neurons, astrocytes, oligodendrocytes, endothelial cells,1,7 and by reactive microglia.
8
It has been proposed that the blockade of the astroglial NCCa-ATP channel by glibenclamide prevents cytotoxic edema after cerebral ischemia.1,7 However, glibenclamide administration resulted in ameliorated reperfusion-derived oxidative stress and proinflammatory cytokine release in the rat hippocampus after cerebral ischemia in rats.
9
In line with this, our previous study demonstrated that glibenclamide administration at 6, 12, and 24 hours after reperfusion caused early neuroprotection and this was accompanied by a better neurological outcome.
8
Although recently Simard
Our previous studies also showed that reactive microglia increase their expression of the KATP-channel components Kir6.1, Kir6.2, SUR1, and SUR2B after brain pathologies, and the
We hypothesized that early blockade of SUR1 by glibenclamide after cerebral ischemia enhances ischemia-induced neurogenesis in the striatum and long-term brain repair, thus leading to an improved functional outcome. To test this hypothesis, we treated rats with low doses of glibenclamide intravenously early after focal cerebral ischemia and evaluated long-term neuroprotection, neurogenesis, angiogenesis, and behavioral recovery. Next, to assess whether microglia could be involved in neurogenesis, we analyzed the expression
METHODS
Transient occlusion of the middle cerebral artery in rats
Male Wistar rats (3 months old and weighing 250–300 g at the beginning of the study; National Laboratory Animal Center, Kuopio, Finland) were used. They were kept on a 12/12 hours day and night cycle and housed individually with free access to food and water (20 ± 1 °C). Animals were handled following European legislation (86/609/EEC) and all efforts were made to minimize the number used and animal suffering, in accordance with the ARRIVE guidelines. The Animal Ethics Committee (Hämeenlinna, Finland) approved the study.
Focal cerebral ischemia was induced by the intraluminal filament technique as described elsewhere. 8 Briefly, the rats were anesthetized with 5% halothane (in 70% N2O/30% O2) and a surgical depth of anesthesia was maintained throughout the operation with 0.5 to 1% halothane delivered though a nose mask. The right common carotid artery was exposed through a midline cervical incision under a surgical microscope and gently separated from the nerves. A heparinized nylon filament (diameter 0.25 mm, rounded tip) was inserted into the stump of the external common carotid artery and advanced into the internal carotid artery 1.8–2.1 cm until it blocked the blood flow into the middle cerebral artery. Corticostriatal damage was induced by an occlusion of the middle cerebral artery (MCAO) of 90 minutes and the blood flow was then restored by removing the filament. Sham-operated control rats underwent the same procedure except that the filament was not inserted. To balance the MCAO groups, the neurological symptoms were assessed before drug administration using two tests: contralateral rotation/circling behavior and impaired contralateral forelimb/hindlimb response to proprioceptive stimulus (i.e., lack of limb withdrawal when hanging over the edge of a table). Animals with no behavioral impairment were excluded from the study.
Drug treatment
We divided 34 rats into the following three groups: sham-operated rats (
Behavioral outcome measures
Immunohistochemistry
Thirty days after MCAO, rats were anesthetized and transcardially perfused with saline for 5 minutes, followed by 4% w/v paraformaldehyde in phosphate buffer. Brains were post-fixed, cryopreserved in 30% w/v sucrose, and frozen on dry ice. Sections (14 μm) covering the entire brain were cut with a cryotome.
The first section between bregma +1.7 and +2.2 of each animal stained with standard hematoxylin–eosin was selected at random. Sequential sections separated by 1 mm were selected for each animal to determine the total infarct volume using the optical microscope software AxioVision 4 AC (Zeiss, GmbH, Jena, Germany) and applying the Cavalieri's principle. This consisted of multiplying the infarct area by the distance between sections (1 mm) and combining the values from each section (5–6 sections/animal). The total infarct volume was calculated by subtracting the area of the remaining tissue in the ischemic hemisphere from the area of the intact contralateral tissue of each section. 23
Immunohistochemistry was carried out with the avidin–biotin peroxidase method as previously described.
8
We used primary antibodies against doublecortin (DCX; clone C-18; 1:150; Santa Cruz, Santa Cruz, CA, USA) to detect undifferentiated neurons and NeuN (1:150; Chemicon, Temecula, CA, USA), calbindin (CB; 1:500; Swant, Bellinzona, Switzerland), parvalbumin (PV; 1:2000; Swant), tyrosine hydroxylase (TH; 1:5000; Abcam, Cambridge, UK), or calretinin (CR; 1:2000; Swant) for differentiated neurons.
26
To detect blood vessels, we used RECA-1 (1:100; Serotec, Oxford, UK). For glial detection, we used antibodies against GFAP (1:400; Sigma-Aldrich, St Louis, MO, USA), CD11b (OX-42 clone; 1:150; Serotec), or the isolectin B4 peroxidase-conjugated (IB4) from
Degenerating neurons were detected by Fluoro-Jade B (FJB, Chemicon) fluorochrome, following the manufacturer's instructions.
Stereological methods were used to quantify the number of cells in the peri-infarct areas as described elsewhere. 8 We applied the optical fractionator method to count labeled cells. Peri-infarct regions were outlined at cortical and subcortical levels at × 2.5 magnification on arbitrary uniform random coronal sections, located throughout the entire brain. Individual cells were viewed at × 40 in arbitrary uniform randomsampled sites chosen by the Mercator Pro 7.0 software (ExploraNova, La Rochelle, France) and then counted.
Cell culture and microglial activation
Cell culture procedures were approved by the Ethics Committee of the Universitat de Barcelona, in accordance with the regulations established by the Catalan autonomous government (Generalitat de Catalunya). Primary microglia-enriched cultures were obtained from post-natal day 1 rat brains, following the protocol described by Saura
These microglial cultures were activated and treated 24 hours after isolation as described below. Based on our preliminary results, cell cultures were pre-treated with 1 nmol/L glibenclamide diluted in culture medium (dimethyl sulfoxide final concentration <0.05%; Sigma-Aldrich, >99% purity).
8
Thirty minutes later, cells were activated with 0.1 μg/mL of recombinant lipopolysaccharide (LPS) and 0.05 ng/mL of recombinant mouse interferon gamma (IFNγ; Sigma-Aldrich). After 48 hours, cells were incubated for 30 minutes at 37 °C in Hank's balanced salt solution and supplemented with 500 nmol/L glibenclamide–BODIPY-FL (green fluorescent dye; Invitrogen). After cell fixation with 4% w/v paraformaldehyde, we performed immunocytochemical analyses with mouse antirat CD11b (OX-42 clone; 1:500; Serotec) antibody and goat antimouse AlexaFluor-555 as a secondary antibody (1:500; Molecular Probes, Eugene, OR, USA). Cultures were directly observed in an AxioObserver Z1 (Carl Zeiss, GmbH, Jena, Germany) inverted microscope equipped with FluoUp and CoLoc image software (ExploraNova, Bordeaux, France) and recorded on a high sensitivity camera (RetigaEXi Fast 1394, QImaging, Surrey, BC, Canada). Image processing was performed using the method described by Jaskolski
Statistical Analyses
Statgraphics 5.0 (STSC, Rockville, MD, USA) and GraphPad Prism4 (La Jolla, CA, USA) statistical packages were used.
RESULTS
Infarct size, gliosis, neurodegeneration and apoptosis
We analyzed the effects of glibenclamide in terms of lesion volume by histological methods 30 days after MCAO. Quantification of the lesion volume of hematoxylin–eosin-stained sections showed that there was no difference in infarct volume size between vehicle- and glibenclamide-treated MCAO rats (Figure 1A). We then performed stereological quantification of the MCAO-induced neuronal loss in the peri-infarcted volume by NeuN immunohistochemistry. Stereological counting showed same number of NeuN-positive cells in the peri-infarct striatum (Figure 1B), but higher number in the peri-infarct cortex of glibenclamide-treated MCAO rats (Figure 1C,
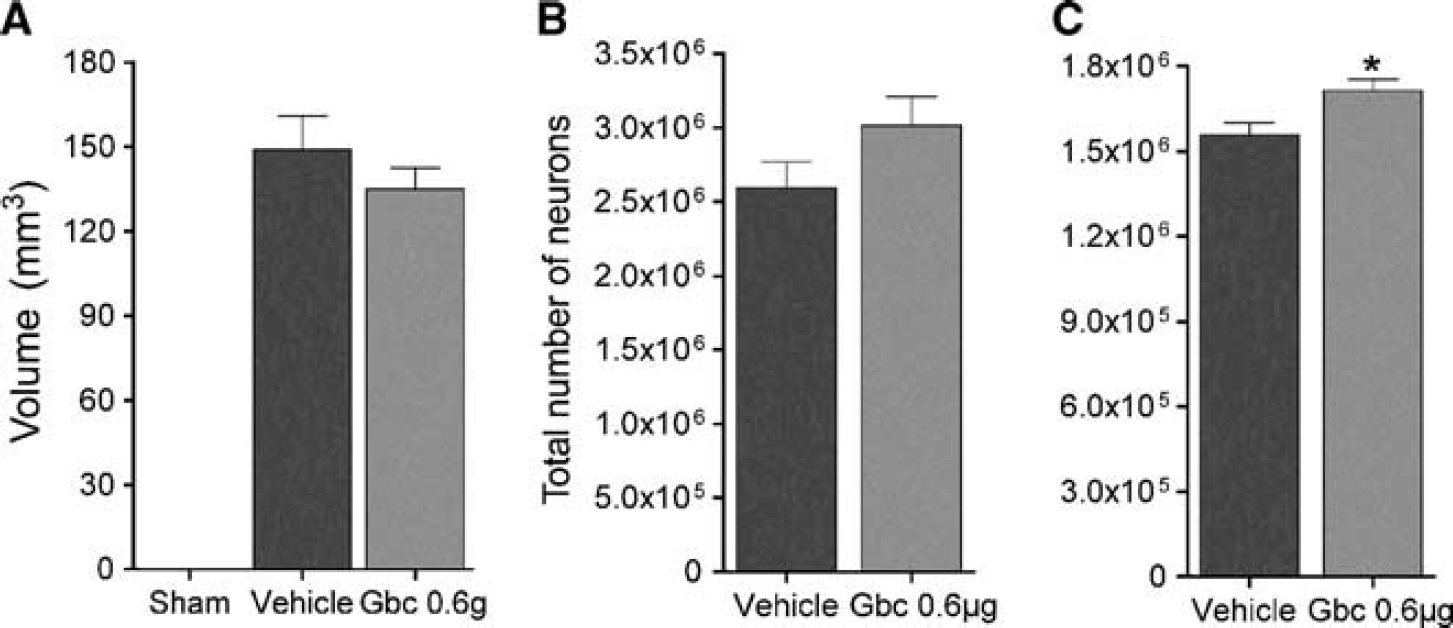
Glibenclamide does not affect corticostriatal infarct size in middle cerebral artery occlusion (MCAO) rats (
The pattern and cell density of microglia/macrophages (IB4-stained) and astrocytes (GFAP-immunopositive) in ischemic animals showed activation, as evidenced by their reactive morphology (Supplementary Figure 1C). In addition, when we quantified the overall staining and cell density for both markers, we found that glibenclamide did not modify MCAO-induced astrogliosis or microgliosis in the cortex (Supplementary Figure 1A,B).
As hypoxia and ischemia causes neuronal death in the peri-infarcted region, and consequently, secondary neurodegeneration in other brain regions, we analyzed whether neurodegenerative processes were still active 30-days after MCAO by FJB staining. Only scattered FJB-positive neurons were detected in the thalamus (Supplementary Figure 2A). We also studied apoptosis using cleaved caspase-3 immunohistochemistry. Cleaved caspase-3 staining did not co-localize with neurons (NeuN) or astrocytes (GFAP) (Supplementary Figure 2B,C). However, cleaved caspase-3 staining co-localized with some cells expressing the microglial/macrophage marker CD11b in the peri-infarct region (Supplementary Figure 2D). These results suggest that 30-days after ischemia secondary neurodegenerative processes had completed.
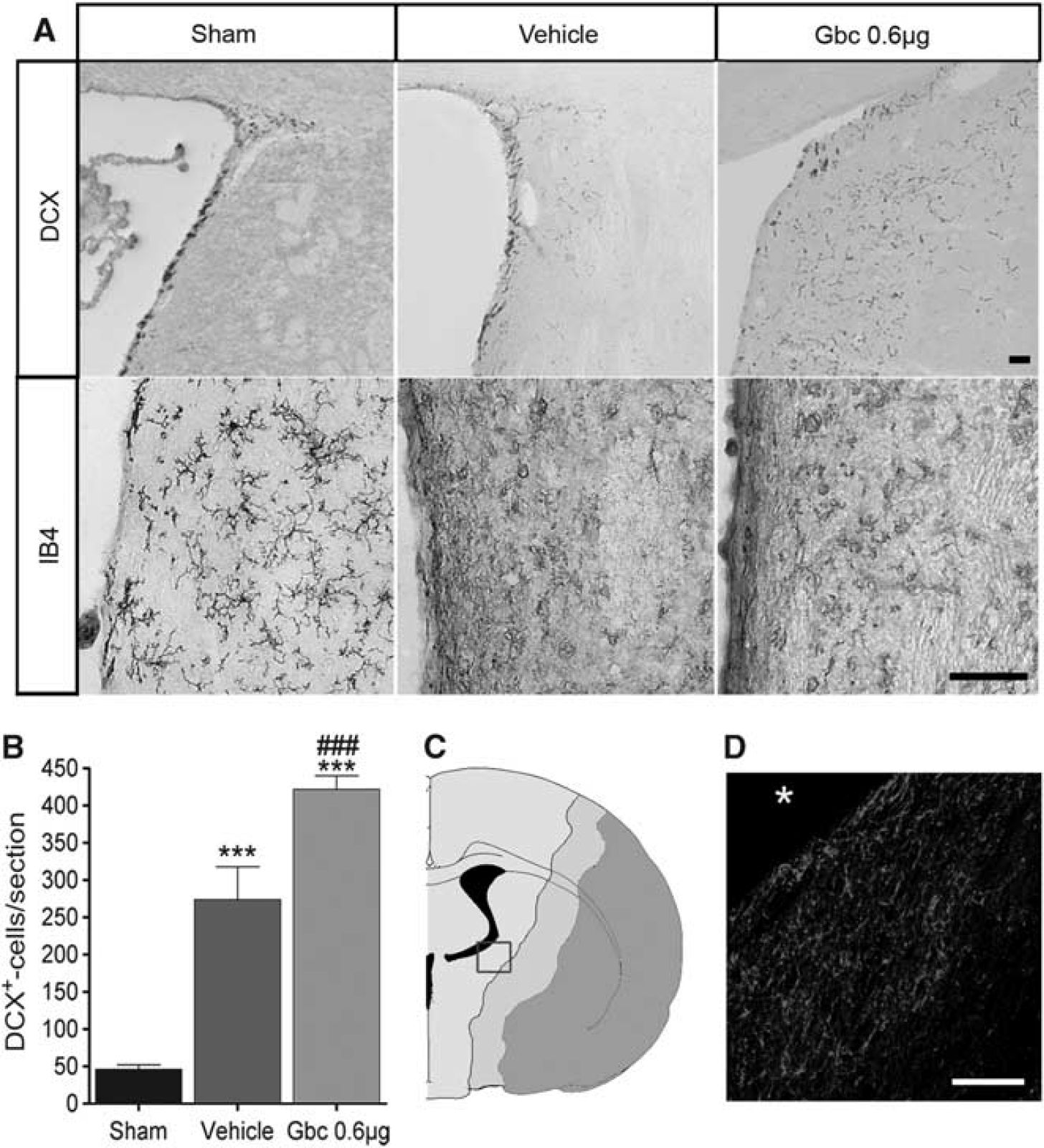
Glibenclamide (Gbc) enhances neuroblast migration toward the ischemic lesion. Representative micrographs of doublecortin (DCX) and microglia/macrophage (IB4) immunostaining in the striatum adjacent to the ventricle of sham-operated rat and vehicle- and Gbc- (0.6 μg) treated middle cerebral artery occlusion (MCAO) rats 3 days after ischemia (
Characterization of neuroblast migration and long-term neurogenesis
We hypothesized that glibenclamide treatment could influence neurogenesis after stroke. First, DCX-immunohistochemistry was carried out to detect neuroblasts in the striatum migrating toward the lesion core to a group of rats killed 3 days after MCAO (Sham
Second, to detect cell proliferation, we perfused a group of rats 30 days after MCAO (sham
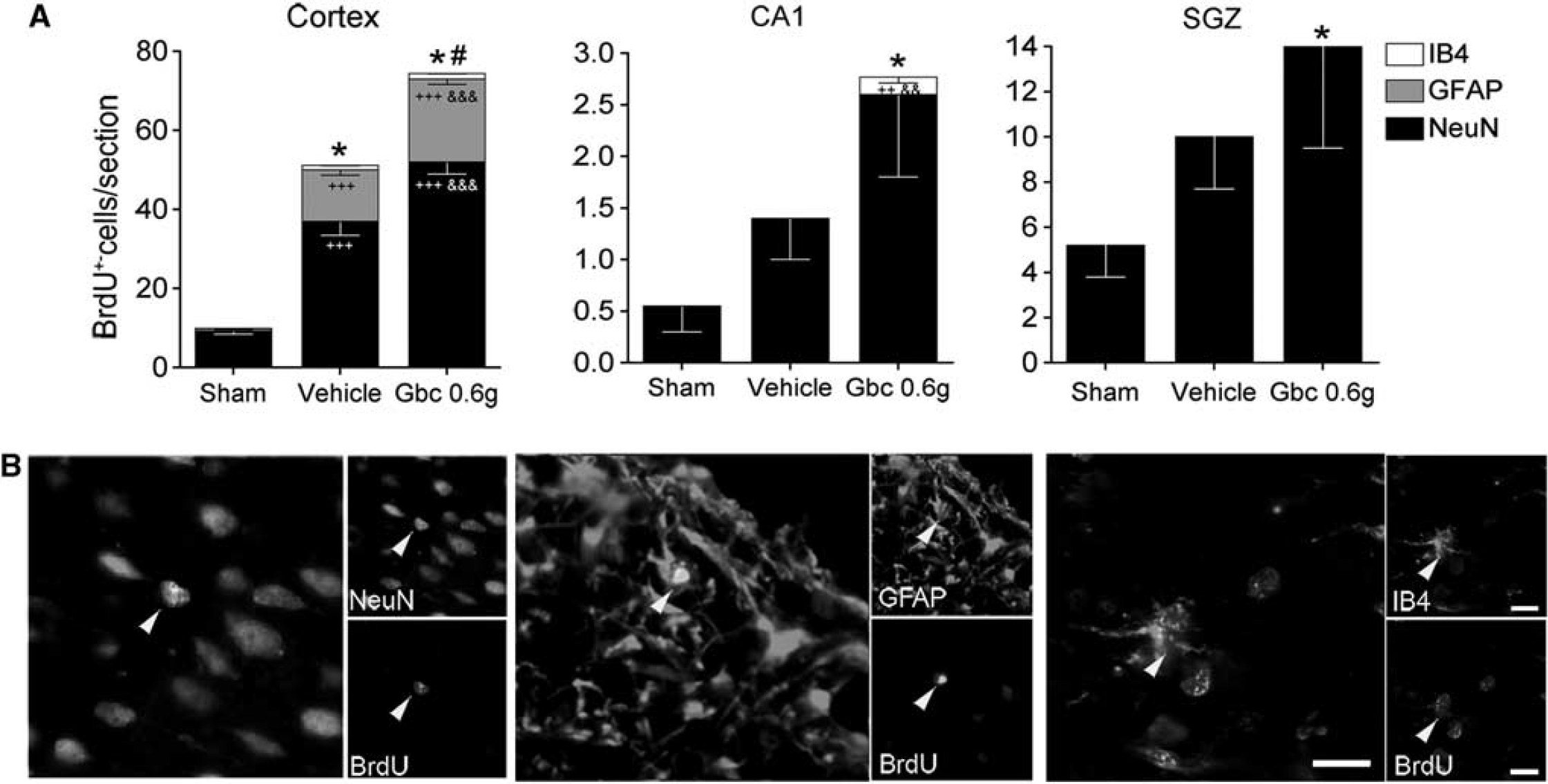
Characterization of proliferative cells into the ischemic brain. Quantitative analysis of the total number of 5-bromo-2-deoxyuridine (BrdU)-positive cells/section and relative number of neurons (NeuN), astrocytes (GFAP), and microglia (IB4) double-stained BrdU-positive cells in the cortex, CA1 pyramidal cell layer, and subgranular zone (SGZ) (
To study whether glibenclamide modifies the fate of the neuroblasts as they migrate through the RMS, we quantified the number of BrdU-positive cells in the olfactory bulb 30 days after MCAO. No differences in the number of these cells were found between MCAO groups in the olfactory bulb (542 ± 26 BrdU-positive cells/section for sham-operated, 518 ± 37 for vehicle-treated group and 580 ± 28 for glibenclamide-treated group). As the total number of proliferative cells in the olfactory bulb is not modified by ischemia or glibenclamide, the most likely the fate of neuroblasts migrating through the RMS is also unaffected. Therefore, these results indicate that glibenclamide strengthens intrinsic neurogenic processes as higher numbers of NeuN/BrdU-positive cells were found in the cortex.
Angiogenesis
To assess another brain repair mechanism, we studied angiogenesis in several brain regions by measuring vessel diameter and RECA-1 immunoreactivity 30 days after ischemia.
29
The diameter of vessels increased in the ipsilateral cortex of glibenclamide-treated MCAO rats (Figure 4A,C). Quantitative analysis of RECA-1 immunoreactivity showed increased microvessel density in the ipsilateral hippocampus of glibenclamide-treated MCAO rats (Figure 4B,C;
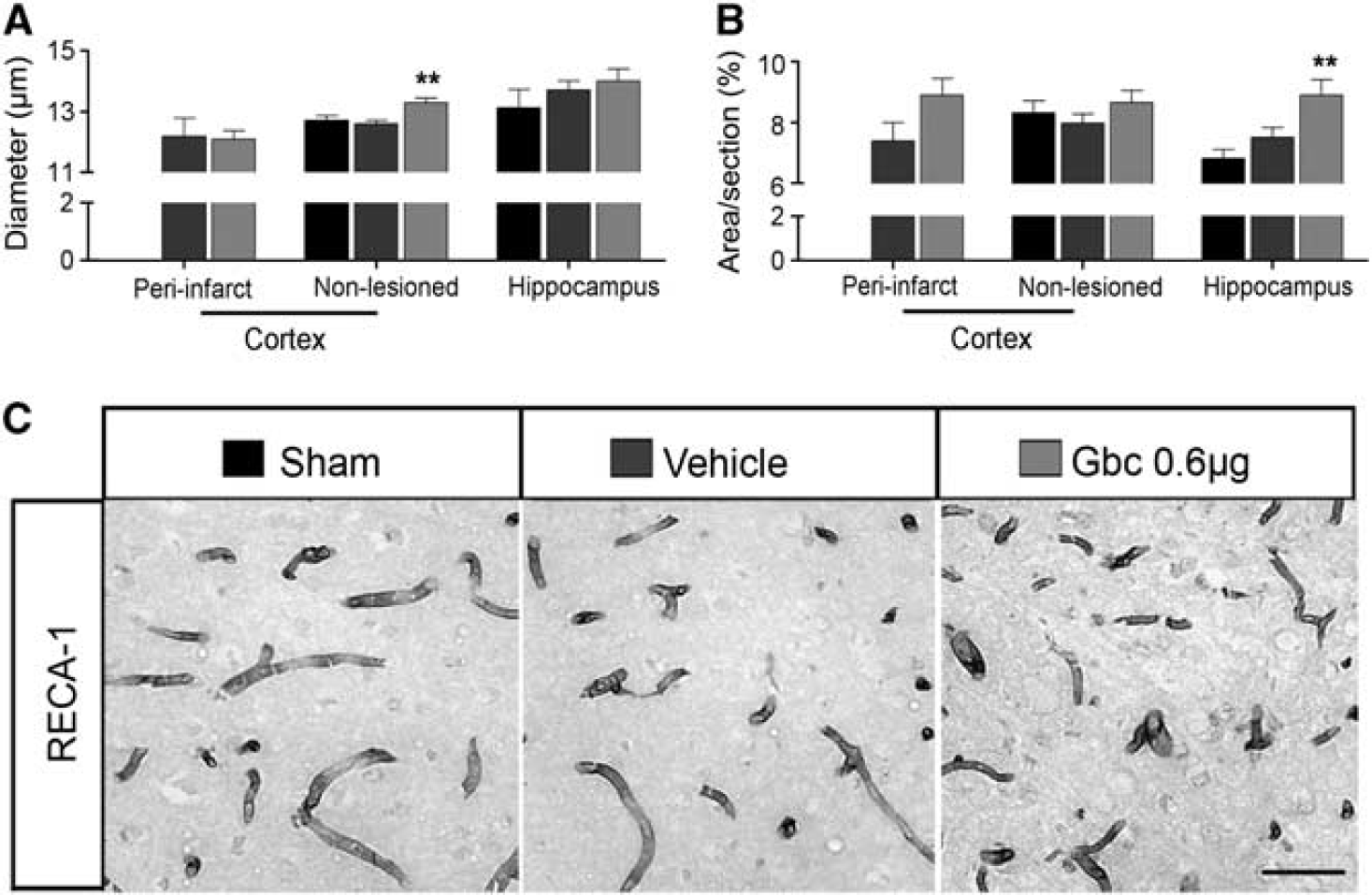
Glibenclamide (Gbc) fosters angiogenesis in the cortex and hippocampus of ischemic animals. Quantitative analysis of the microvessel diameter (
Recovery of sensorimotor and cognitive functions
The effects of ischemia and glibenclamide treatment in the recovery of sensorimotor and cognitive functions were analyzed using a battery of behavioral tests during a 30-day follow-up period. Spontaneous forelimb use was analyzed by the cylinder test (Figure 5A). Analysis of variance for repeated measures showed an overall group effect (
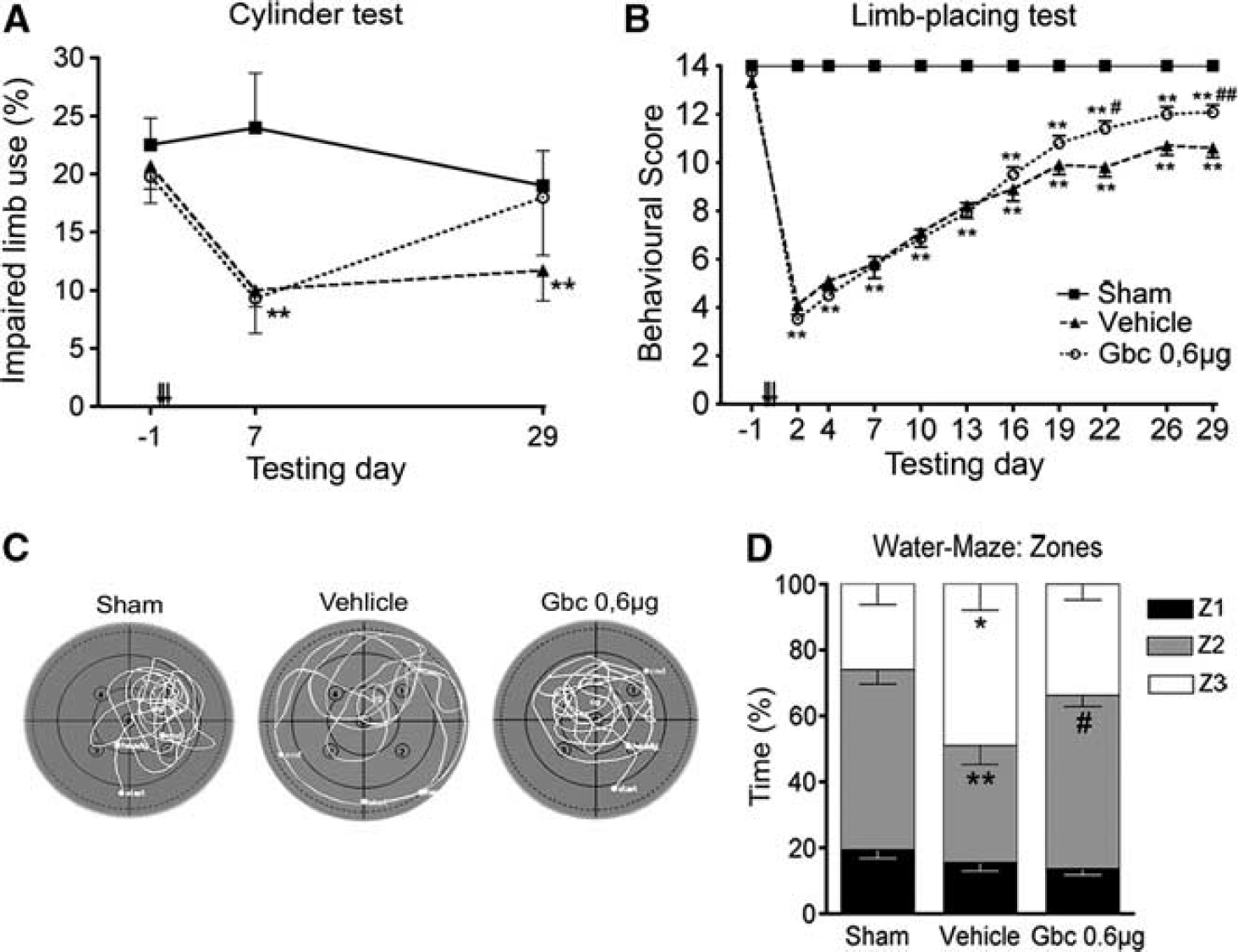
Ischemic animals showed long-term improvement of neurological function after glibenclamide (Gbc) treatment. Contralateral forelimb use (cylinder test) (
We then used the limb-placing test to further analyze recovery of the forelimb function. All ischemic animals presented initial severe impairment in the limb-placing test after MCAO, followed by spontaneous recovery during the follow-up (Figure 5B). Limbplacing scores between sham-operated and MCAO rats were different in all post-operative time points (
The effect of glibenclamide on recovery of hindlimb function in MCAO rats was analyzed by tapered/ledged beam walking.
Analysis of variance for repeated measures showed an overall group effect (
Results from the Morris water-maze on post-operative days 26 to 28 showed that escape latency, length and swimming speed did not differ between groups. No significant differences were observed in the number of passes over the removed platform or the time rats spent in the target quadrant (probe trial). However, a thigmotaxis behavior was observed in vehicle-treated MCAO rats, which they spent more time in the outermost zone (zone 3;
Glibenclamide binds to reactive microglia
To assess whether microglia express the KATP-channel components after ischemia, we performed double immunohistochemical labeling against SUR1 and Kir6.2 combined with the microglia/macrophage marker CD11b. Co-location of striatal CD11b-positive cells adjacent to the ventricle indicated that the reactive microglia of MCAO animals expressed the KATP-channel components SUR1 and Kir6.2 at 72 hours after ischemia (Figure 6A). To determine the specificity of binding to SUR1 expressed by microglia, we analyzed the binding of fluorescently tagged glibenclamide (glibenclamide BODIPY FL; green fluorescence) in primary rat microglial cultures. When microglial cells were in the resting state, glibenclamide labeling was localized to the perinuclear space (Figure 6B). Forty-eight hours after LPS + IFNγ activation, glibenclamide labeling was present in the plasmalemmal membrane, denoting a translocation of SUR1 from its internal reservoir toward the cell surface.
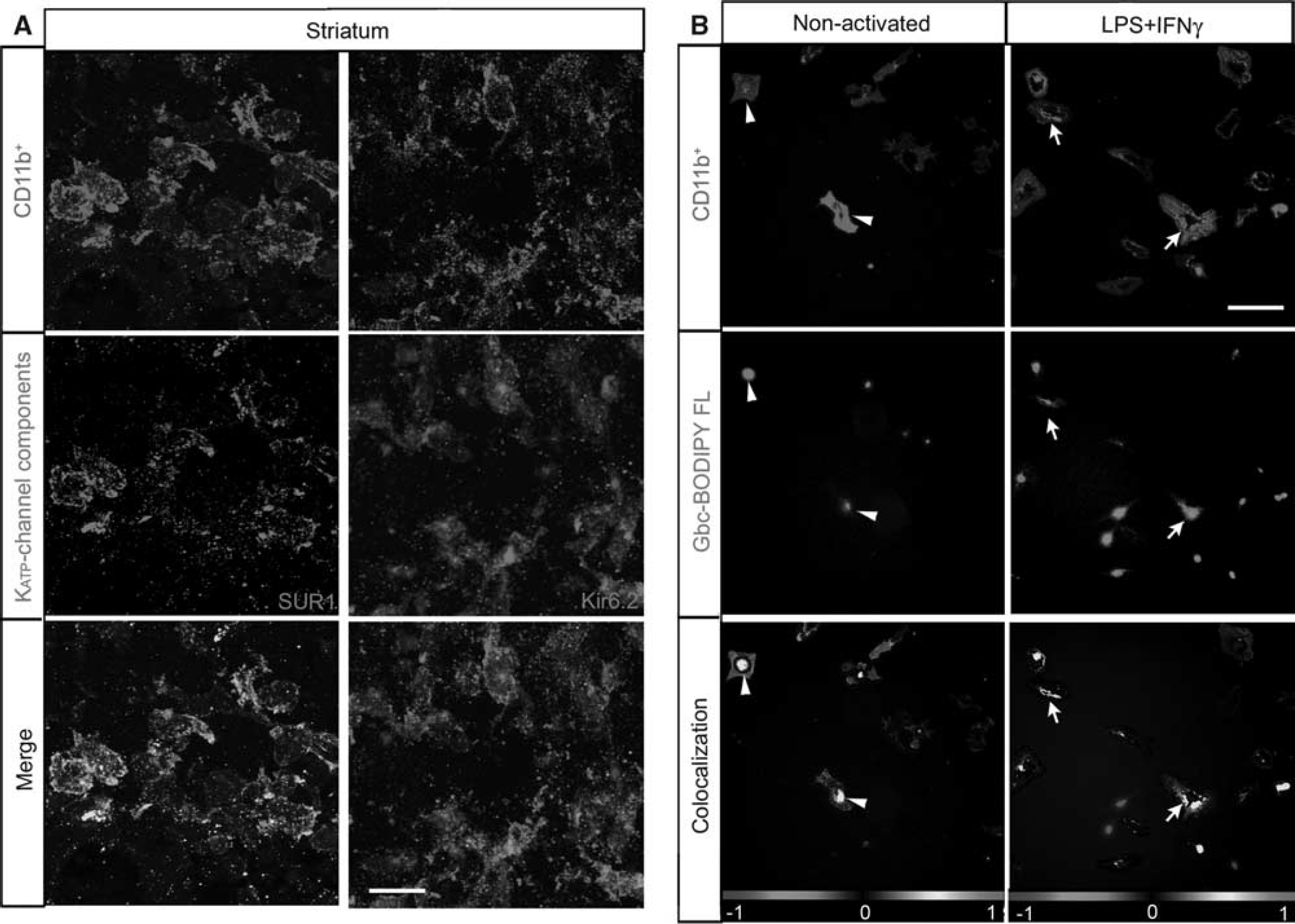
Reactive microglia express and translocate sulfonylurea receptor 1 (SUR1) to the cell surface. (
DISCUSSION
Previous preclinical and clinical data suggest a neuroprotective role for glibenclamide after cerebral ischemia.3,4 Here we showed that glibenclamide also enhanced early migration of neuroblasts toward the striatal lesion core and facilitated long-term brain repair in MCAO rats. Interestingly, this was associated with improved behavioral recovery.
After a stroke, massive necrotic or apoptotic processes are activated in the ischemic core, as well as in distal regions owing to Wallerian neurodegeneration. In our model, we only observed a few scattered FJB-positive neurodegenerative cells, which were mainly located in the thalamus 30-days after ischemia.
In addition, the only cells labeled by cleaved caspase-3 in the peri-infarct zone were some microglia/macrophages. Recent data demonstrated that cleaved caspase-3, a known mediator of apoptosis, regulates microglial activation by a non-apoptotic pathway.
30
Thus, we here found that 30-days after ischemia the majority of secondary neurodegenerative processes have been completed and caspase-3-positive microglia/macrophages may be participating in the tissue repair response to injury instead of undergoing apoptosis. Although glibenclamide did not reduce the size of the infarct, our results are in line with Simard
Focal cerebral ischemia promotes neurogenesis in the subventricular zone and the subgranular zone of the dentate gyrus and induces neuroblast migration toward the striatal ischemic boundary.13,22 Recent data have provided new evidence that the cerebral cortex also has the same capacity 17 and, more importantly, stroke-induced cortical neurogenesis has also been found in the adult human brain.19,20 In the present study, ischemia increased neuroblast migration toward the lesion 3 days after ischemia/reperfusion and this event persisted for up to 1 month. Inhibition of SUR1 using glibenclamide led to a further increase in the number of migrating neuroblasts, thereby indicating that glibenclamide modifies the cell lineage choice or enhances progenitor cell proliferation and migration.
Proliferative cells detected by administration of BrdU from days 4 to 8 after ischemia/reperfusion showed increased co-expression of NeuN in the peri-infarct area of the cortex 30 days after reperfusion and this was potentiated by glibenclamide. Although we cannot exclude the possibility that some neural progenitors migrate from the subventricular zone to establish themselves in the ipsilateral cortical network, we found no co-localization of BrdU-positive cells with the classical RMS-derived neuronal markers (i.e., calbindin, calretinin, tyrosine hydroxylase, and parvalbumin) and no change in the total number of proliferative cells in the olfactory bulb. Thus, these newborn cortical neurons may have originated from potential resident neural stem cells within the cortex. 17 Several authors have proposed that these endogenous quiescent neural stem cells are present in the cerebral cortex and that their proliferation and differentiation to mature neurons is induced by ischemic insults. 18 Although the number of these cells may be small, strategies to foster their intrinsic neurogenic potential would be highly relevant for clinical approaches to facilitate neural repair and functional recovery.
Another purpose of this study was to investigate angiogenesis after cerebral infarction, because angiogenesis is also activated after cerebral ischemia. Generally, tissue repair is activated at 2 to 3 days after reperfusion, and the infarction is eventually encircled by a glial scar a few weeks later. As the glial response, in terms of the reactive area or cell density, was not modified by glibenclamide, we analyzed the microvessel diameter and the surface density as a parameter to quantify angiogenesis.
29
We herein demonstrated that glibenclamide causes microvascular changes, where interestingly, glibenclamide further increased the diameter of microvessels in the non-lesioned cortex and RECA-1 immunoreactivity in the hippocampus, and both regions showed glial proliferation. These observations are consistent with those of Fantin
Several studies using experimental stroke models have indicated the beneficial effects of glibenclamide administration after reperfusion, such as reversing ischemia-reperfusion injury by halting oxidative stress and inflammation in the hippocampus,
9
preventing cytotoxic edema after cerebral ischemia,2,3,1,7 and enhancing neuroprotection, which all eventually lead to a better functional outcome.
8
Thus, although glibenclamide did not modify the size and cell density of microglia nor astroglia after ischemia here, it enhanced long-term brain repair processes and functional recovery outcomes of the rats. We recently reported that 72 hours after MCAO, activated microglia/macrophages in the ischemic hemisphere increase KATP-channel expression, and so does the murine microglial BV2 cell line 48 hours after inflammatory stimuli.
8
We have now confirmed these results in postnatal primary rat microglial cultures. When microglial cells became activated after pro-inflammatory stimuli, they showed higher specific glibenclamide labeling and the signal extended to the plasmalemmal membrane
In summary, we have demonstrated here that early blockade of SUR1 by glibenclamide produces long-term cortical neuroprotection, stimulates neuroblast migration toward the striatal lesion, and strengthens neurogenesis in the cortex after cerebral ischemia. All these results were temporally associated with improved long-term behavioral recovery. Therefore, these data identify SUR1 as a multifaceted target that promotes both long-term neuroprotective processes and brain repair mechanisms. Further experiments are necessary to identify the specific role of the microglial KATP channel and its diffusible mediators that may modulate brain repair, which will help us to shed light on the understanding of the intricate mechanisms underlying neurogenesis and neuroprotection after stroke caused by glibenclamide treatment.
DISCLOSURE/CONFLICT OF INTEREST
NM and MJR hold an EU patent (No. WO2006/000608). The other authors report no disclosures.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.