
Abstract
This review covers the pathogenesis of ischemic stroke and future directions regarding therapeutic options after injury. Ischemic stroke is a devastating disease process affecting millions of people worldwide every year. The mechanisms underlying the pathophysiology of stroke are not fully understood but there is increasing evidence demonstrating the contribution of inflammation to the drastic changes after cerebral ischemia. This inflammation not only immediately affects the infarcted tissue but also causes long-term damage in the ischemic penumbra. Furthermore, the interaction between inflammation and subsequent neurogenesis is not well understood but the close relationship between these two processes has garnered significant interest in the last decade or so. Current approved therapy for stroke involving pharmacological thrombolysis is limited in its efficacy and new treatment strategies need to be investigated. Research aimed at new therapies is largely about transplantation of neural stem cells and using endogenous progenitor cells to promote brain repair. By understanding the interaction between inflammation and neurogenesis, new potential therapies could be developed to further establish brain repair mechanisms.
INTRODUCTION
Stroke is the fourth leading cause of death annually in the United States 1 and is the most common cause of permanent disability in adults worldwide.2,3 Nearly 90% of all stroke victims experience an ischemic event caused by the sudden blockage of blood flow from a thrombus or embolism.1,4 This percentage is even higher if one includes hemorrhagic stroke patients who may experience delayed ischemic events arising from arterial or arteriolar vasospasms associated with subarachnoid hemorrhage. Although the exact mechanism(s) responsible for the brain damage occurring after an ischemic insult is/are not fully understood, there is an increasing amount of evidence suggesting that post-ischemia inflammation is a significant contributing factor to the pathogenic process. Ischemia is accompanied by a rapid increase in inflammatory mediators causing irreversible damage to neurons in the ischemic core, with more long-term consequences ultimately appearing in the ischemic penumbra. However, those pathologies can be largely ameliorated via prompt restoration of blood flow. Clinically, this reperfusion can occur spontaneously or via the use of either pharmacological thrombolysis or endovascular thrombectomy. At the present time, the thrombolytic agent tissue plasminogen activator (tPA) constitutes the only FDA-approved therapy for acute ischemic stroke. However, tPA has several shortcomings including the potential risk of hemorrhagic transformation, therapeutic window limited to 3 hours from time of symptom onset, and limited efficacy, among others. Data obtained from the Get With The Guidelines-Stroke database shows that only 4% to 7% of all the acute ischemic stroke patients are treated with tPA, 5 an observation that highlights the need for novel and effective therapeutic approaches.
The last decade or so has produced large amounts of evidence supporting the existence of neurogenesis in adult mammals. Mammalian brains contain neural stem cells (NSCs) that have the ability to self-renew, proliferate, and differentiate into multiple lineages. 6 The participation of these cells in the process of adult neurogenesis has attracted interest among members of the research community, especially about their use as possible therapeutic options for brain repair. Both transplantation of NSCs and the utilization of endogenous progenitor cells appear to be plausible strategies for brain injury repair. Exploring the interactions between inflammation and neurogenesis has not been fully studied, and gaining a better understanding of the relationship between these two processes could help in the development of new and exciting therapeutic options for brain repair.
ISCHEMIC STROKE
Ischemic stroke causes damage as a result of primary and secondary insults mediated by ischemia and inflammation. Primary injury results from the initial ischemic period, which deprives the brain of oxygen and vital nutrients, such as glucose, critical for survival of central nervous system (CNS) cells. Neurons in the infarcted tissue die as a result of the initial injury whereas cells in the penumbra are affected by the rapid influx of immune cells, reactive oxygen species, and toxic inflammatory mediators. 7 Current therapeutic options for ischemic stroke, and the less damaging transient ischemic attack, are aimed at dissolving the clot causing the blockage of blood flow and reestablishing perfusion of the infarcted area. Although these strategies are beneficial, reperfusion can also further the injury by increasing local inflammation and inducing chronic changes in axonal structure and function, which can increase neuronal damage. 8
There are a number of strategies currently under investigation in the preclinical setting aimed at restoring brain function after ischemic insult. These strategies concentrate not on blood flow restoration but rather on inducing neuroprotection and salvaging brain function after the initial injury. Pharmacologic hypothermia, induced by the TRPV1 channel agonist dihydrocapsaicin, has been shown to be neuroprotective if given within 90 minutes of reperfusion after transient middle cerebral artery occlusion in mice. 9 Furthermore, the pharmacologic agent, LJP-1586, which targets and inhibits leukocyte trafficking has been shown to confer neuroprotection after middle cerebral artery occlusion in a rat model of ischemic stroke. 10 In addition, by preventing interactions of the receptor for advanced glycation endproducts with its ligands (e.g., high-mobility group box-1 and S100B), one can promote corresponding reductions in post-ischemic neuroinflammation and neuropathology. 11
NEUROINFLAMMATION
Inflammation in the Mammalian Brain
In the intact brain, trafficking of cellular and molecular components from the peripheral circulation is regulated by the blood–brain barrier (BBB). However, after brain injury, the tight junctions between endothelial cells of the BBB become permeable, allowing peripheral immune cells to infiltrate brain parenchyma. These cells, along with the proinflammatory cytokines they secrete, contribute to the inflammatory response that follows brain injury. Acute inflammation arises both from responses of resident immune cells, the microglia, as well as from infiltrating immune cells of the peripheral circulation. 12 Inflammation of the mammalian brain, however, is not only a response mechanism that follows CNS injury, such as from traumatic brain injury, spinal cord injury, and stroke. It is also often a large contributing factor to neurologic diseases such as Alzheimer's disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson's disease.13,14 Chronic inflammation and the release of proinflammatory molecules from immune cells have also been implicated as causative factors of neurodegeneration and CNS disorders.14,15 The term ‘inflammation,’ and subsequently the ‘inflammatory response,’ seek to combine many different processes and pathways into one unified theme when, in fact, they are distinct pathways that have different downstream consequences. Therefore, it is imperative to keep in mind that, although all of the aforementioned pathologies and pathologic states have an inflammation-related component to them, the specific inflammatory response could be, and most likely is, very different among the various diseases and injuries.
Susceptibility to stroke and subsequent prognosis are highly influenced by peripheral inflammatory cells. Numerous inflammatory cells, including neutrophils, macrophages, and T cells, are recruited to sites of ischemic injury. 16 The mechanism by which these cells decrease neurogenesis after ischemic injury, and the way in which they affect NSCs, remains largely unknown. Post-ischemic inflammation is characterized by microglia activation followed by infiltration of circulating inflammatory cells.16–20 Acutely, reactive oxygen species and inflammatory mediators cause endothelial cell and leukocyte expression of adhesion molecules, promoting the adhesion and migration of circulating leukocytes that leads to a rapid inflammatory state at the site of injury. These leukocytes further release inflammatory cytokines that lead to tissue damage in the injury site and the ischemic penumbra (Figure 1).
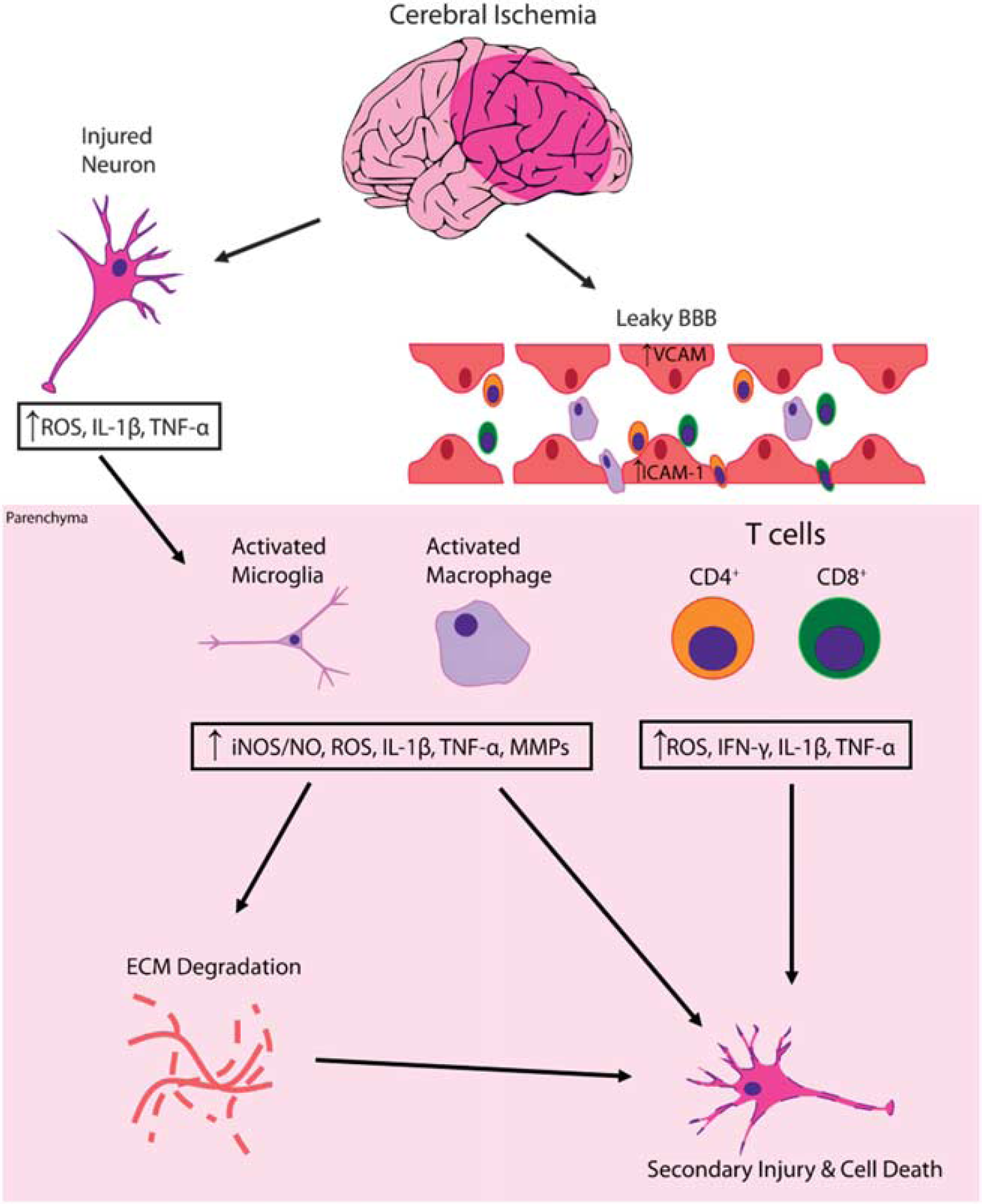
Inflammatory cascade leading to secondary brain inflammation and cell death after ischemic injury. Cerebral ischemia alone leads to both an initial cell necrosis and generation of reactive oxygen species (ROS) and proinflammatory molecules like interleukin-1 beta (IL-1
Like any ischemia–reperfusion injury, a large portion of the damage is due to the inflammatory cascade that follows reperfusion. This response results in an increase in toxic inflammatory mediators including, but not limited to, interleukin (IL)-1
Peripheral Versus Resident Immune Response after Ischemic Injury Under normal physiologic conditions, trafficking of peripheral immune cells into the brain is restricted. 23 This status is maintained by the BBB. Under such conditions, immune surveillance in the CNS is performed by resident microglia. When the BBB becomes dysfunctional, as occurs after injury, circulating immune cells can gain direct access to the brain. The relative contributions to the inflammatory response from peripheral immune cells and resident immune cells after injury, though, need to be further elucidated.
Acutely (4 to 24 hours) after ischemic injury, circulating immune cells begin to adhere to, and migrate along, the damaged endothelium lining the cerebral vasculature and start to infiltrate into brain parenchyma owing to the breakdown of the BBB.
24
Macrophages, which reside in the perivascular space in the glia limitans, are a large driving force behind infiltration of peripheral immune cells into the brain parenchyma.
25
Macrophages exist in multiple states that are phenotypically and functionally distinct. M1-type macrophages are proinflammatory and secrete numerous inflammatory mediators such as TNF-
After macrophage and neutrophil infiltration, lymphocytes begin to infiltrate the brain as well.
24
Under normal conditions, naïve T cells are not capable of migrating through the BBB to enter the brain. However, after injury, activated CD4+ and CD8+ T cells can enter the brain in an antigen- and major histocompatibility complex-independent fashion.29,30 In addition, there is evidence showing the presence of CNS-specific T cells located within the choroid plexus that promotes leukocyte trafficking into the CNS after injury.31,32 Approximately 15% of these choroid plexus T cells are CD4+ TH1 cells, which secrete interferon-
While the peripheral immune response is an extremely important factor in neuroinflammation, the response of activated microglia is of equal, if not more, importance. Resting microglia are the resident immune cells of the brain and their job is to actively survey the brain. Like peripheral antigen-presenting cells, microglia are continuously extending and retracting their processes looking for any signs of damage. Similar to macrophages, microglia also exist in two different states, the M1 and M2 phenotypes.
33
On activation, as occurs in ischemic injury, microglia take on the M1 phenotype and secrete various proinflammatory molecules including IL-1
Signaling molecules associated with the inflammatory cascade
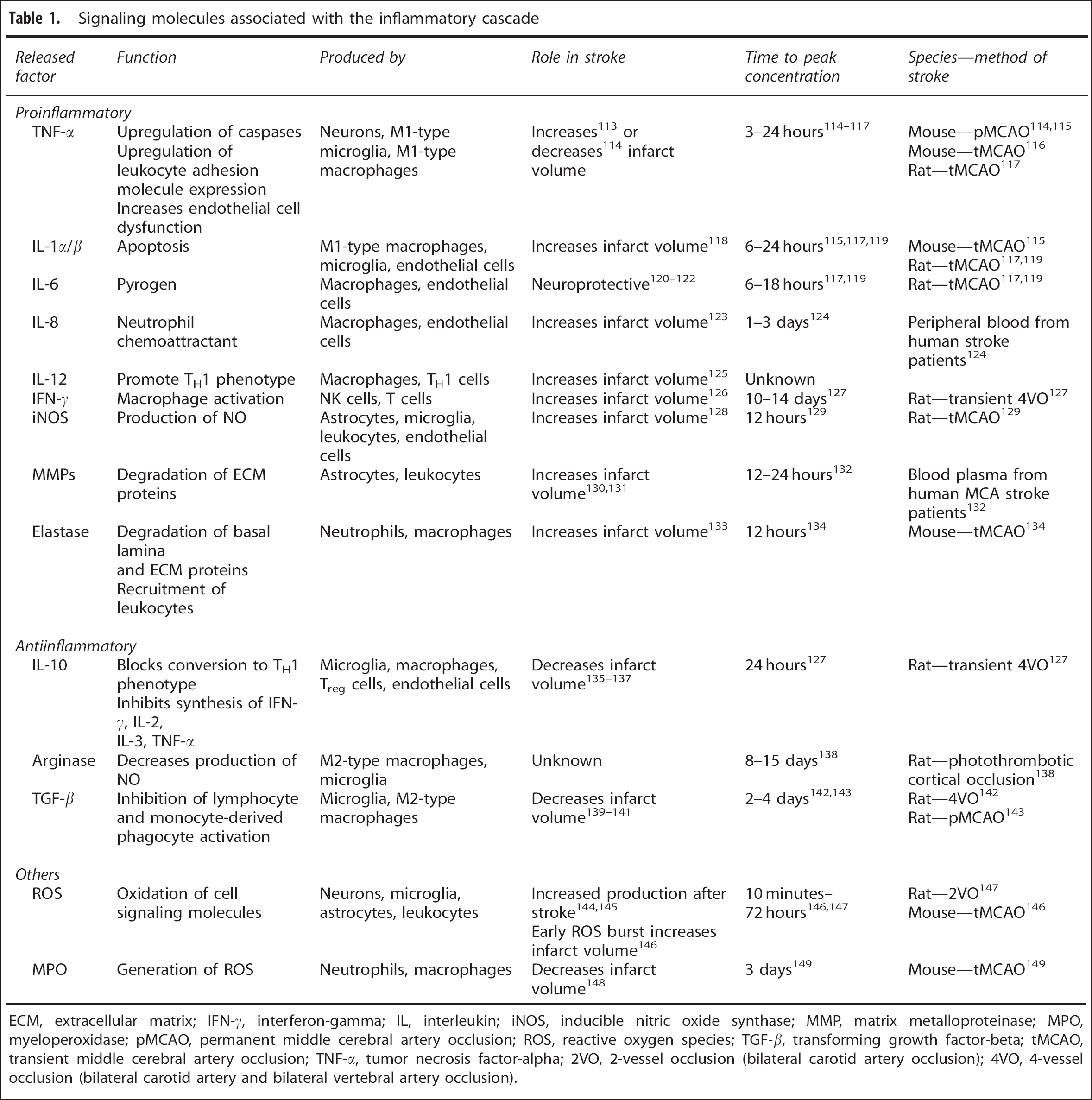
ECM, extracellular matrix; IFN-
Neuroinflammation after ischemic stroke is, for the most part, a self-limiting event. However, unlike systemic inflammation that subsides due largely in part to exhaustion of inflammatory mediators, resolution of inflammation in the brain seems to be an active process in which inflammatory mediators are suppressed by regulatory mechanisms.
26
Among these suppressor molecules are arachidonic acid derivatives such as lipoxins, resolvins, and protectins.
34
Furthermore, resolution of inflammation in the brain, similar to systemic inflammation, involves clearing of dead and dying cells primarily via the phagocytic activity of activated microglia with help from infiltrating macrophages35,36 as well as production of antiinflammatory, proregenerative signaling molecules like IL-10 and TGF-
Current research aimed at the inflammatory state after ischemia is targeted at reducing infarct size after ischemia.16–20 While this approach is important, it does not address a crucial aspect of repair: the neural stem cell. Neural stem cell numbers increase after ischemic stroke; however, they rapidly die 37 thereby preventing successful brain repair. By understanding how inflammation increases the susceptibility toward premature NSC death, we can better design therapies to promote brain repair.
NEUROGENESIS IN THE ADULT BRAIN
Traditionally, neurogenesis was thought to only occur during embryonic and perinatal development; 38 however, over the last decades, it has become clear that neurogenesis occurs throughout adulthood in distinct regions of the brain. These regions consist of the subventricular zone (SVZ), located adjacent to the lateral ventricle, and the subgranular layer (SGL) of the dentate gyrus (DG) in the hippocampus. Furthermore, these two regions of the brain maintain distinct populations of NSCs that exhibit multipotency and greater plasticity compared with other cellular components of the central nervous system. 6 The NSCs located in the SVZ, referred to as radial glia-like B cells, are quiescent, glial fibrillary acidic protein (GFAP) positive, and have characteristics in common with astrocytes.6,39 These type B cells give rise to the so-called transit-amplifying type C cells, which are GFAP negative. From there, type C neural progenitor cells (NPCs) give rise to doublecortin (DCX)-expressing neuroblasts. In rodents and nonhuman primates, these neuroblasts travel through the rostral migratory stream (RMS) to the olfactory bulb where they ultimately become periglomerular neurons or granule cells (Figure 2).6,40 However, while there is a comparable SVZ region in the human brain, there exists a unique hypocellular layer that lacks cellular bodies. This region is thought to be the site for the regulation of neuronal function, metabolic homeostasis, and/or NSC proliferation and differentiation. 41 Furthermore, the existence of an RMS or any pathway leading to the olfactory bulb is controversial with previous studies lacking demonstration of a similar RMS track in humans.40,42 However, more recent studies43,44 describe the presence of NSCs in the human striatum 43 and also the presence of an RMS in humans with neuroblast migration to the olfactory bulb (Figure 3).44,45 These studies illustrate the ongoing discussion in the scientific community about the presence or absence of an RMS in humans and demonstrate the need for further research to fully elucidate the extent of adult human neurogenesis.
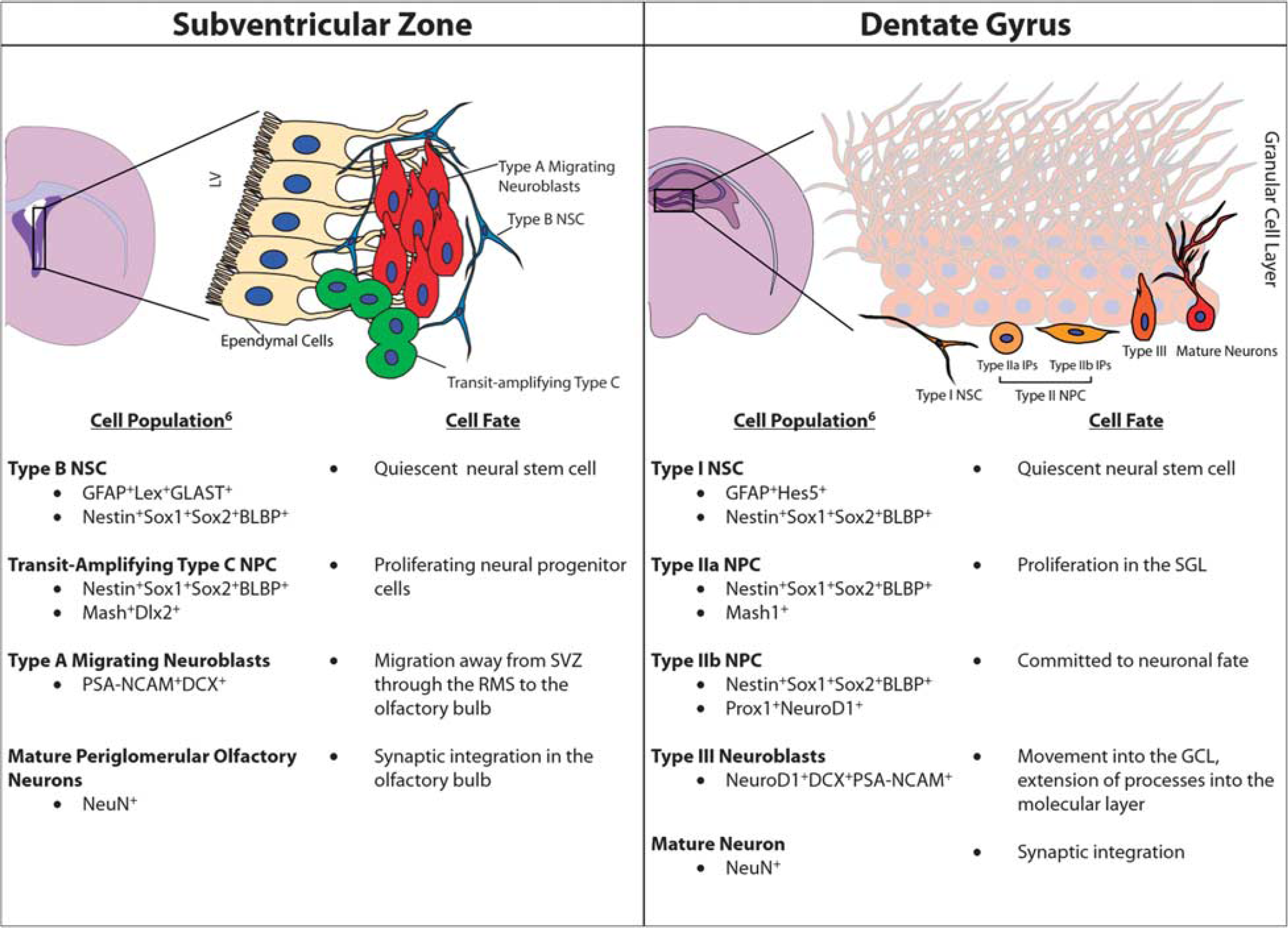
Adult neurogenesis in rodents. Coronal sections of the rodent show the neurogenic environments of the adult brain: the subventricular zone (SVZ; left panel) and the subgranular layer (SGL; right panel). The SVZ contains type B neural stem cells (NSCs), which give rise to transit-amplifying type C cells followed by type A migrating neuroblasts. These cells migrate along the rostral migratory stream (RMS) toward the olfactory bulb before terminal differentiation. The SGL contains type I NSCs, which are similar to the type B cells in the SVZ. Type I NSCs give rise to type II NPCs (or intermediate progenitors; IP), which can be further classified as type IIa and type IIb. These type IIb cells are early committed neuronal progenitor cells, which give rise to type III neuroblasts, which migrate into the granular cell layer where they exit the cell cycle and become mature neurons. All cell types described here can be identified based on a unique set of molecular and morphologic markers, as described in the cell population column. GCL, granular cell layer; GFAP, glial fibrillary acidic protein.
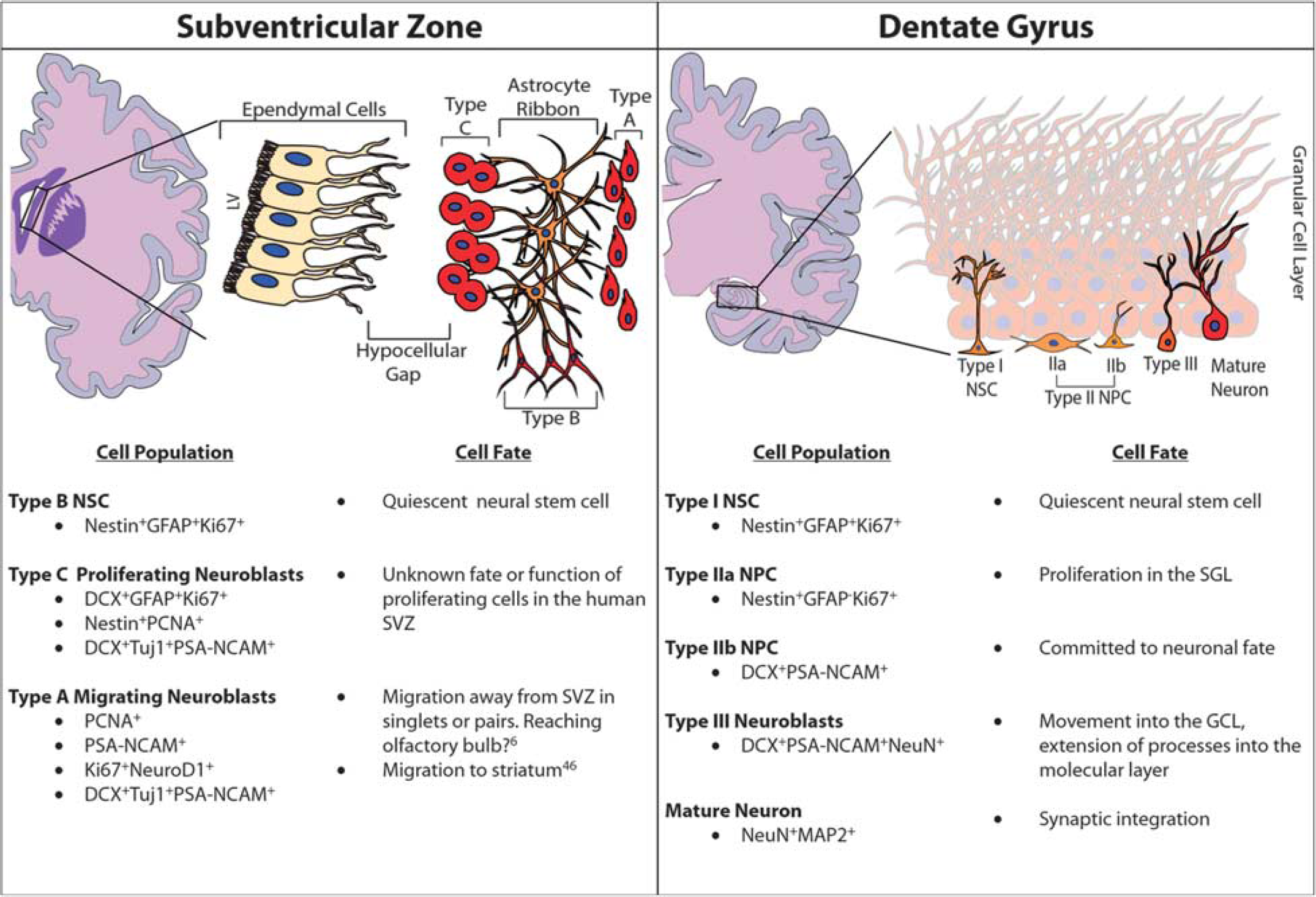
Adult neurogenesis in humans. Coronal sections of the human brain show the neurogenic environments of the adult brain: the subventricular zone (SVZ; left panel) and the dentate gyrus (DG) of the hippocampus (right panel). Cell types unique to these regions as well as the fate of these cells are listed. Cell types described here can be identified based on a unique set of molecular and morphologic markers, as noted in the cell population column. GCL, granular cell layer; LV, lateral ventricle; SGL, subgranular layer.
Neural stem cells in the DG give rise to new neurons which reside within the granular cell layer (GCL). In both rodents and humans, two distinct subpopulations can be identified in the DG that are unique in their morphologies and molecular marker expression profiles. Similar to the type B cells of the SVZ, type I cells express GFAP and have a similar radial glia-like morphology. Type II cells are more similar to the type C cells of the SVZ and are also GFAP negative. Type II cells can be further subdivided into two subpopulations: (1) type IIa cells, which express Mash1 and continue to express the NSC marker Sox2, and (2) type IIb cells, which are early committed NPCs, which express the transcription factors Prox1, NeuroD1, and DCX. Like in the SVZ, NSCs in the DG proceed through similar levels of differentiation from type I/II NSCs to NPCs to DCX-positive neuroblasts and ultimately into new, mature neurons in the GCL.
46
These newly formed neurons project axons into the CA3 region of the hippocampus and dendrites into the molecular layer of the DG, where they form synapses with entorhinal terminals much like neurons formed during embryogenesis.6,47 The rate at which this happens
Interaction between Inflammation and Neurogenesis
Thoughtful research has demonstrated the active role of neuroinflammation in both secondary brain injury and neurorepair after stroke. The effect of the post-ischemic immune response on neurogenesis is not well understood. However, studies done in different models of disease demonstrated that an active cross talk exists between inflammation and neurogenesis. Invertebrate models of brain injury and inflammation showed that inflammation is both sufficient and necessary to increase endogenous neurogenesis after injury.
48
Data obtained in vertebrate models of chronic neuroinflammation induced by stereotaxically injected lipopolysaccharide into the DG, for example, demonstrated the functional integration of new, adult born, highly plastic hippocampal neurons.
49
Also, the presence of chronic inflammation induced by electrical induction of status epilepticus was linked to a sevenfold increase in the number of mature neurons in the dentate GCL formed during the first 2 weeks after seizure induction with the majority of these newborn cells replacing dead granule cells. Significantly, 6 months after seizure activity, there was evidence of continued neuron formation in the hilus of the DG supporting the long-term effect of inflammation on neurogenesis.
50
These reports are promising for utilization of endogenous neurogenesis as a repair mechanism after brain injury. However, the interplay of inflammation and neurogenesis is complex and there is substantial evidence obtained in vertebrate models of brain injury that show that inflammation impairs not only basal neurogenesis levels but also attenuates the increased neurogenesis seen after injury via increased activated ED1+ microglia
51
and via secretion of numerous proinflammatory cytokines including IL-6, TNF-
Both pro- and antiinflammatory cytokines can influence the proliferation and migration of neural progenitors.58,59 The link between the immune response that occurs after ischemic stroke, neurogenesis, and subsequent functional recovery has not been well established; however, hypotheses regarding both the beneficial
60
and detrimental61,62 effects of inflammation have been proposed. Some of the beneficial effects have been attributed to the interaction of T cells with microglia, which stimulates proliferation of SGL and SVZ progenitor cells
60
and directs both NPC migration and differentiation
Effect of Ischemia on Neurogenesis
There are increasing amounts of evidence suggesting that ischemic injury drastically increases neurogenesis in the rodent SVZ and SGL70–73 as well as in the primate SGL.74,75 Furthermore, studies in mice76,77 and rats78–82 demonstrate an increase in NPC proliferation in the DG peaking around 7 days after ischemic injury.70,77,80 However, the increased number of NSCs quickly returns to baseline with most of the NSCs dying shortly after proliferation. In fact, the number of NPCs that survive and become mature neurons are approximately equal in number in animals undergoing ischemia compared with animals without injury. The cells that do survive, however, migrate from the SGL into the GCL, have increasing dendritic length, and begin to both lose expression of immature neuronal markers and gain expression of mature neuronal markers with increasing time after ischemia after a similar time course to normal neuronal maturation. 83 Despite the increase in NPC numbers after an ischemic injury, these cells prematurely die without the ability to repair dead tissue. The mechanisms by which these NPCs die early are as yet undetermined.
Similar to the SGL, NPCs in the SVZ have a short-lived proliferation again peaking around 7 days after ischemia.70,84 Also like the cells in the SGL, those NPCs that do survive migrate out of the SVZ into the striatum and neocortex and mature into 15 functional neurons.73,85–89 In addition, it has been demonstrated that transient angiogenesis after stroke allows neuroblasts to migrate along these newly formed blood vessels aiding in their migration out of the SVZ. 89 While some of these newly generated neurons survive up to 5 weeks after ischemia, the majority seem to die with only ~0.2% of the cells surviving. 73 Furthermore, these surviving cells have been shown to be spiny neostriatal projection neurons73,85 and small, calretinin+ interneurons. 90 Despite these promising data demonstrating functional maturation of NPCs in both the SVZ and SGL after ischemic injury, much work still needs to be done to determine why the majority of these cells die, why the initial NPC proliferation fails to promote brain repair, and how neurogenesis can be manipulated for the support of functional recovery.
PROSPECTIVE THERAPIES AFTER ISCHEMIC STROKE
Over the last decade, significant advances have been made in our understanding of the pathogenesis of brain injury from stroke. The knowledge gained, however, failed to translate into novel treatment strategies. Our current armamentarium to treat cerebral ischemia relies mainly on the use of pharmacological thrombolytics (tPA) in acute cases and antithrombotic therapy along with correction of the modifiable vascular risk factors for recurrent stroke prevention. The time window for initiating the treatment with tPA is limited to 3 hours after stroke symptom onset. The short therapeutic window, stroke severity, concern of the occurrence of major or fatal hemorrhage, severe hypertension, and other variables greatly limit the number of patients that can benefit from this treatment. The development and implementation of hospital-based protocols with the goal of facilitating the identification of ischemic stroke patients and the early treatment with tPA has increased the use of thrombolytics from 4% in the period 2003–2005 to 7% in 2010–2011 (
Antiinflammatory Treatment Strategies
As discussed previously, a large effector of stroke outcome is the post-ischemia inflammatory environment that exists. Therefore, large efforts are underway to target these inflammatory pathways in an attempt to reduce their effect on the brain after the initial ischemic injury. Various research studies as well as clinical trials have been conducted to examine the effect of various antiinflammatory treatments after ischemic stroke (Table 2). While two of these studies report better outcomes with their antiinflammatory treatment,91,92 the remaining studies report worse outcomes,93,94 no difference between treatment and placebo,95–98 or were simply safety studies99–101 with all of them failing to make it beyond clinical phase trials. Importantly, some of these studies were underpowered to detect a meaningful clinical effect. However, the dual effect of inflammation on stroke outcome and particularly on neurogenesis highlights that antiinflammatory strategies might be a difficult avenue to pursue in ischemic stroke treatment. Further studies, however, need to be conducted to hone in on more specific targets in the inflammatory cascade that can hopefully be effective targets for future therapeutic trials. One such target is vascular adhesion protein-1, which is expressed by endothelial cells and aides in neutrophil transmigration from the vasculature into the brain parenchyma. The pharmacologic agent LJP-1586, and its predecessor LJP-1207, are highly selective vascular adhesion protein-1 inhibitors and prevent neutrophil transmigration into the brain parenchyma. Furthermore, when these drugs are given to rodents subjected to transient forebrain ischemia 102 or transient middle cerebral artery occlusion 10 6 to 12 hours after reperfusion, there is a profound antiinflammatory action, which is linked to neuroprotection.10,102 These studies, and others currently in preclinical development may identify clinical targets for antiinflammatory therapeutic options.
Clinical studies utilizing antiinflammatory agents in acute ischemic stroke
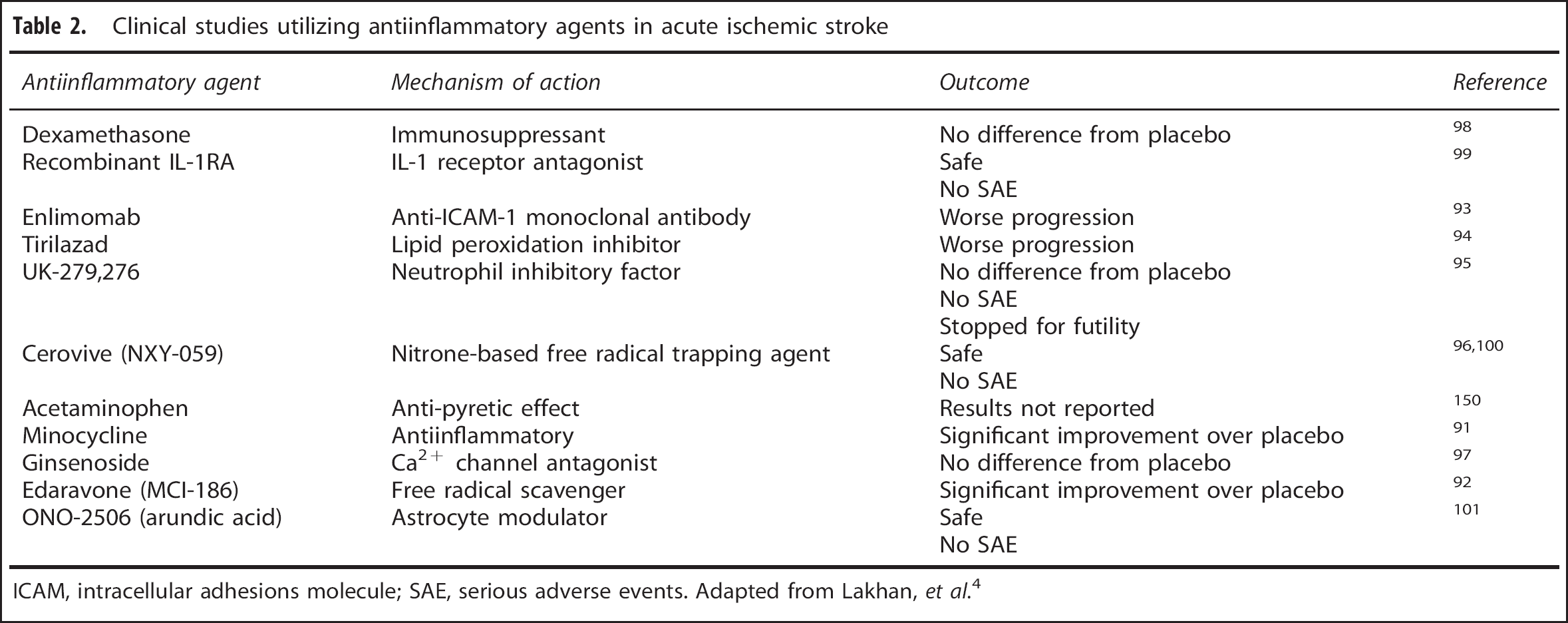
ICAM, intracellular adhesions molecule; SAE, serious adverse events. Adapted from Lakhan,
However, while these studies offer promising avenues for future research, the fact that blocking post-ischemia inflammation could also eliminate any beneficial effects of inflammation cannot be overlooked. Although the consensus thus far that inflammation is largely detrimental to neurogenesis after stroke there is evidence, as discussed above, showing that chronic neuroinflammation supports maturation of highly plastic hippocampal NSCs.49,50 Therefore, investigators must weigh the advantages and disadvantages when considering these types of treatment approaches, as they could potentially inhibit potent proregenerative effects.
Regenerative Strategies
Current therapies involving regenerative strategies have mostly involved two separate ideas: (1) the ability to repopulate infarcted tissue via transplantation of stem cells and (2) utilizing endogenous progenitor cells to repopulate the damaged brain. Transplantation of stem cells has been attempted in rodent models of disease103–105 with some degree of success, showing benefit to the animals receiving stem cell transplantation. In both rodents and primates with damage to the hippocampus, functional recovery was seen after transplantation of both fetal and immortalized neuroepithelial stem cells. 103 In addition, when compared with young animals, aged animals (which naturally experience decreased endogenous neurogenesis) demonstrated increased performance on hippocampus-based memory tasks after transplantation of immortalized neuroepithelial stem cells. 104 Furthermore, human NSCs injected into aged rat brains successfully integrated into brain parenchyma and increased performance on hippocampus-based memory tasks. 105 Stem cell transplantation seems to be beneficial; however, getting stem cells to engraft with a high degree of efficiency and to differentiate into mature cells is highly variable. Furthermore, the translation of these studies in animal models to humans poses significant technical hurdles that would make the use of cell transplantation difficult in clinical practice. Consequently, utilizing endogenous neurogenesis mechanisms provides an alternative approach to regenerative therapies after ischemic injury.
Not only has transplantation of stem cells been shown to be directly beneficial to the injured brain, but it has also been shown to stimulate endogenous neurogenesis. 106 Endogenous neurogenesis, though, is not a one-step process. It is a series of events involving the proliferation of NSCs/NPCs, migration of these cells to the damaged area, survival of these cells, differentiation of progenitor cells into different lineages of neural cells, and functional integration of these mature cells. Therefore, therapies aimed at each of these steps could prove beneficial in successful brain repair after ischemic stroke. As discussed earlier, proliferation of NSCs does not seem to be impaired after stroke. Rather, it is the survival of these progenitor cells that seems to be hindered with up to 80% of newborn neurons dying within 2 weeks after their birth. 69 It would therefore seem that therapies aimed at increasing the survival of NSCs and NPCs would be the most advantageous. These strategies, though, are not without complication. The existence of large numbers of NSCs is not seen in humans to the same extent as in rodents.70,107 Consequently, manipulation of endogenous NSCs alone may not be sufficient to adequately repopulate damaged brain tissue.
Furthermore, the pathologic environment created after ischemic stroke pose numerous hurdles for newborn neurons that makes the utilization of endogenous repair mechanisms difficult. For instance, aged and injured brains have reduced neurogenic signaling making it even more difficult to direct the migration and differentiation of endogenous NSCs needed to repair infracted tissue.6,108 In addition, unlike other areas of the body, the brain is unique in its response to hypoxic cell death in that it ultimately leads to gliotic scar formation and liquefactive necrosis, which will negatively impact all aspects of endogenous repair. Not only will NPCs and neuroblasts be unable to migrate but any surviving newborn neurons will have difficulty integrating into existing circuits and forming functional synaptic connections. Because of these issues, successful recruitment and differentiation of NSCs into cortical and subcortical structures may be difficult to attain, making the use of endogenous progenitor cells an unlikely source of independent brain repair.
Despite these obstacles, newborn neurons have several advantages compared with mature cells that can help overcome these barriers. The most obvious of these advantages is that, unlike the postmitotic mature neurons, NSCs and NPCs can undergo asymmetric cell division and not only produce mature daughter neurons but can maintain the stem and progenitor pools allowing for continued brain repair. In addition, it has been shown that young, newborn neurons in the GCL of the DG are more active than old granule cells and that the young cells are necessary for enhanced pattern separation. 109 Furthermore, immature and newborn neurons are more excitable than mature cells. Consequently, they fire more action potentials in a given time than their mature counterparts.110–112 Thus, overall circuit excitation will be increased, which could help ameliorate some of the loss of excitation after cell death after ischemia. Therefore, in combination with other strategies such as stem cell transplantation, the use of endogenous neurogenesis mechanisms could prove to be extremely beneficial for brain repair.
CONCLUSIONS
Ischemic stroke is a devastating disease that affects millions of people every year. The basic mechanisms leading to the full extent of damage are still not fully understood. Furthermore, strategies to repair the brain after ischemic injury have left hopes for successful treatment largely unfulfilled. It is becoming more and more evident that inflammation has a substantial role in the insult obtained after ischemia and that neurogenesis is in some way affected by this inflammation. How these two processes are related is still highly debated, but it seems that their interaction is critical in understanding at least part of the mechanism behind the ischemic injury. By elucidating the relationship between inflammation and neurogenesis after stroke, it is possible that better, more efficient therapies could be developed to aid in brain repair after ischemic injury. However, only by understanding this relationship can we hope to successfully implement therapies that may be developed in the future.
Footnotes
The authors declare no conflict of interest.