
Abstract
Extracellular ATP, which is released from damaged cells after ischemia, activates P2 receptors. P2Y1 receptors (P2Y1R) have received considerable attention, especially in astrocytes, because their activation plays a central role in the regulation of neuron-to-glia communication. However, the functions or even existence of P2Y1R in microglia remain unknown, despite the fact that many microglial P2 receptors are involved in several brain diseases. Herein, we demonstrate the presence and functional capability of microglial P2Y1R to provide neuroprotective effects following ischemic stress. Cerebral ischemia resulted in increased microglial P2Y1R expression. The number of injured hippocampal neurons was significantly higher in P2Y1 R knockout (KO) mice than wildtype mice after forebrain ischemia. Propidium iodide (PI) uptake, a marker for dying cells, was significantly higher in P2Y1R KO hippocampal slices compared with wildtype hippocampal slices at 48 h after 40-min oxygen–glucose deprivation (OGD). Furthermore, increased PI uptake following OGD was rescued by ectopic overexpression of P2Y1R in microglia. In summary, these data suggest that microglial P2Y1R mediate neuroprotective effects against ischemic stress and OGD insult.
Introduction
Microglia are the principal immune cells of the central nervous system (CNS), monitoring and rapidly responding to alterations in the CNS microenvironment. 1 Once brain ischemia occurs, microglia are activated and migrate toward the ischemic lesion, acting as a potent modulator of CNS repair and regeneration. However, these immune cells are thought to be double-edged swords in the recovery process following ischemia. Namely, activated microglia promote brain recovery by clearing cellular debris and releasing a plethora of neuroprotective factors, including interleukin-6 (IL-6), transforming growth factor-β, and plasminogen.2,3 Conversely, microglia hinder CNS repair and expand tissue damage; brain inflammatory responses dominated by microglia contribute to the exacerbation of neuronal damage and degeneration by secreting neuropathic factors such as glutamate, tumor necrosis factor α, and/or nitric oxide.4,5 These conflicting microglial roles are not ‘all-or-none’ processes, but rather a continuum that depends on encountered stimuli. 2 The basic molecular mechanisms of the dual roles that microglia play after brain ischemia remain obscure, and a better understanding could promote the advancement of treatments for ischemic brain injury.
Adenosine 5'-triphosphate (ATP) is a well-known intracellular energy currency, but also is released into the extracellular space and functions as a neurotransmitter that mediates intercellular communications. It has been demonstrated that ATP is released from both neurons and glial cells to regulate a wide variety of brain functions, including neuronal excitability in normal or pathological situations.6–10 Under normal conditions, ATP acts on specific receptors – P2 receptors that consist of ligand-gated ionotropic receptors (P2X1-7) and G-protein coupled metabotropic receptors (P2Y1,2,4,6,11–14). 11 Once invasive stimuli such as ischemia, trauma, and inflammation occur, ATP release or leakage from neurons or glia is dramatically induced, thereby accentuating ATP/P2 receptor-mediated signals in the brain. 12 Recent studies have revealed that these signals control activated microglia functions, such as chemotaxis, proliferation, and reactivity, via the specific microglial P2 receptor.2,3,11 For example, P2Y12 and P2X4 receptors are involved in microglial migration and extension of processes after neuronal apoptosis, respectively,13,14 P2Y6 receptors evoke phagocytosis for injured neurons,15,16 and P2X7 receptors drive microglial activation and proliferation.17–19
In cerebral ischemia, microglial P2 receptors, such as P2X1,2,4,7 and P2Y6,12, have been implicated in injury pathogenesis.14,15,20,21 However, the roles of other microglial P2 receptors, such as P2Y1 receptors (P2Y1R), remain to be elucidated, although the role of astrocytic P2Y1R has been well established. Under normal physiological conditions, astrocytic P2Y1R expression controls a variety of brain functions by regulating neuron-to-glia communications, including synaptic transmission.3,22,23 When astrocytes are activated by extracellular ATP released from damaged neurons or glial cells, astrocytic P2Y1R induces neuroinflammatory responses by releasing neuroprotective cytokines, such as IL-6. 24
With regard to microglial P2Y1R expression, the function or even existence remains a matter of debate, despite the fact that several lines of literature have reported on their existence or functional consequence.10,14,15,25–27 The role of microglial P2Y1R expression under pathological conditions has been reported in a swab injury model, which showed receptor upregulation and the possible involvement with microglial activation. 11 Therefore, we investigated P2Y1R expression in microglia, as well as the role of P2Y1R expression in neuroprotection following hypoxic/ischemic injury to further the development of novel therapeutic approaches for ischemic brain injury.
Materials and methods
Animals
All experimental procedures using animals were performed in accordance with National Institutes of Health (NIH) guidelines and with the approval of the Animal Experimentation Committee of the University of Yamanashi, Japan. Every effort was made to minimize the number of experimental animals used and their suffering. Reporting of this work complies with ARRIVE guidelines. P2ry1 (−/−) (P2Y1R knockout; KO) mice with a C57BL/6 background and their wildtype (WT) littermates (Japan SLC, Shizuoka, Japan) were used. 28 All animals were housed with 12-h light/dark cycles and had access to food and water ad libitum.
To determine the specific role of microglial P2Y1R, microglial P2Y1R expression was ectopically controlled using a P2Y1R tetracycline operator (tetO) knockin and ionized calcium-binding adapter molecule 1 (Iba1)-tetracycline-controlled transcriptional activator (tTA) mouse system. Detailed procedures for generation of these transgenic mice have been previously described.29,30 In brief, two transgenic mouse models were created, as follows. (1) TetO-P2Y1R mice (TetO-P2Y1R (Tg/Tg)::Iba1-tTA (+/+); termed tetO-P2Y1R): in these mice, P2Y1R expression is the same as WT mice throughout the entire body. (2) TetO-P2Y1R mice × Iba1-tTA mice: in these mice, with P2Y1R expressed normally throughout the entire body but selectively overexpressed in microglia (TetO-P2Y1R (Tg/Tg)::Iba1-tTA (+/Tg); termed tetO-P2Y1RmOE). All mice were housed as described above and underwent genotyping when they were two to three days old.
Preparation of cultured microglia
Microglia were obtained from primary cell cultures of neonatal murine brain tissues as previously described. 31 Cerebral cortices dissected from newborn WT (n = 5) or P2Y1R KO (n = 5) mice were digested with 0.1% trypsin-ethylenediaminetetraacetic acid, and cells (4 × 105 cells/cm2) were placed into 75-cm2 culture flasks in culture medium (10% fetal calf serum-Dulbecco’s Modified Eagle’s Medium, Life Technologies, Carlsbad CA) and maintained at 37℃ in a humidified atmosphere of 10% CO2. After 10 days in culture, the microglia were prepared as floating cell suspensions by shaking the growth flasks. Aliquots of cell suspensions were transferred to 100-mm Petri dishes and allowed to adhere at 37℃ for 60 min. Unattached cells were removed by rinsing with culture medium.
Measurements of intracellular Ca2+
An increase in intracellular Ca2+ concentration ([Ca2+]i) in microglia was measured using the fura-2 method with minor modifications. 31 Culture medium was replaced with balanced salt solution (BSS) consisting of the following: 150 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 10 mM D-glucose (Wako Pure Chemical, Osaka, Japan). Cells were loaded with fura-2 by incubation with 10 µM acetoxymethyl fura-2 (fura-2-AM) in BSS for 40 min at room temperature. After loading, the samples were washed with BSS and mounted on an inverted microscope (ECLIPSE TE2000-U, Nikon, Tokyo, Japan) equipped with a 75-W xenon lamp and band-pass filters of 340 and 380 nm wavelength to measure Ca2+-dependent signals. The ratio of fluorescence intensities at 340 nm and 380 nm (F340/380 ratio) was recorded as an indication of [Ca2+]i. To compare Ca2+ responses to the specific P2Y1R agonist MRS2365, the amplitude of Ca2+ response in a single cell was calculated as the ratio of F340/380 at peak response minus the baseline ratio of F340/380 (average calculated over 2 min before stimulation). Recording and data analysis were performed using Aqua Cosmos software (Hamamatsu Photonics, Shizuoka, Japan).
Microglial isolation and flow cytometry
Male WT (n = 6), TetO-P2Y1R (n = 3), and TetO-P2Y1RmOE (n = 3) mice were used. Briefly, after perfusion with ice-cold phosphate-buffered saline (PBS) into the left ventricle, brains were dissected and enzymatically digested using Neural Tissue Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) for 30 min at 37℃. Tissue debris was removed by passing the cell suspension through a 70 µm cell strainer. Cells were stained with CD11b microbeads (Miltenyi Biotec) in PBS supplemented with 0.5% bovine serum albumin for 30 min. CD11b+ cells were separated in a magnetic field using MS columns (Miltenyi Biotec). Isolated microglia were re-suspended in fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 10% fetal bovine serum) and stained with rabbit anti-P2Y1R antibodies (1:200, Alomone Labs, Jerusalem, Israel) for 2 h at 4℃. Cells were then washed and stained with Alexa-488 conjugated goat anti-rabbit antibodies (1:1000, Life Technologies) for 1 h at 4℃. After washing, cells were examined with a FACS Caliber (Beckman Coulter, Brea, CA) and analyzed using Cell Quest Pro (Beckman Coulter).
Mouse model of transient forebrain ischemia
Male WT (n = 19) and P2Y1R KO (n = 13) 8–12-week-old male mice were used. Anesthesia was induced by inhalation of 4% halothane (Takeda Pharmaceutical Company, Osaka, Japan) and maintained with 1–2% halothane in 70% nitrous oxide/30% oxygen via a facemask. Rectal temperature was recorded and maintained at 36.5–37.5℃ during the procedures using a homeothermic blanket. Transient forebrain ischemia was induced by 20-min bilateral common carotid artery occlusion (BiCCAO). Regional cerebral blood flow was monitored by laser-Doppler flowmetry (FLO C1; Omegawave Inc., Tokyo, Japan). A reduction in blood flow of at least 90% from baseline in the first 2 min was considered to be successful forebrain ischemia and was used for analyses. 32 Sham groups were subjected to a similar procedure without occlusion of the carotid arteries (n = 3). Mice were euthanized with diethyl ether and were transcardially perfused with 4% paraformaldehyde (PFA) at 12 (WT, n = 3), 24 (WT, n = 4) and 72 h (WT, n = 12 and P2Y1R KO, n = 13) after ischemia. The brains were removed, post-fixed in 4% PFA for 24 h, and sectioned on a vibratome (VT1200; Leica, Solms, German) into coronal slices (30 µm thick). In a separate group of mice subjected to 20 min of ischemia, blood pressure was measured through a PE-10 cannula inserted into the left femoral artery, and arterial blood samples were analyzed (n = 4).
Immunohistochemistry
Brain slices from the mice at 72 h after sham operation or ischemia were used for immunohistochemistry. After washing with 0.3% Triton-X in 0.01 M phosphate-buffered saline (PBS) and blocking with 3% normal goat serum in 0.01 M PBS, free-floating sections were incubated with an antibody solution containing mouse anti-Iba1 (1:500, Wako Pure Chemical) and rabbit anti-P2Y1R antibodies (1:50, Santa Cruz, Dallas, TX) with Can Get Signal solution A (Toyobo, Osaka, Japan) for 48 h at 4℃. The secondary antibodies used for visualization of the two primary antisera were Alexa Flour 488 goat anti-mouse IgG (1:1000, Molecular Probes, Eugene, OR) and Alexa Flour 546 goat anti-rabbit IgG (1:1000). Sections were washed three times for 10 min in 0.3% Triton-X in 0.01 M PBS and then incubated for 2 h at room temperature in a solution containing secondary antibodies with Can Get Signal solution A. After three intensive wash steps for 10 min each with 0.3% Triton-X in 0.01 M PBS, the stained sections were covered with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunofluorescent sections were examined using a fluorescent microscope (Axio Imager, Carl Zeiss, Oberkochen, Germany). Fluorescent images were analyzed with Image J software (NIH, Bethesda, MD) and their intensity quantified.
Histological analysis of hippocampal injury after transient forebrain ischemia
Histological analysis of injured hippocampi was performed at 72 h after ischemia using terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end labeling (TUNEL) staining. TUNEL staining was performed using a commercial kit (#11684817910; Roche Molecular Biochemicals, Pleasanton, CA) according to the manufacturer’s protocol. Nuclei were counterstained with Mayer’s hematoxylin solution (Wako Pure Chemical, Osaka, Japan). Two subregions (CA1 and CA3) were quantified for TUNEL staining.
Preparation of organotypic hippocampal slice cultures
WT (n = 10), P2Y1R KO (n = 10), tetO-P2Y1R (n = 8; P2Y1R is expressed normally throughout the entire body), and tetO-P2Y1RmOE (n = 8; P2Y1R is ectopically expressed in only microglia) mice were used. Organotypic hippocampal cultures were prepared according to the standard interface method previously described, with minor modifications.33,34 Hippocampal slices were prepared from five- to six-day-old neonatal mice by removing the brain, dissecting the hippocampal formation, and sectioning transverse slices (350 µm) using a McIlwain Tissue Chopper (Mickle Laboratory Engineering, Surrey, UK). Individual slices were then carefully separated and transferred onto 30-mm Millicell-CM culture plate membrane inserts (pore size, 0.4 µm; Millipore, Burlington, MA). Inserts were placed into 6-well culture plates (BD Biosciences, San Jose, CA) with 1.0 ml growth medium consisting of 50% minimum essential medium, 18% Hanks’ balanced salt solution, 25% heat-inactivated horse serum, 4 mM L-glutamine, D-glucose to a final concentration of 6.0 g/l, and 2% B27 supplement (Life Technologies, Carlsbad, CA). Penicillin (50 U/ml) and streptomycin (50 µg/ml) were added to the medium. Cultures were then grown at 37℃ in a 90–100% humidified atmosphere with 5% CO2. The culture medium was changed every two days.
All cultures used in this study were grown for seven days in vitro. Propidium iodide (PI; Sigma-Aldrich, St. Louis, MO), which rapidly enters cells with damaged membranes and becomes brightly fluorescent after binding to nucleic acids, was added (1.0 µg/ml) to the culture medium as an indicator of neuronal death.
Oxygen–glucose deprivation
Slice cultures were exposed to oxygen–glucose deprivation (OGD) using an anaerobic chamber that was pre-equilibrated to 37℃ with an atmosphere of 0% O2, 5% CO2, and 95% N2. 35 Slices on membrane inserts were transferred into 6-well plates with artificial cerebral spinal fluid consisting of 120 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 25 mM NaHCO3, 20 mM HEPES, and 25 mM sucrose. Plates were then placed into a modular incubator chamber flushed with 5% CO2 and 95% N2 according to the manufacturer’s instructions (Billups-Rothenberg, San Diego, CA) at 37℃ for 40 min. Upon removal of the plates from the anaerobic chamber, the membranes were transferred to pre-warmed normal growth medium. The growth medium also contained PI at a final concentration of 1.0 µg/ml. Cultures were transferred back to an incubator for 48 h at 37℃ before evaluation of cell death. After 48 h, the cultures were examined using an Axio Scope A1 (Carl Zeiss, Oberkochen, German) inverted fluorescent microscope. PI fluorescence in the CA1, CA3, and dentate gyrus (DG) subfields of the hippocampal cultures was used as an index of cell death.
Magnetic cell separation method
Microglia were isolated from tetO-P2Y1R (n = 5) and tetO-P2Y1RmOE (n = 5) mouse brain, as described previously. After enzymatic digestion of brain tissue and myelin removal, primary microglia were isolated using a magnetic cell separation (MACS) method with a Neural Tissue Dissociation Kit (Miltenyi Biotec). Cells were stained with phycoerythrin (PE)-conjugated anti-CD11b antibodies in separation buffer for 10 min, followed by incubation with anti-PE magnetic beads for 15 min. CD11b+ cells were separated in a magnetic field using MS columns. Both CD11b+ and CD11b− fractions were collected and used for further analyses.
Quantitative RT-PCR
Total RNA was extracted from microglia using RNeasy. Reverse transcription-polymerase chain reaction (RT-PCR) was performed using a one-step PrimeScript RT-PCR kit (Takara-bio, Shiga, Japan), according to the manufacturer’s protocol. Reaction mix contained 40 ng of total RNA, 200 nM primers, 100 nM TaqMan probe, TAKARA EX TaqH HS, and PrimeScript RT enzyme mix. RT-PCR amplification and real-time detection were performed using a 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Reverse transcription was performed at 42℃ for 5 min, followed by inactivation at 95℃ for 10 s. The temperature profile consisted of 40 cycles of denaturation at 95℃ for 5 s with annealing/extension at 60℃ for 30 s. The primers and probes for P2ry1 (Mm02619947-s1) and Cd68 (Mm03047343_m1) were purchased from Applied Biosystems (Foster City, CA). PCR was performed with 0.4 ml RT product in a final volume of 40 μl containing primers (0.2 mM), 0.2 mM dNTPs (Bioline, Bio Rad, Hercules, CA), 1.5 mM MgCl2, 1 U DNA polymerase (GibcoBRL, Life Technologies), and the corresponding buffer. The reaction mixture was heated to 95℃ for 2 min then cooled to 80℃, with DNA polymerase added at this temperature. PCR amplification was performed as follows: denaturation at 95℃ for 50 s, annealing at 65℃ for 50 s, and extension at 72℃ for 50 s, for two cycles. Subsequently, the annealing temperature was lowered by 0.5℃ every cycle for 18 cycles until it reached 56℃. Finally, 12 additional cycles were run at this annealing temperature. The resulting PCR products were analyzed on 1.8% agarose gels in 0.5 × TBE, purified using a Wizard PCR Preps DNA purification system (Promega, Madison, WI), and sequenced using Sequenase version 2.0 enzyme (Sequenase kit; Amersham Pharmacia Biotech, Little Chalfont, UK) to confirm specificity of amplification.
Statistical analysis
Two observers blinded to experimental treatments quantified results for all experiments. Data were expressed as means ± standard deviation (S.D.). Statistical analyses were performed using Student’s t-test and Mann–Whitney tests. A P-value < 0.05 was considered statistically significant.
Results
In vitro and in vivo microglia express P2Y1R
The increase in [Ca2+]i in cultured microglia was assessed using the fura-2 method, and was expressed as changes in the ratio of F340/380. BSS perfusion had no effect on [Ca2+]i in the WT and P2Y1R KO microglia. Traces in Figure 1(a) show representative increases in [Ca2+]i in microglia evoked by the selective P2Y1R agonist MRS2365. It evoked an increase of [Ca2+]i in the WT microglia in a concentration-dependent manner over a concentration range from 1 to 10 µM. MRS2365 at 1 μM increased [Ca2+]i in microglia up to 112.9% of the baseline (n = 50). Moreover, 3, 5, and 10 μM MRS2365 significantly increased [Ca2+]i up to 120.9%, 128.9%, and 148.5%, respectively (P < 0.001 compared with the baseline, n = 48 (3 μM), 54 (5 μM), and 52 (10 μM)). The MRS2365 (3–10 µM)-evoked increases in [Ca2+]i in the P2Y1R KO microglia were significantly less than in WT microglia (Figure 1(b), P < 0.001 compared with WT, n = 52 (1 μM), 46 (3 μM), 55 (5 μM), and 51 (10 μM)), suggesting that microglia have functional P2Y1R. To determine whether cultured microglia express abnormally higher levels of P2Y1R than normal microglia in situ, we performed flow cytometry analysis. Expression of P2Y1R was detected in 7.43% of cultured microglia and 8.53% of acutely isolated microglia (Figure 1(c) and (d)). This suggests that microglia express P2Y1R in vivo, and that the P2Y1R-mediated increase in [Ca2+]i detected in cultured microglia is not simply an artifact of culture but reflects the microglial response in vivo.
Increased [Ca2+]i in microglia was mediated by P2Y1R. Increased [Ca2+]i in microglia was measured as a change in the F340/380 ratio. The specific P2Y1R agonist MRS2365 evoked an increase in [Ca2+]i in microglia obtained from WT mice in a concentration-dependent manner over a concentration range from 1 to 10 µM. However, the MRS2365-evoked [Ca2+]i increase was almost completely abolished in microglia obtained from P2Y1R KO mice. Traces in (a) show MRS2365-evoked increases in [Ca2+]i in microglia obtained from WT mice (upper) and P2Y1R KO mice (lower), which are summarized in (b). Values represent mean ± S.D., n = 50 (1 μM), 48 (3 μM), 54 (5 μM), and 52 (10 μM) for WT microglia, and n = 52 (1 μM), 46 (3 μM), 55 (5 μM), and 51 (10 μM) for P2Y1R KO microglia. *P < 0.001 compared with WT. (c, d) Comparable expression of P2Y1R in cultured microglia and acutely isolated microglia. Values represent mean ± S.D., n = 3.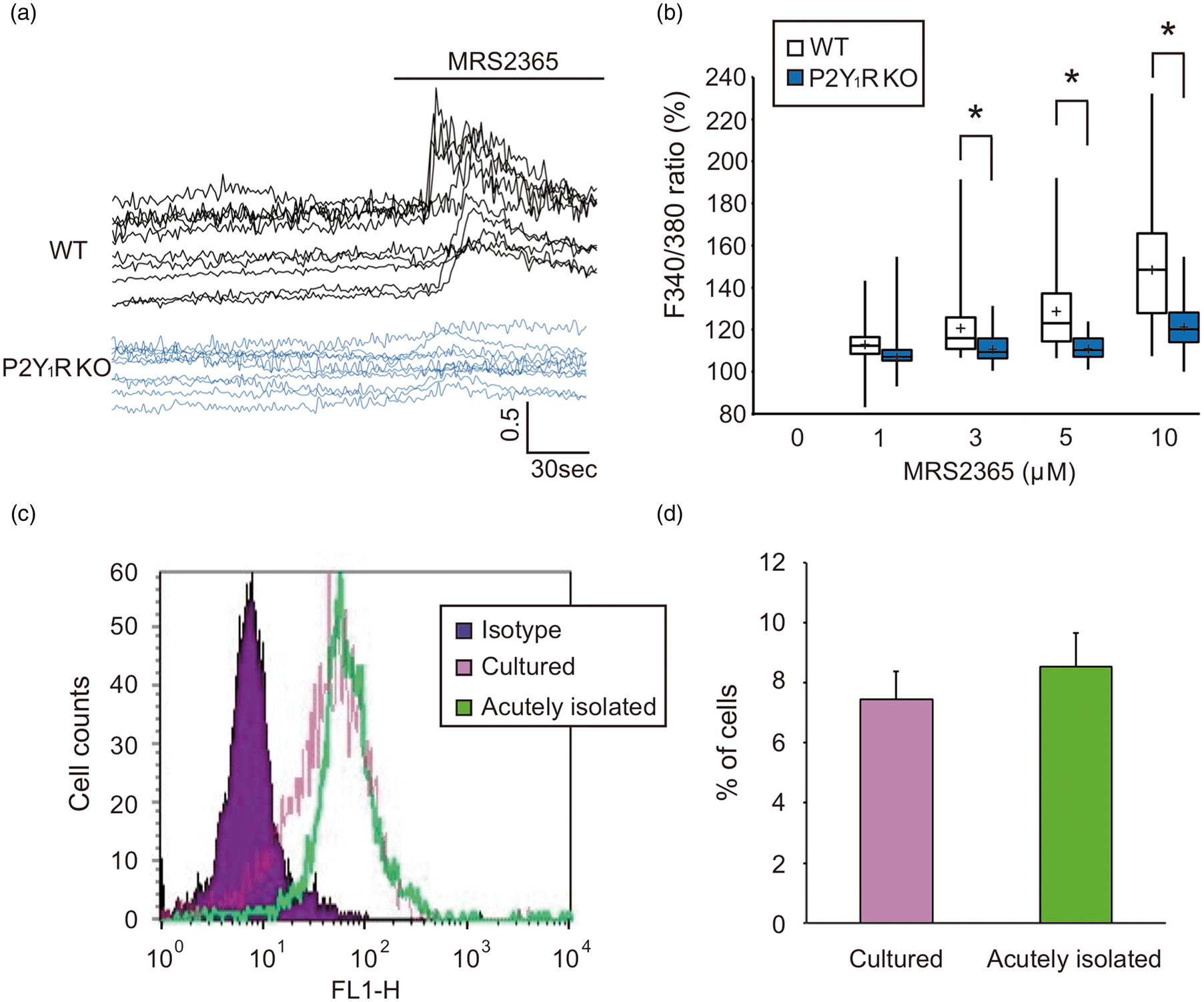
P2Y1R expression in hippocampal microglia after transient forebrain ischemia
Physiological parameters and regional cerebral blood flow.
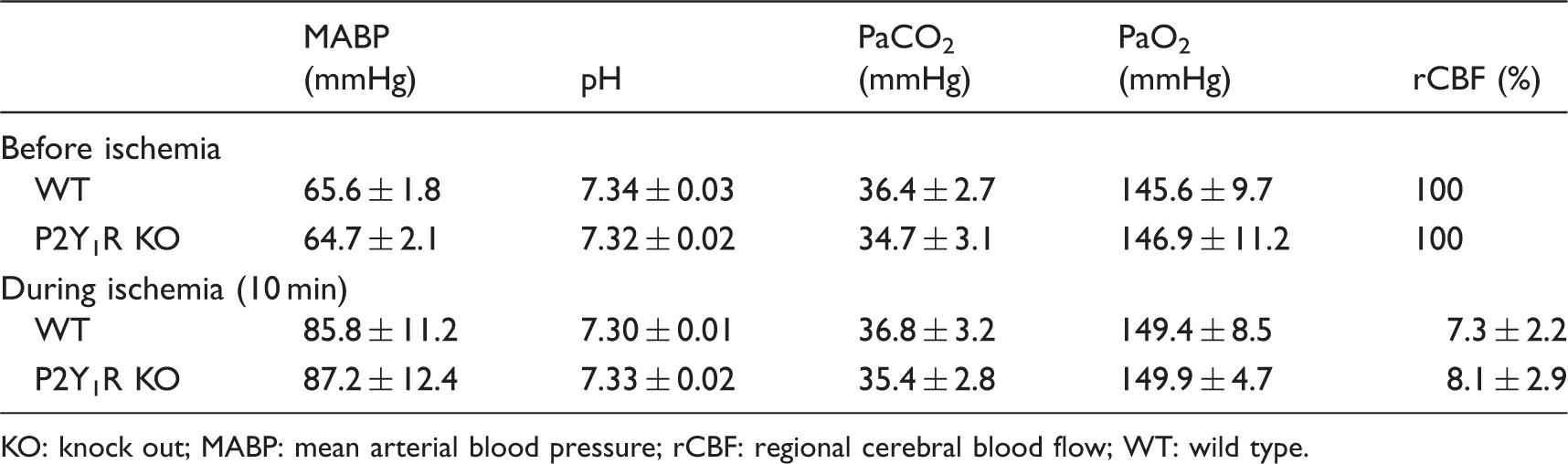
KO: knock out; MABP: mean arterial blood pressure; rCBF: regional cerebral blood flow; WT: wild type.
To investigate P2Y1R expression in hippocampal microglia, we performed immunohistochemical analysis. The hippocampal sections were doubly stained with anti-P2Y1R and Iba1-antibodies. P2Y1R-positive signals were observed, but were faint, in the sham-operated WT mice, but were dramatically higher by transient forebrain ischemia. Double immunofluorescence labeling revealed co-localization of P2Y1R and Iba1-positive cells and GFAP-positive cells (data not shown). However, P2Y1R-positive signals were not observed in the hippocampus of P2Y1R KO mice (Figure 2(a)). Intensity of P2Y1R-positive signal (per unit area) was significantly higher in hippocampi of WT mice compared with P2Y1R KO mice (P < 0.001, Figure 2(b)). Microglial activation was confirmed in a whole hippocampus uniformly after ischemia, and there was no specific localization of P2Y1R-positive microglia or non-P2Y1R microglia.
Immunohistochemical analysis of microglial P2Y1R. (a) Hippocampal brain sections were stained with antibodies specific to P2Y1R and Iba1, as well as DAPI. P2Y1R-positive signals were observed, but were faint, in the sham-operated WT mice. Double immunofluorescence labeling revealed co-localization of P2Y1R with Iba1-positive signals. Further, this signal was significantly higher by 72 h after 20-min of transient forebrain ischemia. Scale bars = 100 µm. (b) Quantified bar chart of intensity per unit area of immunohistochemical analysis. (c, d) Flow cytometry analysis shows differences in P2Y1R expression in sham-operated and post-ischemic mice. Number of P2Y1R-expressing microglia was significantly higher in post-ischemic than sham-operated mice. *P < 0.05 compared with sham-operated.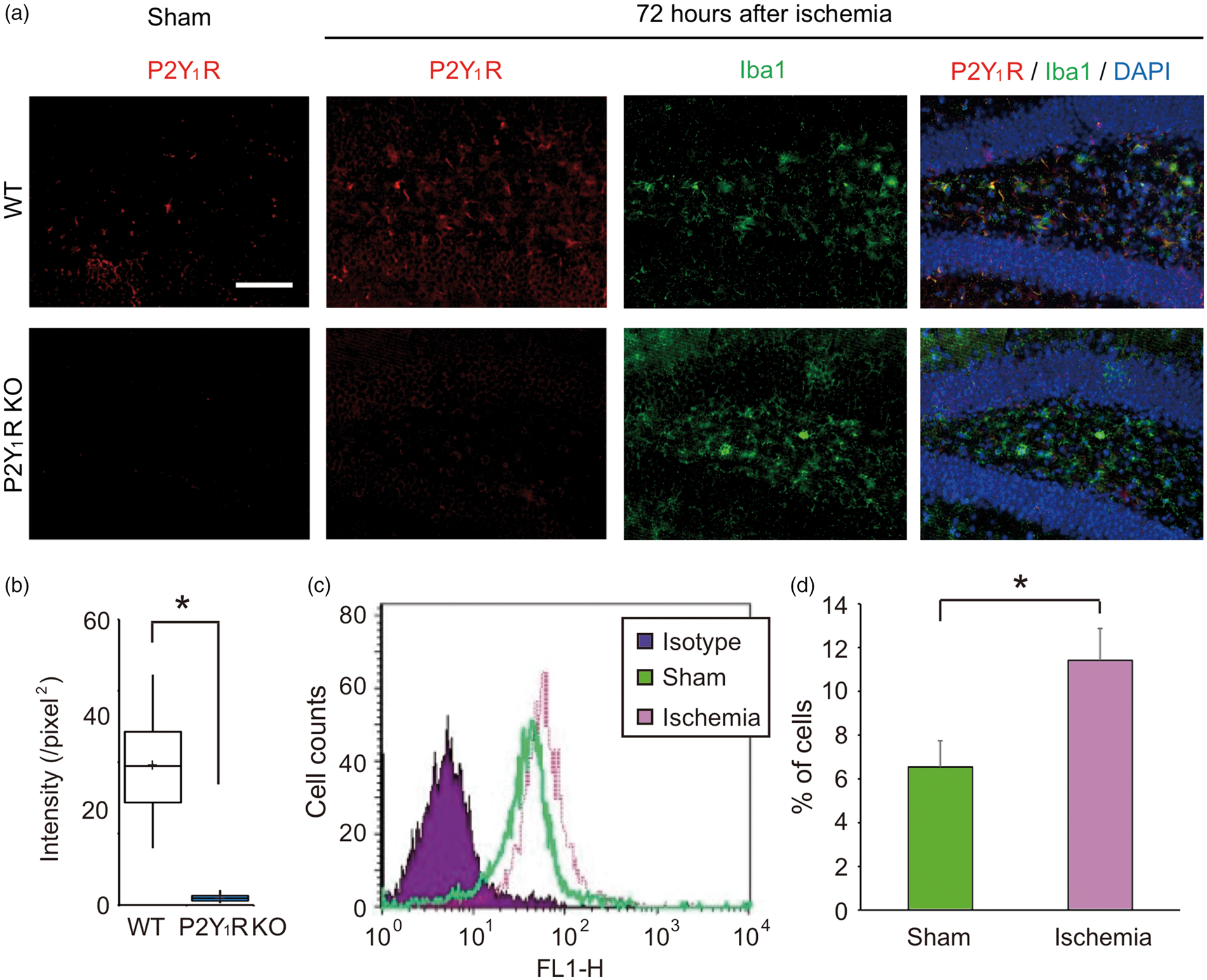
We also performed flow cytometry analysis for quantitative investigation of P2Y1R-expressing microglia in sham-operated and post-ischemic mice in vivo. In sham-operated mice, 6.54% of microglia expressed P2Y1R, whereas 11.41% of post-ischemic microglia expressed P2Y1R, which is significantly higher than sham-operated mice (P < 0.05, Figure 2(c) and (d)). These results reveal that microglia in vivo express P2Y1R, and that it is increased in response to ischemia.
P2Y1R KO exacerbates neuronal cell injury after transient forebrain ischemia
In samples from sham WT and P2Y1R KO mice, neuronal injury was not observed in the hippocampal subregions. Seventy-two hours after transient forebrain ischemia, TUNEL-positive neurons were observed in the pyramidal cell layer of the injured hippocampi. The number of TUNEL-positive neurons in P2Y1R KO mice was significantly higher than in the WT mice, especially in the CA1 (P < 0.001) and CA3 (P < 0.01) subregions, suggesting that ischemic injury is exacerbated by deletion of P2Y1R (Figure 3(a) and (b)).
P2Y1R knockout exacerbates neuronal cell injury after transient forebrain ischemia. (a) Representative pictures of TUNEL staining in the hippocampus 72 h after transient forebrain ischemia. TUNEL-positive signals were absent in sham-operated mice but were present mainly in the hippocampal CA1 and CA3 subregions in the transient ischemia-treated mice. Scale bar = 200 µm. (b) Summary of the number of TUNEL-positive cells in the hippocampus in WT and P2Y1R KO mice exposed to ischemia. Values show the ratio of TUNEL-positive cells in the hippocampal subregions in WT and P2Y1R KO mice. The ratio in the hippocampal CA1 and CA3 subregions in P2Y1R KO mice was significantly greater compared with WT mice. Values represent mean ± S.D., n = 9, *P < 0.001, **P < 0.01.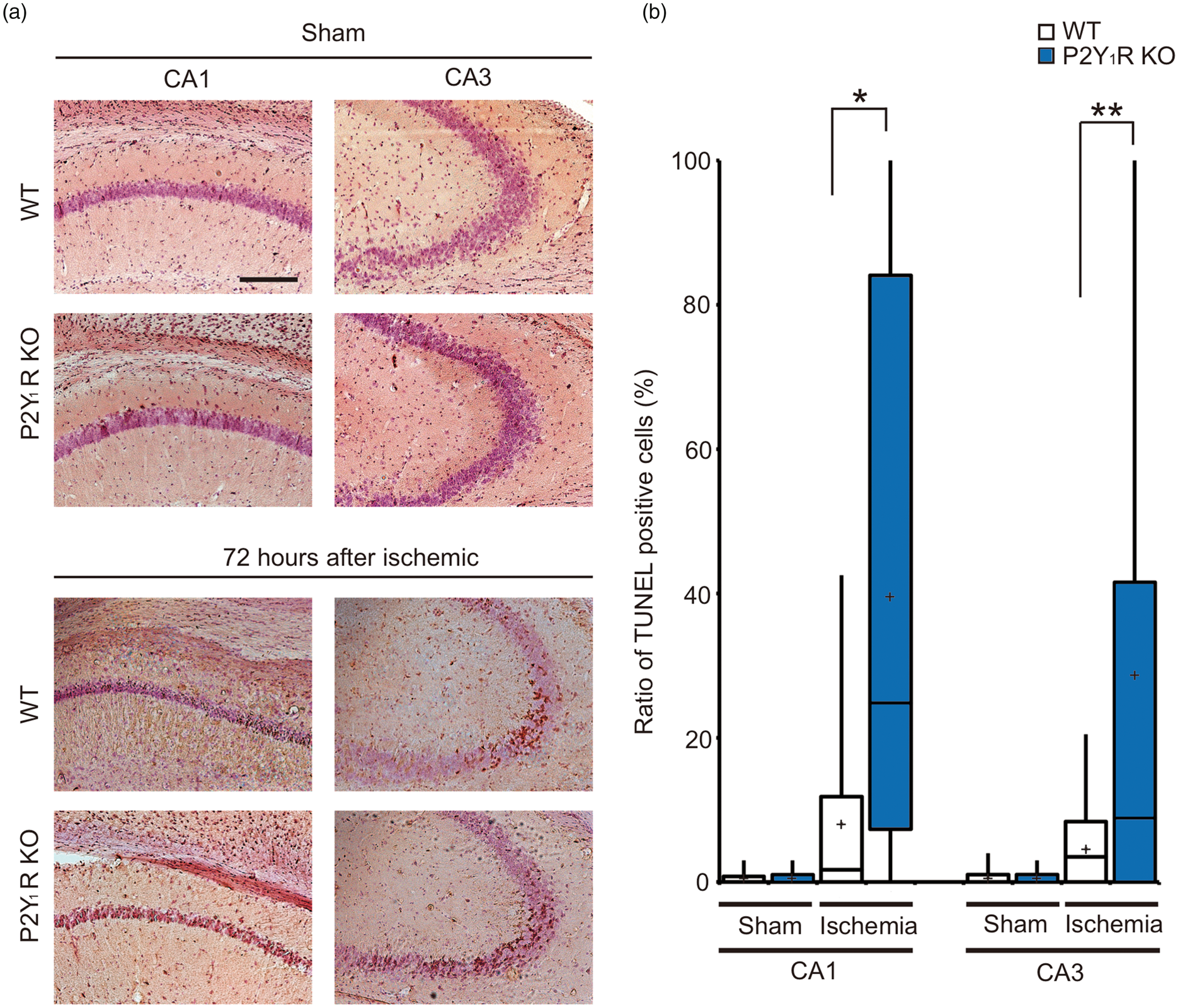
P2Y1R KO exacerbates hippocampal injury after OGD
Using organotypic hippocampal slice cultures, we established an in vitro ischemic model to investigate the effects of P2Y1R on ischemic brain injury. Neuronal injury was assessed by PI uptake, although PI fluorescent signal was hardly detected in the control normoxic (sham) hippocampal slice cultures obtained from the WT and P2Y1R KO mice (Figure 4(a), sham); 48 h after 40-min OGD, PI uptake was observed in the hippocampal slices (Figure 4(a), OGD). PI fluorescent signals were significantly greater in P2Y1R KO hippocampal slices compared with WT hippocampal slices (Figure 4(b), P < 0.001).
In vitro ischemia model using organotypic hippocampal slice cultures. Using organotypic hippocampal slice cultures, we established an in vitro ischemic model. The hippocampal slice cultures were incubated with oxygen–glucose deprivation (OGD) medium for 40 min, and then re-perfused with normal control medium for another 48 h. Neuronal damage was assessed by PI uptake. (a) Representative PI fluorescent images of hippocampal slice cultures obtained from the WT and P2Y1R KO mice. Scale bar = 500 µm. (b) Box plot shows densitometric measurements of PI uptake in the hippocampal. PI fluorescent intensity was significantly greater in P2Y1R KO mice compared with WT mice following OGD. Values represent mean ± S.D., n = 40, *P < 0.001.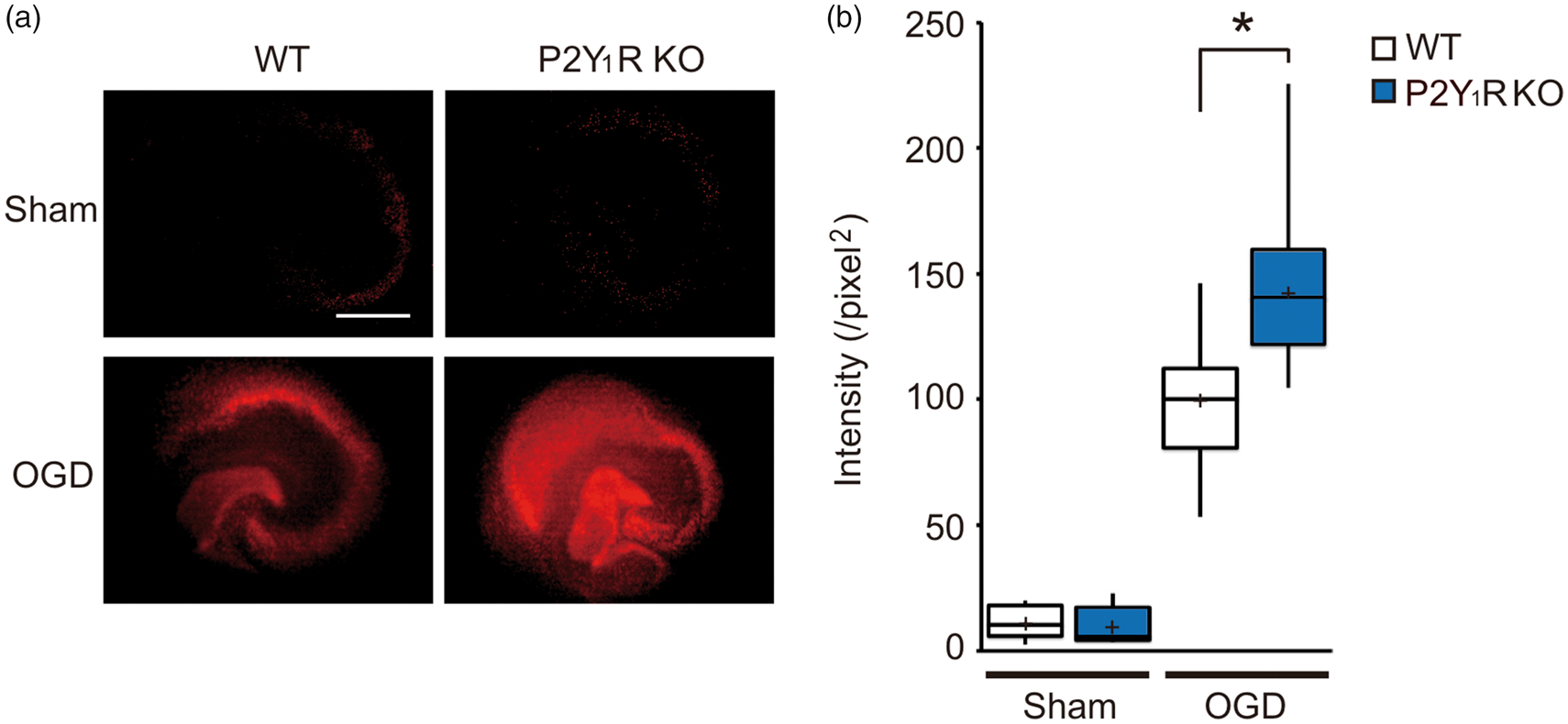
Ectopic expression of P2Y1R in microglia rescues hippocampal injury after OGD
Expression levels of P2Y1R were compared between tetO-P2Y1R and tetO-P2Y1RmOE microglia by flow cytometry analysis and quantitative RT-PCR. Flow cytometry analysis showed that the microglial expression rate of P2Y1R was 15.46% and 22.89% in tetO-P2Y1R and tetO-P2Y1RmOE mice, respectively. This confirms that tetO-P2Y1RmOE mice have a significantly higher number of P2Y1R-expressing microglia compared with tetO-P2Y1R mice (Figure 5(a) and (b), P < 0.05). The extent of this observed increase in P2Y1R-positive microglia of tetO-P2Y1RmOE mice is similar to that observed after ischemia (Figure 2(d)), suggesting that microglial P2Y1R overexpression in tetO-P2Y1RmOE mice is within a viable range in post-ischemic brain, rather than due to an artificial increase.
Rescue by ectopic P2Y1R microglial expression in an ischemia-induced in vitro model. (a, b) Flow cytometry analysis shows greater number of P2Y1R-expressing microglia in tetO-P2Y1RmOE mice than tetO-P2Y1R mice. (c) The majority of CD68 mRNA appeared in the CD11b-positive fraction (and not the CD11b-negative fraction) in both tetO-P2Y1R and tetO-P2Y1RmOE mice. The MACS method successfully separated microglia. (d) Overexpression of P2Y1R mRNA in the CD11b-positive fraction of tetO-P2Y1RmOE mice, but low expression in the CD11b-negative fraction. Values represent mean ± S.D., n = 3, *P < 0.001. (e) Representative PI fluorescent images of hippocampal slice cultures obtained from tetO-P2Y1R and tetO-P2Y1RmOE mice. Scale bar = 500 µm. (f) Summary of OGD-induced neuronal damages assessed by PI uptake. Neuronal damage (increase in PI uptake) was rescued by ectopic microglial P2Y1R expression. PI fluorescent intensity of tetO-P2Y1RmOE mice was significantly less compared with tetO-P2Y1R mice. Values represent mean ± S.D., n = 30, *P < 0.001.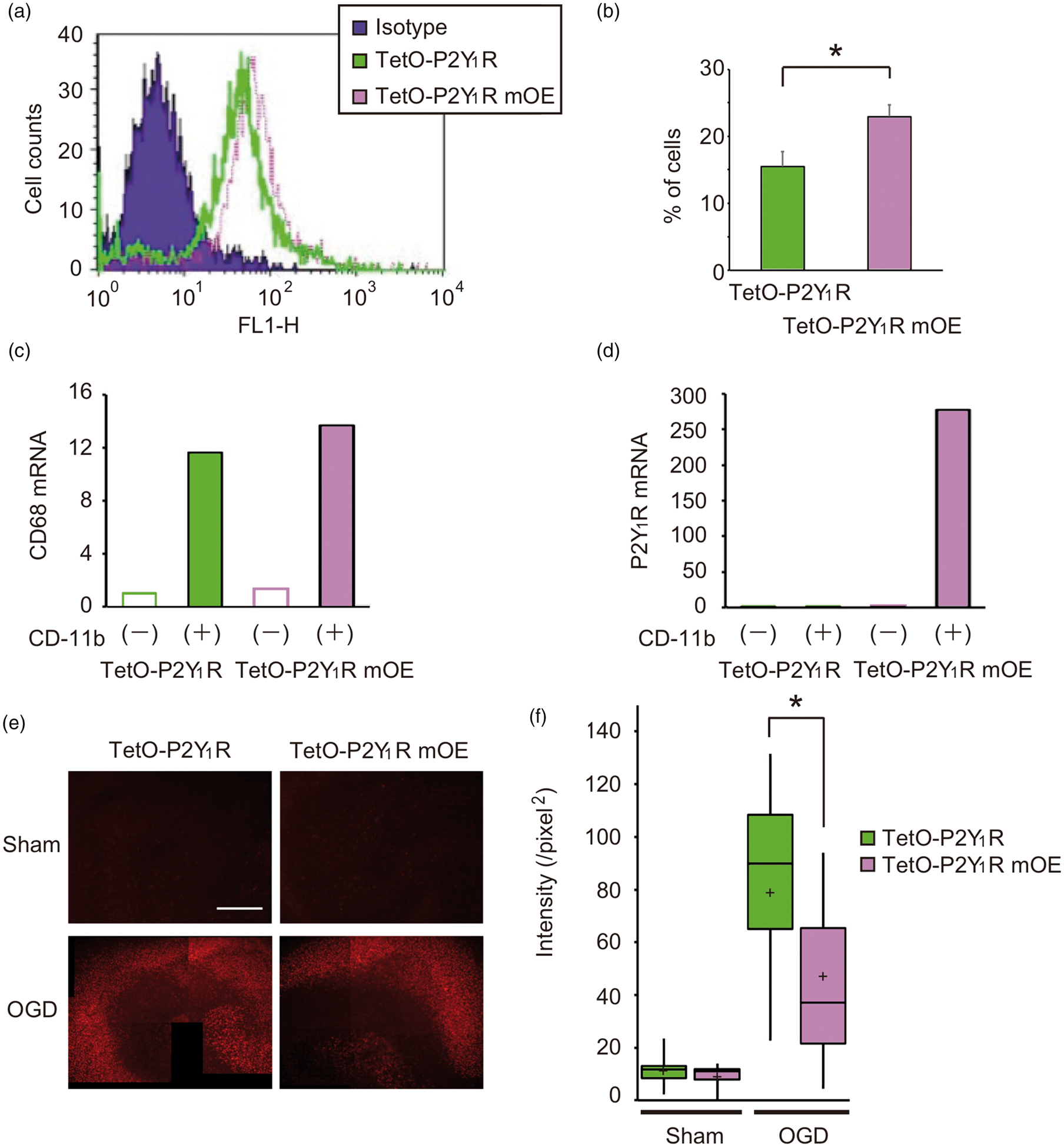
Using the MACS method, we separated cells into CD11b-positive and CD11b-negative fractions. The majority of CD68 mRNA (a marker for macrophages and activated microglia) was observed in the CD-11 b-positive fraction, indicating that microglia are selectively separated by the MACS method. In the CD11b-positive fraction, P2Y1R mRNA was higher in tetO-P2Y1RmOE mice compared with tetO-P2Y1R mice. While in the CD11b-negative fraction, no increase in P2Y1R was observed in tetO-P2Y1RmOE mice, suggesting that P2Y1R overexpression should be induced in only CD11b-positive cells, i.e. microglia (Figure 5(c) and (d)).
Without OGD, PI fluorescent signals were faint in hippocampal slices from both tetO-P2Y1R and tetO-P2Y1RmOE mice. Similar to Figure 4, treatment of the slice cultures with OGD (40-min OGD, 48-h reperfusion) resulted in greater PI uptake in both slice cultures, although PI fluorescent intensities were significantly less in the tetO-P2Y1RmOE slices compared with corresponding regions in the tetO-P2Y1R slices (Figure 5(e) and (f), P < 0.001). These results suggest that ectopic expression of microglial P2Y1R rescues hippocampal injury after OGD.
Discussion
Results from the present study demonstrated that functional microglial P2Y1R expression was induced following transient forebrain ischemia. The existence of microglial P2Y1R expression was required for neuroprotection against ischemia, because neuronal injury was exacerbated by the deletion of P2Y1R in in vivo (transient forebrain ischemia) and in vitro (OGD in hippocampal slice cultures) ischemic models. Additionally, this exacerbation was rescued by ectopic microglial expression of P2Y1R in an in vitro ischemic mouse model, suggesting that microglial P2Y1R protect neurons against ischemic/hypoxic insults at least within 72 h after ischemia.
Results from the Ca2+ imaging experiments demonstrated functional expression of P2Y1R in microglia. Furthermore, immunohistochemical analysis of the hippocampus revealed that P2Y1R-positive signals were present and co-localized with Iba1-positive signals. P2Y1R-positive signals were low or faint under normal conditions, but dramatically higher in Iba1-positive cells at 72 h after transient forebrain ischemia. Our flow cytometry analysis also showed significantly higher P2Y1R expression in microglia from post-ischemic WT mice compared with sham-operated mice. These results suggested that P2Y1R are likely upregulated in activated microglia after ischemic stress and could play a pivotal role in pathophysiological conditions. In this study, upregulation of P2Y1R in microglia was not limited in the CA1 subregion, but was confirmed in a whole hippocampus uniformly after ischemia. Although the CA1 subregion is well known as the most vulnerable to ischemia, not only the CA1 but also other hippocampal subregions such as the CA3, 4 and dentate gyrus are damaged in a mouse transient forebrain ischemic model. 32 Therefore, ischemic stress induced microglial activation in a whole hippocampus, and P2Y1R-positive signals were not limited in the CA1 subregion in this study.
Under in vitro conditions, microglia exhibit an ameboid shape, known as the activated form, instead of the rhomboid form during resting conditions. These results suggested that cultured microglia are likely activated by environmental changes within the culture. Indeed, the cultured microglia demonstrated P2Y1R-mediated Ca2+ responses, indicating upregulated P2Y1R, despite the lack of stress or injury. Previous studies have reported no observed microglial P2Y1R expression, while recent studies and the present findings clearly demonstrated that P2Y1R expression was present and functional in microglia.11,14,36 Since activated microglia are subclassified into different subtypes, these discrepancies may arise from differences in properties of activated microglia, probably depending on intensity of injury, timing from injury, and injured region. However, we must await further studies to clarify them.
Results from this study demonstrated significantly greater neuronal cell death in the hippocampus after transient forebrain ischemia in P2Y1R KO mice than WT mice. In a preliminary study, we were unable to generate stable transient forebrain ischemic models using tetO-P2Y1R and tetO-P2Y1RmOE mice. As the background of these mice is different from WT and P2Y1R KO mice, we could not induce and directly compare forebrain ischemic stress. In future, we will compare both mouse strains using other ischemic models including a focal infarction model. Additionally, we did not evaluate behavior analysis in this study since our transient forebrain ischemia model did not induce any motor functional disability. We will also evaluate functional outcomes using other ischemic models in future studies. Increased cell death was also confirmed by the in vitro experiment in the present study, in which OGD-induced PI uptake was greater in hippocampal slice cultures obtained from P2Y1R KO mice compared with WT mice (Figure 4). Previous studies using OGD experiments and hippocampal slice cultures reported that PI uptake is most notable by two days after OGD; there was a strong correlation between PI fluorescence and histological damage.37,38 Additionally, the present study also revealed upregulated P2Y1R in hippocampal microglia following transient ischemia. Taken together, these results strongly suggest that P2Y1R expression in microglia plays a neuroprotective role against cerebral ischemia.
However, because P2Y1R are expressed in several different cell types in the hippocampus other than microglia, including pyramidal cells and astrocytes, the exacerbation of ischemic injuries following P2Y1R deletion might not always be due to microglia-specific deletion.36,38 The reason for less neuronal damage in OGD experiments compared with in vivo studies might be due to the influence of P2Y1R expression on cells other than microglia. Therefore, to determine whether ischemic vulnerability can be counteracted by ectopic expression of P2Y1R on microglial specifically, we used a P2Y1R tetO knockin and Iba1-tTA mouse system.29,30 We established two transgenic mouse lines: (1) tetO-P2Y1R mice, with P2Y1R expressed throughout the entire body; and (2) tetO-P2Y1RmOE mice, with P2Y1R ectopically overexpressed in only microglia but normally expressed in other cell types (as for tetO-P2Y1R mice). We confirmed selective overexpression of P2Y1R in microglia from tetO-P2Y1RmOE mice, with expression levels within a viable range for occurrence of ischemia and not in an artificial range (Figure 5). These transgenic mice allowed us to evaluate the effects of microglial P2Y1R expression following brain ischemia. However, P2Y1R are not only ectopically expressed in microglia, but also in macrophages, as Iba1 is also expressed in macrophages. To exclude the influence of peripheral macrophages, we used an in vitro ischemic model in hippocampal slice cultures, where the influence of peripheral macrophages was likely very limited. As shown in Figure 5, ectopic expression of microglial P2Y1R improved the OGD-induced vulnerability in hippocampal slice cultures, suggesting that upregulation of microglial P2Y1R provides neuroprotection against ischemic injury.
Microglial activation not only increases inflammatory responses, but also exacerbates brain injury. The majority of other P2 receptors in microglia, such as P2X4, P2X7, and P2Y6 receptors, also facilitate brain disease or neuronal damage, but recent studies have reported the neuroprotective effects of microglia.13,16–18,39 Previous reports showing the effects of P2Y1R on ischemic brain damage have been inconsistent. Stimulation with the P2Y1R agonist 2MeSADP reduces and partially reverses neuronal damage induced by photothrombosis, while the P2Y1R specific agonist MRS2365 increases infarct volume in a transient middle cerebral artery occlusion (tMCAO) model.21,40 In addition, other researcher and we reported that P2Y1R controls proliferation of neural stem cells in the developmental and adult stages.41,42 Microglia also contribute to neurogenesis in the early developmental stages.43,44 Thus, microglial P2Y1R might be involved in neurogenesis after brain ischemia, and thus, reveal neuroprotection against neuronal ischemic injury. However, further studies are required to clarify this. The complex and conflicted results suggest that P2Y1R expression in the CNS may have multiple roles or functions that are dependent on cell type, brain region, timing of stimulation, and injury severity.3,27,38 Accordingly, the present in vivo (transient forebrain ischemia) and in vitro (OGD in hippocampal slice cultures) experiments suggest that microglial P2Y1R expression exhibits a protective role against ischemic neuronal injury. However, the mechanisms or molecules responsible for microglial P2Y1R-mediated neuroprotection remain poorly understood.
Previous studies have shown that astrocytic P2Y1R expression is responsible for cellular protection against several different types of injuries, including ischemia, in which P2Y1R-mediated IL-6 production is involved. 24 Although it is not yet confirmed whether microglia are involved in the same protective mechanisms as astrocytes, it is clear that P2Y1R expression in microglia and astrocytes is involved in neuroprotection. Considering a dynamic and rapid change of microglia to ischemic stress and ATP-mediated intracellular signaling mechanism, microglia must play an important role and contribute a lot on neuroprotection as well as astrocytes. Although previous studies from our group and others have shown that several microglial P2 receptors contribute to brain diseases, such as neuropathic pain and excitatory neurotoxicity, the present study provides a better understanding about the beneficial role of microglial P2 receptors in conditions of ischemia-related brain injuries, as well as the complex behaviors of glial cells.13,15
In conclusion, we demonstrated the existence and function of microglial P2Y1R, which were upregulated and activated in the pathophysiological condition of brain ischemia. Using transgenic mice where the P2Y1R gene was deleted throughout the entire body or ectopically expressed in only microglia, we also demonstrated that microglial P2Y1R played an essential role in neuroprotection following brain ischemia. Although the mechanisms underlying the microglial P2Y1R-mediated neuroprotection remain unclear, further studies will contribute to the development of novel therapeutic approaches for stroke.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Frontier Brain Research Grant of University of Yamanashi (to SK), AMED (to SK), CREST (to SK), KAKENHI on Innovative Areas (25117003 & 18H05121) (to SK), Challenging Exploratory Research (25670622) (to SK), Scientific Research (B) (16H04669) and Scientific Research (C) (24592119) (to HK), Takeda Science Foundation (to SK), Kurata Grants (to SK), Takahashi Industrial and Economic Research Foundation (to SK), and SENSHIN Medical Research Foundation (to SK).
Acknowledgments
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
YF and SK planned and designed the experiments. KFT and KI made conditional P2Y1R overexpression mouse, CG made P2Y1R knockout mouse. YF performed main experiments, BP performed FACS analysis, KS performed validation studies, YF, HY and KK analyzed data. YF, HY, SK and HK drafted the manuscript.