
Abstract
Ketone bodies are important alternate brain fuels, but their capacity to replace glucose and support neural function is unclear. In this study, the contributions of ketone bodies and glucose to cerebral cortical metabolism were measured in vivo in halothane-anesthetized rats fasted for 36 hours (n=6) and receiving intravenous [2,4-13C2]-
Introduction
Although brain ketone oxidation can be substantial, the quantitative contribution of ketone bodies to neuronal and astroglial oxidative metabolism, and their capacity to replace glucose in support of neural function, remains unclear. In a study using [U-13C]BHB, Kunnecke et al (1993) reported greater oxidation of ketone bodies in astrocytes than in neurons. Increased astroglial acetate metabolism has been observed in mice maintained on a ketogenic diet possibly reflecting adaptation to prolonged ketonemia (Melo et al, 2006; Yudkoff et al, 2005). In contrast, a study of acute hyperketonemia in overnight-fasted humans (Pan et al, 2001, 2002) infused intravenously with [2,4-13C2]-
Although brain utilization of ketone bodies is increased by fasting, contradictory findings have appeared on the effects of ketone bodies on brain glucose consumption. Early studies using [14C] 2-deoxyglucose (2-DG) autoradiography showed no effects on glucose uptake and oxygen consumption during acute infusion of BHB or after 3 days of fasting (Corddry et al, 1982), whereas later studies reported that ketone bodies administered acutely or increased over time by the ketogenic diet inhibited brain glucose utilization (LaManna et al, 2009; Melo et al, 2006).
In this study, we investigated the effects of hyperketonemia on cortical glucose and ketone metabolism of anesthetized rats subjected to a prolonged (36 hours) fast. Using [2,4-13C2]-
Materials and methods
Animal Preparation
Experiments were conducted using male Sprague–Dawley rats (weighing 185±15 g) fasted for 36 hours before study (n=6). Rats were anesthetized with halothane (2% induction), tracheotomized, and ventilated (30% oxygen; ∼68% nitrous oxide). After surgery, halothane was reduced to ∼1% and
Rats received [2,4-13C2]-
A separate small group (n=3) of 36 hour-fasted rats was infused with [2-13C]acetate (instead of 13C-BHB) for 2 hours using a three-step infusion protocol (Jiang et al, 2009) and the brain tissue was studied ex vivo. Cerebral cortical extracts were prepared and 13C metabolite enrichments determined to calculate the steady-state value of the flux ratio, Vcyc/VtcaN, as described previously (Lebon et al, 2002; Patel et al, 2005), and subsequently used to constrain the fits of the metabolic model to the 13C-enrichment time courses.
At the end of each animal experiment, the head and brain of the anesthetized animal were frozen in situ with liquid N2 with continued mechanical ventilation.
All animal experiments were conducted under protocols approved by the Yale Animal Care and Use Committee.
In Vivo Nuclear Magnetic Resonance Spectroscopy
In vivo1H-[13C] NMR spectra were obtained at 9.4 Tesla (Bruker ADVANCE NMR spectrometer/imager, Bruker BioSpin Corp., Billerica, MA, USA) (400.5 MHz for 1H and 100.71 MHz for 13C). The 13C-edited 1H NMR spectra were acquired using a circular surface coil of 14 mm diameter for 1H detection, in which an outer butterfly coil was used for 13C decoupling. Magnetic-field homogeneity was optimized using FASTMAP (Gruetter, 1993). The 1H-[13C] NMR spectra were obtained from a localized volume (6 × 6 × 6 mm3) using a polarization transfer pulse sequence in which the 90° and 180° pulses were replaced with adiabatic half- and full-passage pulses, respectively. Suppression of signals outside the localized volume was achieved with localization by adiabatic selective refocusing (de Graaf, 2007). The 1H-[13C] spectra were acquired with a 2.5-second repetition time in blocks of 512 scans with alternate on/off (180°) inversion of 13C. Free induction decays were saved independently and processed in Matlab7.0 (MathWorks, Natick, MA, USA) for analyzing time-dependent changes. The time-dependent labeling of metabolites was analyzed by fitting the 1H-[13C] total and difference spectrum using an in-house written version of the linear-combination model algorithm implemented in Matlab (MathWorks). The concentrations of the metabolites were also determined in the brain extracts at the end point of the labeled isotope infusions.
Preparation of Blood Plasma and Brain Extracts and Nuclear Magnetic Resonance Spectroscopy
Plasma samples were prepared for NMR spectral analysis by the addition of 200 μL of 2.5 mmol/L formic acid and 0.25 mmol/L TSP (3-trimethylsilyl tetradeuterated sodium propionate) and 400 μL 100 mmol/L phosphate buffer (pH 7) to 50 μL blood plasma. The solution was passed through a microcentrifuge filter tube (10,000 Da cutoff, Nanosep Centrifugal Devices, VWR, Batavia, IL, USA). Concentrations and 13C enrichments of BHB and other substances (such as acetoacetate, acetone, acetate, lactate) were determined using 1H-[13C] NMR spectroscopy with 20-second repetition time to achieve full spin-lattice T1 relaxation of the measured metabolite resonances. Plasma and tissue extract samples were measured at 500 MHz (Bruker AVANCE high-resolution NMR spectrometer, Bruker BioSpin Corp., Billerica, MA, USA). Both TSP and formic acid were used as chemical shift and concentration references, respectively.
Ethanol extracts were prepared from the frozen frontoparietal cortex (100 to 150 mg wet weight) using the procedure described in the study by Patel et al (2001). [2-13C]glycine (50 μL, 5 mmol/L) was added as an internal concentration reference at the beginning of tissue extraction. After centrifugation and lyophilization of the supernatant, the extract powder was suspended in 600 μL of a buffer solution containing 50 mmol/L phosphate (pH 7), 0.8 mmol/L formic acid, and 0.08 mmol/L TSP in D2O/H2O (2:1). The concentrations and 13C enrichments of metabolite resonances in the extracted brain samples were measured fully relaxed (repetition time=20 seconds) with 1H-[13C] NMR spectroscopy.
Description of the Metabolic Model Incorporating d -β-Hydroxybutyrate Oxidation
The time courses of brain amino-acid 13C enrichments were fitted to a two-compartment metabolic model (Figure 1), using as input the plasma time courses of 13C-enriched BHB, glucose, and lactate. In addition, nonlabeled sources (set to natural abundance) were included in the model to represent dilution flows. The metabolic model was fitted to the time courses of Glu-C4, Gln-C4, and Glx-C3 enrichment, along with the end-point enrichments for Gln-C3 and Glu-C3 (determined in the extracts). The differential equations describing the model (mass and isotope balance) were generated using CWave software (GF Mason, New Haven, CT, USA; Mason et al, 2003) and are given in Table 1. The differential equations were solved in Matlab using a first/second-order Runge–Kutta algorithm, and data were fitted using the Levenburg–Marquardt algorithm. Tissue glutamine was assumed to be present entirely in astrocytes, whereas glutamate was divided between neurons (90%) and astrocytes (10%) (Patel et al, 2005; Storm-Mathisen et al, 1983). Pyruvate carboxylase flux (Vpc) was assumed to be 20% of the rate of glutamine synthesis (Vgln) (Sibson et al, 2001). To improve the reliability of flux estimates, the fits were constrained by the value of the ratio, Vcyc/VtcaN (Patel et al, 2005). The flux ratio was calculated from steady-state Glu-C4 and Gln-C4 13C enrichments (fe ss ) measured in a separate group of 36 hour-fasted animals receiving an infusion of [2-13C]acetate according to:
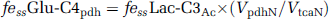
where ‘c’ is a correction to remove label incorporation arising from 13C-labeled plasma glucose (C1,6), lactate (C3), and BHB (C2,4) by peripheral metabolism of the [2-13C]acetate. The contribution of 13C-labeled plasma glucose and/or lactate to Glu-C4 was determined from the steady-state enrichment of brain lactate C3 (fe ss Lac-C3), which was assumed to be equivalent to the enrichment of pyruvate-C3, multiplied by the fraction of acetyl-CoA 13C labeling derived through pyruvate and pyruvate dehydrogenase and given by
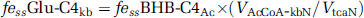
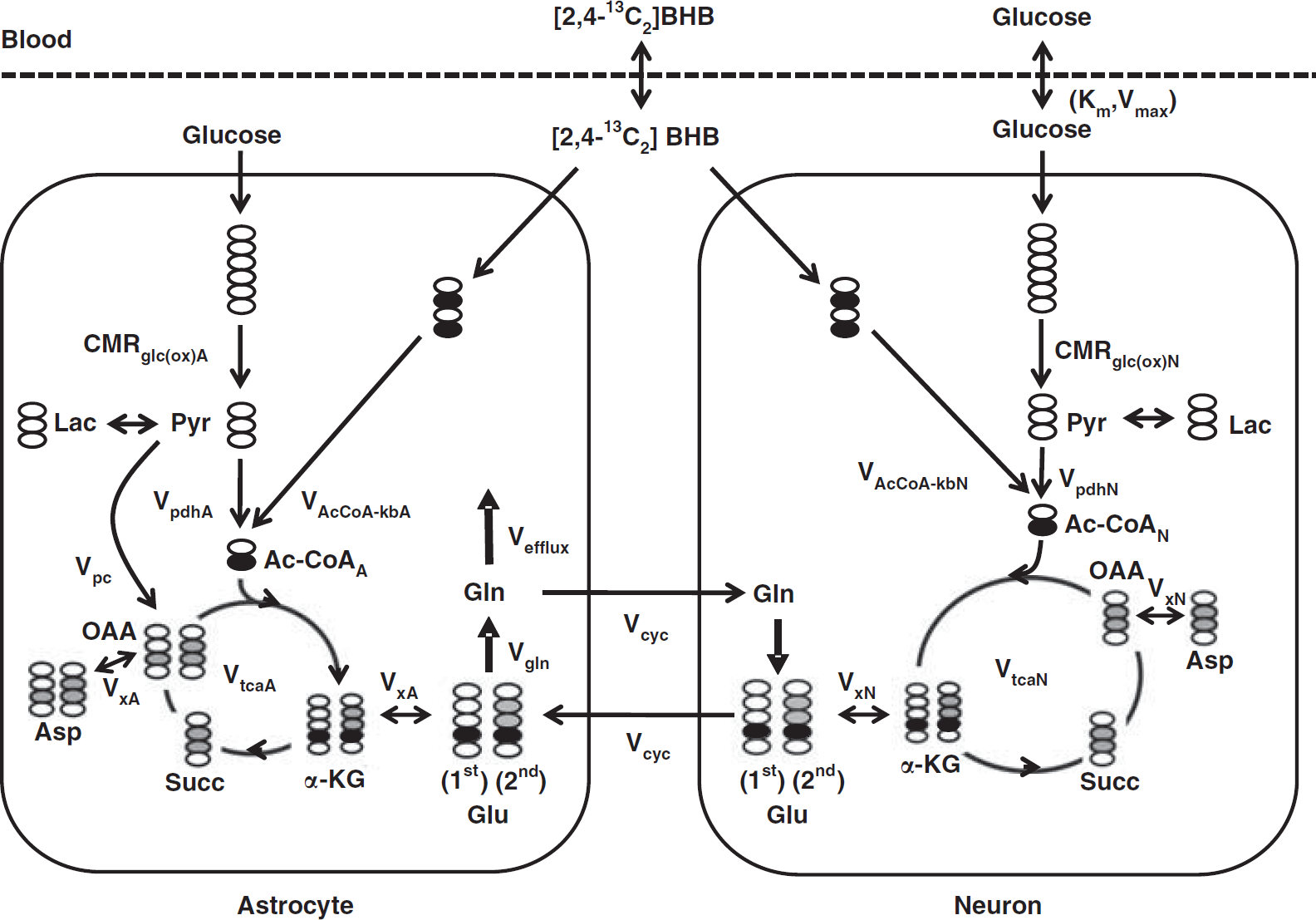
Two-compartment (neuron and astrocyte) model used to illustrate the metabolic pathway of label flow from BHB. CMRkb=(VAcCoA−kbA+VAcCoA−kbN)/2, CMRglc(ox)=(VpdhA+VpdhN+Vpc)/2. The OAA label distribution in astrocytes is shown for the pathway from succinate (equal labeling of OAA at C2 and C3) and pyruvate carboxylase (unlabeled carbon entry and dilution of OAA at C2). BHB,
Differential equations describing the two-compartment model

BHB,
The contribution to Glu C4 enrichment from ketone bodies (BHB) is given by
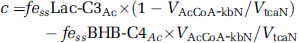
Assuming that the neuronal acetyl-CoA pool is derived only from pyruvate and BHB, the value of ‘c’ can be expressed as:
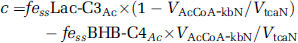
where the value for VAcCoA-kbN/VtcaN was estimated from BHB-infused fasted animals and scaled downward proportionally to reflect the lower plasma BHB levels (3.8±0.2 mmol/L, 13C enrichment 16%±5%) in fasted [2-13C]acetate-infused animals.
Statistics
Metabolites concentrations, enrichments, and metabolic fluxes were reported as group average±s.d. Monte Carlo analysis of each animal's data set was performed using CWave to assess the distribution of uncertainties in the model parameters for individual animals. The uncertainties in Monte Carlo fitting were smaller than the group variability for each parameter reported in this study, indicating that uncertainties were dominated by inter animal variation.
Results
Effects of Acute [2,4-13C2]-d -β-Hydroxybutyrate Infusion on Blood and Brain Ketone Bodies
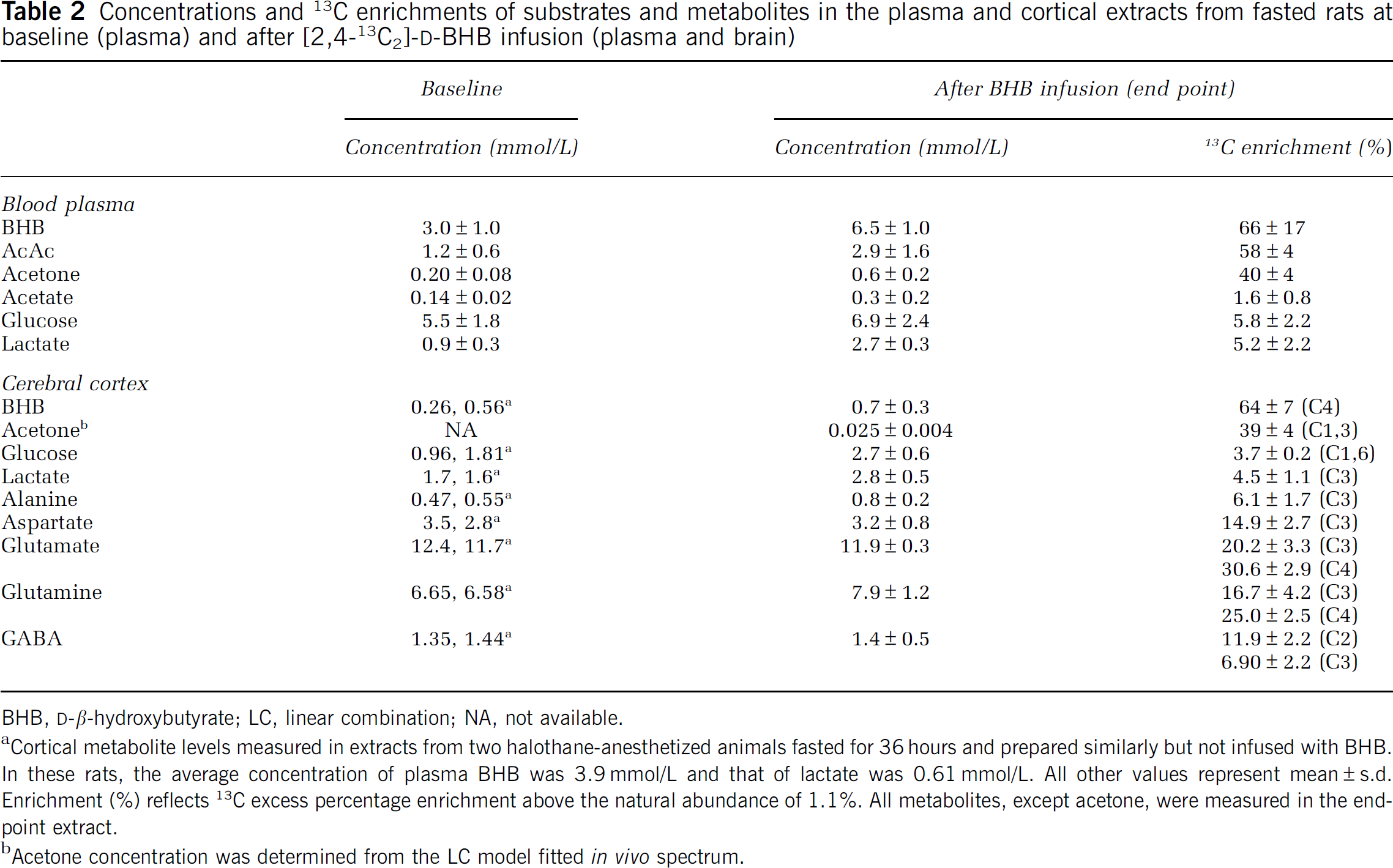
Table 2 presents the concentrations and enrichments of blood ketone bodies before and during 13C-BHB infusion. The preinfusion plasma BHB concentration was 3±1 mmol/L, which was quickly increased to 6.5±1 mmol/L giving an average enrichment of 66%±17% (Figure 2). The BHB infusions led to a small elevation in arterial blood pH (∼0.15 Units) compared with the preinfusion baseline.
Concentrations and 13C enrichments of substrates and metabolites in the plasma and cortical extracts from fasted rats at baseline (plasma) and after [2,4-13C2]-
BHB,
Cortical metabolite levels measured in extracts from two halothane-anesthetized animals fasted for 36 hours and prepared similarly but not infused with BHB. In these rats, the average concentration of plasma BHB was 3.9 mmol/L and that of lactate was 0.61 mmol/L. All other values represent mean±s.d. Enrichment (%) reflects 13C excess percentage enrichment above the natural abundance of 1.1%. All metabolites, except acetone, were measured in the end-point extract.
Acetone concentration was determined from the LC model fitted in vivo spectrum.
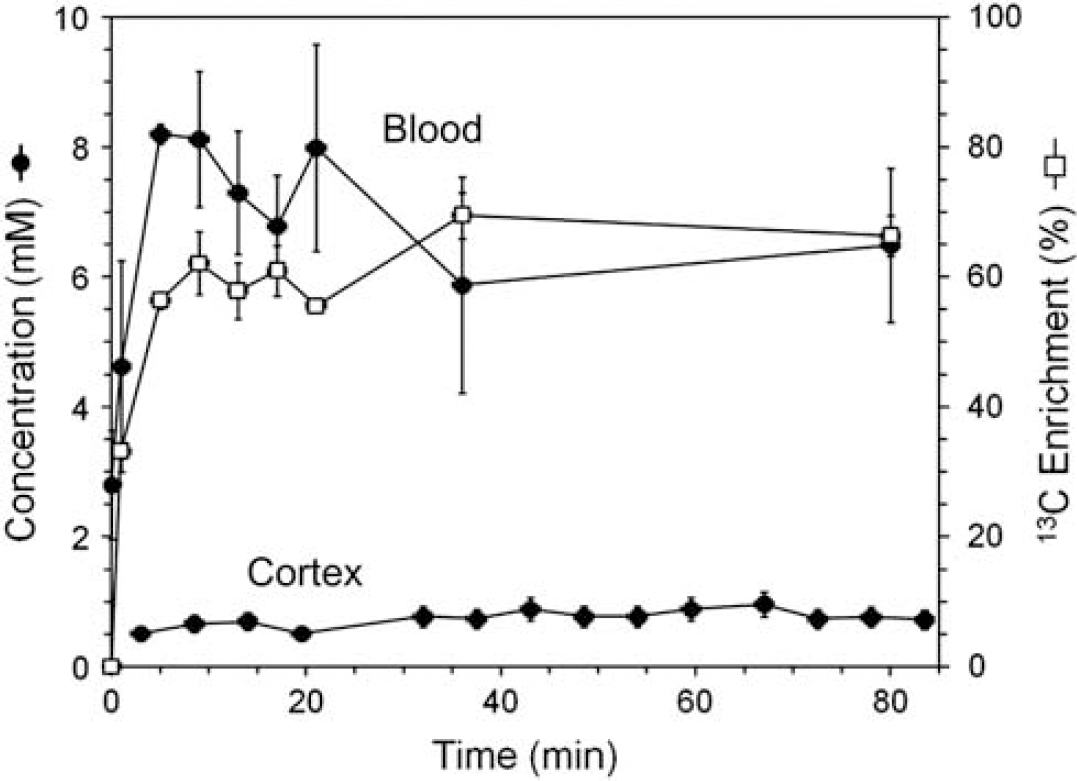
Time courses of BHB concentrations in the plasma and brain and 13C enrichment (plasma). BHB,
Blood levels of other ketone bodies were also increased and enriched with 13C during 13C-BHB infusion. Acetoacetate comprised ∼40% to 44% of the plasma BHB level (Table 2) and was similarly enriched, but was not detected in brain spectra in vivo or in the extract, likely because of both low brain concentration and overlap with GABA-C2. Plasma levels of acetone increased ∼3-fold during 13C-BHB infusion and were highly enriched with 13C (∼40%). Acetone was observed in plasma and brain spectra in vivo, but not in brain extracts because of its high volatility and loss during lyophilization. Plasma acetate levels increased similarly to BHB and acetoacetate (2.1-fold) during 13C-BHB infusion, although 13C enrichment remained relatively low (1.6%, Table 2), indicating limited conversion of the infused 13C-BHB to free acetate.
Enrichment of Brain Amino Acids From [2,4-13C2]-d -β-Hydroxybutyrate
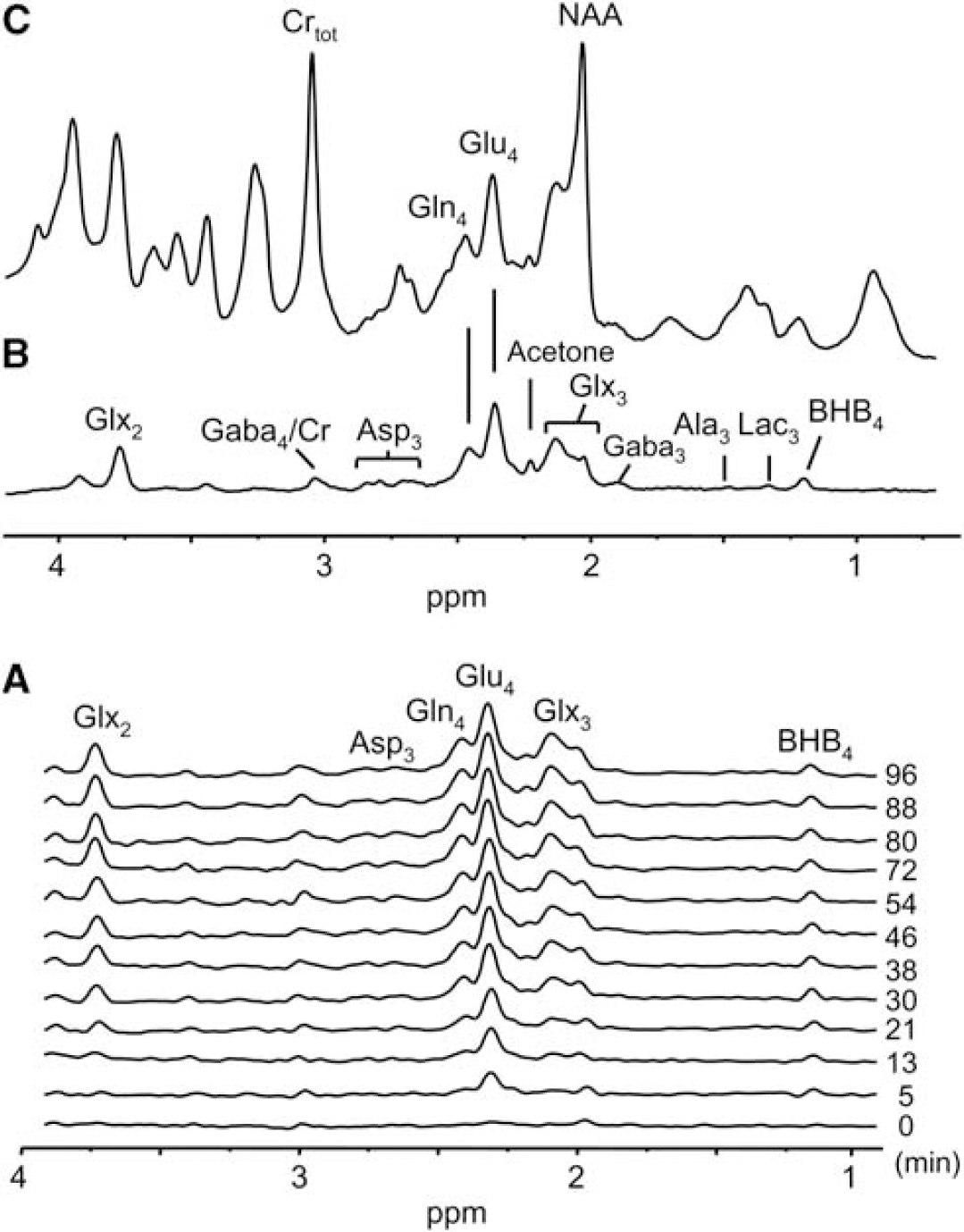
Spatially localized in vivo1H-[13C] spectra of the rat cerebrum acquired during an intravenous infusion of [2,4-13C2]-
Table 2 depicts the total concentrations and end-point 13C enrichments of cortical amino acids measured after 100 minutes of infusion of [2,4-13C2]-
Assessment of Metabolic Fluxes
Figure 4 depicts time courses of cortical 13C enrichments of Glu-C4 and Gln-C4 for a typical animal during [2,4-13C2]-
Estimates of cerebral metabolic fluxes (μmol/g per min) during hyperketonemia and [2,4-13C2]-
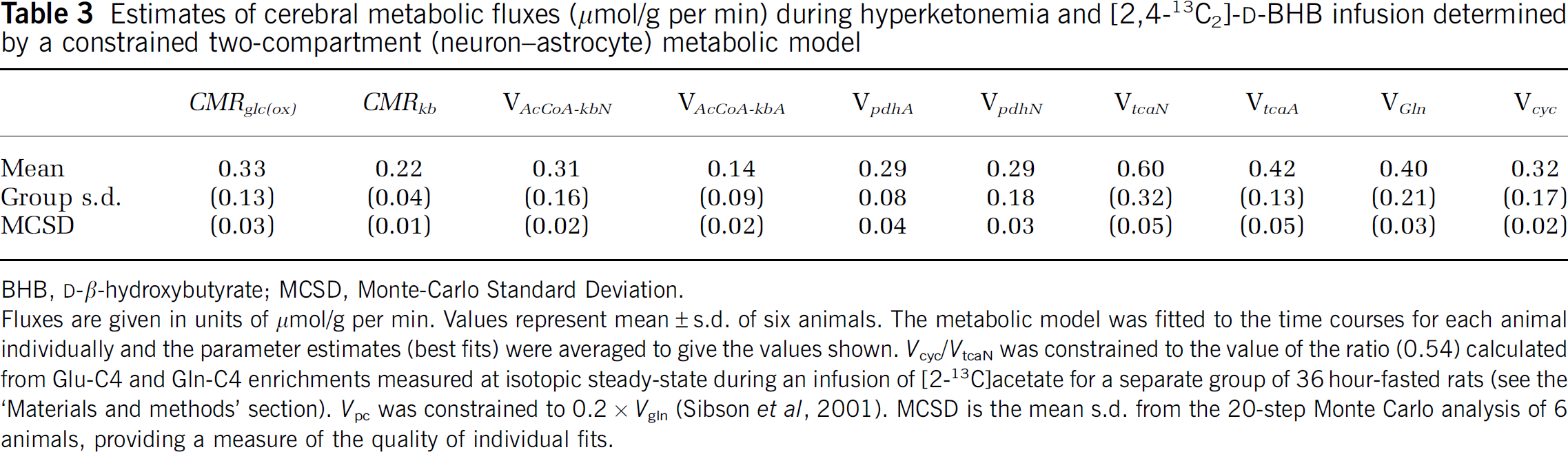
BHB,
Fluxes are given in units of μmol/g per min. Values represent mean±s.d. of six animals. The metabolic model was fitted to the time courses for each animal individually and the parameter estimates (best fits) were averaged to give the values shown. Vcyc/VtcaN was constrained to the value of the ratio (0.54) calculated from Glu-C4 and Gln-C4 enrichments measured at isotopic steady-state during an infusion of [2-13C]acetate for a separate group of 36 hour-fasted rats (see the ‘Materials and methods’ section). Vpc was constrained to 0.2 × Vgln (Sibson et al, 2001). MCSD is the mean s.d. from the 20-step Monte Carlo analysis of 6 animals, providing a measure of the quality of individual fits.
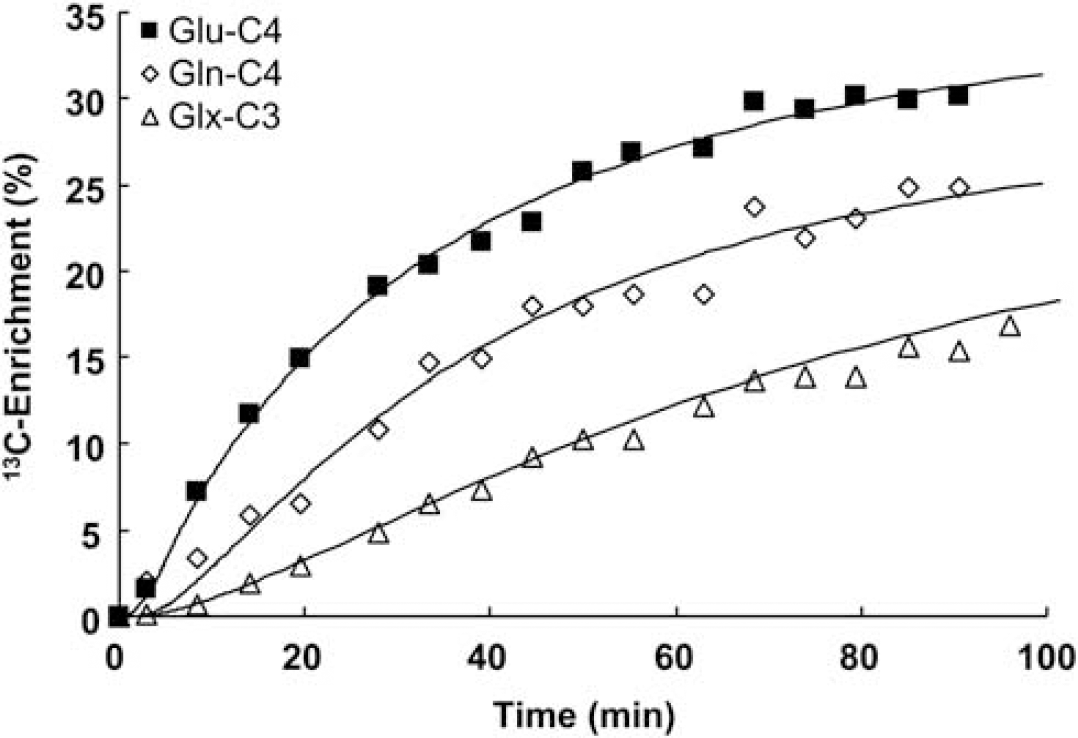
Time courses of 13C enrichments of Gln-C4, Glu-C4, and Glx3, and best fits of the metabolic model for a 36 hour-fasted rat during [2,4-13C2]-BHB infusion. The brain amino-acid 13C enrichments reflect the measured values (not normalized by plasma BHB 13C enrichment). The average blood BHB 13C enrichment was 65% for this animal. BHB,
The measured neuronal TCA cycle rate in 36 hour-fasted rats is similar to that seen in overnight-fasted, halothane-anesthetized rats infused with [1,6-13C] or [U-13C]glucose (0.47 to 0.55 μmol/g per min, van Eijsden et al, 2010; Jiang et al, 2009; Patel et al, 2004). In the latter studies, glucose accounts for >90% of oxidized substrate, such that VpdhN≈VtcaN. In contrast to glucose-infused rats, VpdhN (0.29 μmol/g per min) was ∼40% to 50% lower in hyperketonemic rats indicating that BHB oxidation is compensated by an equal reduction in oxidation of glucose-derived acetyl-CoA.
Effects of Parameter Constraints on the Calculated Fluxes
The reliability of the estimated value of the glutamate/glutamine cycling flux (Vcyc) when iterated as a free parameter depends on the difference in the time courses of Glu-C4 and Gln-C4 (Shen et al, 2009). In contrast to the well-separated Glu-C4 and Gln-C4 enrichment curves seen in anesthetized rats infused with [1-13C]glucose (Patel et al, 2005), the enrichment curves were closer together in [2,4-13C]BHB-infused rats, decreasing reliability in the value of Vcyc as an iterated parameter. To improve reliability in flux estimates, the model fits were constrained by the ratio, Vcyc/VtcaN, determined in 36 hour-fasted rats infused with the astroglial substrate, [2-13C]acetate (see the ‘Materials and methods’ section) (Supplementary material, Table S1).
Fitting the metabolic fluxes with the Vcyc/VtcaN ratio constrained (0.54) compared with Vcyc freely iterated improved the individual fits, reducing %MCSD (% Monte Carlo Standard Deviation) of Vcyc ∼40% without significantly altering variance in VtcaN and VAcCoA-kb, effects consistent with previous analysis (Shen et al, 2009).
[2,4-13C]BHB labels the TCA cycle and associated amino acids through [2-13C]acetyl-CoA, and is relatively insensitive to glial anaplerosis (pyruvate carboxylase flux). Therefore, Vpc was constrained to a value equal to 0.2 × Vgln based on a previous study of anesthetized rats infused with [2-13C]glucose (Sibson et al, 2001). To assess the sensitivity of the calculated fluxes on the value of this parameter, a sensitivity analysis was performed by fitting the metabolic model to the 13C time courses using different Vpc/Vgln ratio values, 50% below and 50% above the assumed value (Supplementary Material, Table S2). The analysis estimates the potential error in the calculated fluxes from their nominal values should the true value of the parameter be lower or higher than assumed. Relatively small differences from the nominal values were seen for lower values (Vpc/Vgln=0.1, −3% to −10%), but differences became larger for higher values (Vpc/Vgln=0.3, −44% to +35%), whereas fit quality was poor.
Discussion
In this study, we measured the contributions of ketone bodies and glucose to neuronal and astroglial oxidation in anesthetized rats made hyperketonemic by prolonged (36 hours) fasting and infused acutely with [2,4-13C]-BHB. The following main conclusions can be drawn: (1) BHB oxidation in the fasted anesthetized rat cortex is extensive, accounting for ∼40% of total TCA cycle flux; (2) quantitatively, BHB oxidation is greater in neurons (70% of total ketone consumption) than in astrocytes; and (3) comparisons with glucose-infused rats from previous studies show that ketone body oxidation is compensated by an equal reduction in glucose oxidation.
Comparison of Present Results With Previous Studies
Prolonged fasting is well known to enhance ketone body transport (Gjedde and Crone, 1975; Hasselbalch et al, 1996). The prolonged fast effectively produced a stable steady state of hyperketonemia before 13C-labeled BHB infusion, thus avoiding the major brain metabolic/regulatory shifts in transitioning between glucose- and BHB-fed conditions. The brain-to-blood plasma ratio for BHB in 36 hour-fasted rats of 0.11±0.04 agrees well with values from 48 hour-fasted rats (0.12, Hawkins et al (1971)), and is higher than that seen in fed rats infused acutely with ketone bodies (0.075, Hawkins et al (1971)), consistent with a fasting-induced enhancement in ketone body transport.
The rate of cortical ketone body oxidation, CMRkb (=1/2VAcCoA-kb) of 0.22±0.04 μmol/g per min, can be compared with previous estimates based on cerebral arteriovenous difference or 14C-autoradiography. Assuming a net extraction ratio for BHB of ∼0.04 to 0.07 (Hawkins et al, 1971; Daniel et al, 1971; Gjedde and Crone, 1975; Conn et al, 1983) and for acetoacetate of ∼0.1 (Hawkins et al, 1971), and a brain blood flow of ∼0.62 mL/g per min (Dahlquist and Persson, 1976), the corresponding rates per unit blood concentration (mmol/L) is given by the product of the extraction ratio and cerebral blood flow, yielding ∼0.02 to 0.04 and ∼0.06 μmol/g per min per mmol/L, respectively. For plasma BHB and acetoacetate concentrations of 6.5 and 2.9 mmol/L, respectively (Table 1), CMRkb would be ∼0.30 to 0.43 μmol/g per min. This value likely represents an upper estimate because extraction is assumed to be unidirectional with ketone bodies metabolized immediately upon entry. However, brain concentration of BHB (after correction for an assumed 3% blood volume) is not zero but is substantial (0.51 mmol/L), suggesting that net extraction (metabolism) is less than the values used above. Using clearance data from the 14C-autoradiography study of Hawkins et al (1986) for the parietal cortex of 2 day-starved unanesthetized rats, ketone body influx (μmol/g per min) can be computed from the BHB clearance rate (0.025 mL/g per min), the ratio of acetoacetate and BHB clearance (1.46), and the present concentrations of BHB (6.5 mmol/L) and acetoacetate (2.9 mmol/L), and is given by: CMRkb=CMRBHB+CMRAcAc=(0.025 × 6.5)+(0.025 × 1.46 × 2.9)=0.27 (±0.08) μmol/g per min. Considering the differences between methods, conditions (awake versus anesthetized), brain tissue represented in the measurements (whole brain versus cortex), and rat strains/gender (Wistar females and Long-Evans males versus Sprague–Dawley males), the Magnetic Resonance Spectroscopy (MRS)-derived value for ketone body consumption in vivo is in reasonably good agreement with other approaches.
β-Hydroxybutyrate Oxidation in Cortical Neurons and Astrocytes
Glutamate is concentrated primarily in neurons, whereas glutamine is produced and concentrated in astrocytes. [2,4-13C2]
However, our findings contrast with those of Kunnecke et al (1993) who reported a higher labeling ratio in brain Gln-C4 relative to Glu-C4 (Gln/Glu >1) during acute hyperketonemia in fed rats receiving infusions of [U-13C]BHB, and was interpreted as a greater metabolism of ketone bodies in astrocytes than in neurons. It is not clear whether dietary status alone (fed versus fasted) might produce this effect. Enhanced glutamine synthesis has been reported in mice fed the ketogenic diet after intraperitoneal injection of [1,2-13C]acetate and [1-13C]glucose (Melo et al, 2006; Yudkoff et al, 2005), suggesting that astrocytes may adapt to prolonged ketosis, but this condition would have been anticipated in fasted rats rather than in fed rats. The influence of dietary status (fed, fasted, ketogenic diet) and species on astrocyte metabolism during hyperketonemia in vivo merits further study.
Effects of Hyperketonemia on Glucose Utilization in the Cerebral Cortex
The neuronal TCA cycle rate of hyperketonemic-fasted animals in our study (0.60±0.32 μmol/min per g) is similar to our previously reported values from overnight-fasted, halothane-anesthetized rats infused with [1,6-13C]glucose, as measured from a comparable brain region and spectroscopic volume (0.52 to 0.53 μmol/min per g (Patel et al, 2004; van Eijsden et al, 2010)). This observation is consistent with previous studies of rats showing that brain oxygen consumption (which is proportional to TCA cycle activity) is unaffected by hyperketonemia whether by acute BHB infusion (Linde et al, 2006) or by starvation for 48 to 72 hours (Dahlquist and Persson, 1976; Hawkins, 1971).
As total rates of substrate oxidation (TCA cycle flux) in neurons and astrocytes seemed similar to their respective values in nonketonemic, 13C-glucose-infused, anesthetized rats (Patel et al, 2005), we conclude that BHB oxidation was compensated by an equivalent reduction in acetyl-CoA oxidation from glucose. Previous studies have reported inconsistent findings regarding the effects of hyperketonemia on glucose utilization. In an early study of brain slices/minces, Openshaw and Bortz (1968) reported that ketone body elevation decreased glucose oxidation, whereas others reported increased lactate formation with no change in glucose oxidation (Ito and Quastel, 1970; Rolleston and Newsholme, 1967). Studies reporting brain glucose utilization by arterio/venous difference (Linde et al, 2006) or 14C-2-deoxyglucose phosphorylation (Corddry et al, 1982; Crane et al, 1985) in rats during hyperketonemia, whether induced by fasting (2 to 3 days) or by acute BHB infusion, consistently show no change in CMRglc compared with the fed condition, unless fasting is more prolonged when a decrease in CMRglc occurs (Crane et al, 1985). The findings that increased oxidation of ketone bodies occurs without altering either glucose or oxygen consumption suggest that glucose metabolism during hyperketonemia is diverted to lactate production and efflux to the blood, which may occur transiently, as reported by Hawkins et al (1971).
Brain lactate was elevated in end-point extracts when compared with rats fasted for a comparable time (36 hours) but not infused with BHB (Table 2), although animal numbers were too few at baseline for statistical assessment. As plasma lactate levels also increased during 13C-BHB infusion, we cannot say whether the higher brain lactate reflects blood level changes, or possibly reduced intracellular clearance caused by the reduction in pyruvate/lactate oxidation and efflux as described by Pan et al (2000) in 2 to 3 day-fasted human subjects. As a cautionary note, extract measurements are susceptible to postmortem effects that can increase brain lactate, and whereas rats in our study were ventilated during brain freezing to reduce the likelihood of agonal change, this possibility cannot be excluded entirely.
Few in vivo studies are available that report glucose oxidation, as opposed to glucose uptake (arterio/venous difference) or phosphorylation (14C-2-deoxyglucose autoradiography). One exception is the study by Mans et al (1987), who found a small decrease in glucose oxidation (∼12% averaged over the whole brain) in unanesthetized rats fasted for 2 days compared with fed rats. These authors used quantitative autoradiography with short time infusion (5 minutes) of [6-14C]glucose, which is metabolized and trapped mainly as 14C-labeled amino acids, a technique most closely related to the NMR approach in this study. The larger reduction in glucose oxidation assessed in this study would be expected based on the much greater plasma BHB level (6.5 mmol/L) compared with the study by Mans et al (0.85 mmol/L).
Rodents subjected to prolonged hyperketonemia through a high-fat (ketogenic) diet show similar effects on glucose metabolism to those seen after short periods of fasting (<3 days), in that glucose oxidation is reduced but glucose uptake (phosphorylation) is unaltered. al-Mudallal et al (1995) found no alteration in regional CMRglc based on 14C-2-deoxyglucose phosphorylation in rats fed a ketogenic diet for 6 to 7 weeks. In contrast, in mice fed a ketogenic diet for 2 weeks followed by injection of [1-13C]glucose, significant reductions were reported in 13C labeling of brain metabolites and amino acids (Yudkoff et al, 2005; Melo et al, 2006).
Interestingly, in contrast to findings in rodents, prolonged fasting or acute BHB infusion in humans is associated with reduced brain glucose utilization (26% to 33%) as measured by positron emission tomography with [18F]2-fluoro-2-deoxy-
Potential Impact of Other 13C-Labeled Ketones and Nonketone Substrates on the Analysis
The 13C enrichments of acetoacetate and acetone were not considered in the present analysis, although both were highly enriched (Table 2). Acetoacetate is an intermediate in BHB catabolism and is in fast equilibrium with BHB; hence, its time course of labeling would be expected to be proportional to BHB. As acetoacetate was not detected in the brain spectra, and the transport kinetics of acetoacetate across the blood–brain barrier is uncertain, the use of blood or brain acetoacetate levels as a driver was precluded. We also did not account for the labeled acetone in the metabolic model. As acetone is highly volatile, it is likely to be exhaled and may not contribute significantly as a fuel substrate. However, acetone can be metabolized by the liver and labels plasma glucose in fasted humans (Reichard et al, 1979) and in rats under normal and diabetic ketotic conditions, as well as BHB in ketosis (Kosugi et al, 1986).
Any labeling of BHB-C4,2 by acetone-C1,3 would not be distinguished from the infused 13C-BHB used as the input.
Blood glucose 13C enrichment (5.8% in C1) was detected in fasted/13C-BHB-infused animals at ∼90 minutes, which would not be distinguished from the 13C-BHB because both substrates lead to the same labeling pattern for acetyl-CoA. Although we cannot specifically address its origin, we included the labeled glucose as an additional driver (input) in the modeling (Table 1) by assuming that glucose enrichment increased linearly over the period of the 13C-BHB infusion and that glucose-C1 and glucose-C6 were equally enriched. Although acetyl-CoA derived from glucose represented ∼56% of total acetyl-CoA (the rest supplied by ketone bodies), the low plasma glucose enrichment of 5.8% would be expected to result in a relatively small contribution (0.56 × 0.058 × 100=3.2%) to the glutamate-C4 end-point enrichment at isotopic steady state.
Ketone Body Utilization in Relation to Aspartate and GABA
The steady-state labeling of aspartate (Asp) and GABA relative to glutamate or glutamine from [2,4-13C]-
Conclusions
In summary, our results show that in anesthetized rats fasted for 36 hours, ketone bodies can support as much as 40% of total substrate oxidation in the cerebral cortex. Neurons and astrocytes oxidize BHB, although neurons consume the most and in a pattern similar to glucose. The oxidation of ketone bodies is compensated by reduced oxidation of glucose resulting in an apparently unaltered TCA cycle flux. Further experiments are required to define the range over which ketone body oxidation can be increased. This is important because of recent work indicating that transport and utilization of acetate (and possibly other monocarboxylic acids) is increased in humans with type 1 diabetes (Mason et al, 2006). The capacity of brain cells to oxidize substrates other than glucose (e.g., acetate, ketone bodies, free fatty acids, etc.) could provide significant therapeutic opportunities to enhance neuronal and astroglial function during times of hypoglycemic stress.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.