
Abstract
As ischemic stroke is associated with an excessive release of glutamate into the neuronal extracellular space, a decrease in blood glutamate levels could provide a mechanism to remove it from the brain tissue, by increasing the brain-blood gradient. In this regard, the ability of glutamate oxaloacetate transaminase (GOT) to metabolize glutamate in blood could represent a potential neuroprotective tool for ischemic stroke. This study aimed to determine the neuroprotective effects of GOT in an animal model of cerebral ischemia by means of a middle cerebral arterial occlusion (MCAO) following the Stroke Therapy Academic Industry Roundtable (STAIR) group guidelines. In this animal model, oxaloacetate-mediated GOT activation inhibited the increase of blood and cerebral glutamate after MCAO. This effect is reflected in a reduction of infarct size, smaller edema volume, and lower sensorimotor deficits with respect to controls. Magnetic resonance spectroscopy confirmed that the increase of glutamate levels in the brain parenchyma after MCAO is inhibited after oxaloacetate-mediated GOT activation. These findings show the capacity of the GOT to remove glutamate from the brain by means of blood glutamate degradation, and suggest the applicability of this enzyme as an efficient and novel neuroprotective tool against ischemic stroke.
Keywords
Introduction
It is well established that during ischemia, glutamate acts as an important mediator of neuronal degeneration, being released in large amounts from neurons and astrocytes, causing cellular overload of calcium, mainly through its action on calcium-permeable NMDA receptors. This overload of calcium leads to necrosis and breakdown of cellular structures including proteins, DNA, and membrane phospholipids (Lipton, 1999). In fact, studies from our group have demonstrated that neurologic deterioration of patients with acute ischemic stroke is associated with higher glutamate levels in blood and cerebrospinal fluid (Castillo et al, 1996, 1997).
Under normal physiological conditions, glutamate release in the synaptic cleft is quickly removed by the excitatory amino-acid transporters (EAATs), located on astrocytes and neurons (Hazell, 2007). Apart from astrocytes and neurons, the presence of EAATs has also been observed in endothelial cells from the brain vasculature, showing an important role of these cells in the maintenance of glutamate levels in the extracellular medium (O'Kane et al, 1999). It has been described that endothelial cells have facilitative carriers for glutamate in the luminal membranes, and EAATs in the abluminal membrane. This localization allows a unidirectional export of glutamate from the brain to the blood torrent following a gradient of concentration. Therefore, the decrease of blood glutamate levels leads to a larger glutamate gradient between the brain and blood, facilitating the lowering of extracellular levels of brain glutamate (Gottlieb et al, 2003; Teichberg et al, 2009).
As ischemic stroke is associated with an excessive release of glutamate into the neuronal extracellular space (Castillo et al, 1996, 1997), a decrease in the levels of blood glutamate could provide a mechanism to reduce extracellular glutamate at early times, with possible therapeutic implications after an ischemic insult (Gottlieb et al, 2003; Hawkins, 2009; O'Kane et al, 1999; Teichberg et al, 2009).
In this regard, recent studies have reported that glutamate oxaloacetate transaminase (GOT), a blood-resident enzyme, can be used as an important neuroprotective tool for brain diseases associated with elevated levels of glutamate (Gottlieb et al, 2003; Teichberg et al, 2009; Zlotnik et al, 2007, 2008). The potential applicability of GOT is based on the fact that this enzyme, using oxaloacetate as cosubstrate, is able to transform glutamate to 2-ketoglutarate and aspartate, leading to a decrease of glutamate levels in the blood.
On the above premises, we aimed to demonstrate if the reduction of glutamate levels in blood, by means of oxaloacetate-mediated GOT activation, induces a neuroprotective effect in a rat model of cerebral ischemia.
Materials and methods
Animals
Experimental protocols were approved by the local Animal Care Committee according to the European Union (EU) rules (86/609/CEE, 2003/65/CE, and RD 1201/2005). Male Sprague-Dawley rats (Harlan Laboratories, Udine, Italy) with a weight of 300 to 330 g were used. Rats were watered and fed
Surgical Procedures
Transient focal ischemia was induced in rats by using the occlusion of the middle cerebral artery (MCAO). Middle cerebral arterial occlusion model of brain ischemia is considered one of the best models to mimic human ischemic stroke and has been used in numerous studies in rats (Carmichael, 2005). Middle cerebral arterial occlusion was performed as previously described (Longa et al, 1989) with some modification. In brief, under operating microscope, the left common, external, and internal carotid arteries were dissected from connective tissue through a midline neck incision. The left external carotid artery and pterygopalatine artery of the internal carotid artery were separated and ligated by 5-0 silk sutures. A 23-mm segment of 3-0 nylon monofilament suture with the tip rounded by heat was inserted into the stump of the left common carotid artery and advanced into the internal carotid artery ∼19 to 20 mm from the bifurcation to occlude the origin of the MCA. The suture was removed after 90 minutes of occlusion. A laser-Doppler flow probe (tip diameter 1 mm) attached to a flowmeter (PeriFlux 5000; Perimed AB, Stockholm, Sweden) was located over the thinned skull in the MCAO territory (4 mm lateral to bregma) to obtain a continuous measure of relative cerebral brain flow during the experiment. Only animals with a cerebral blood flow reduction over 60% and with reperfusion after occlusion were included in the study.
Animal Experimental Procedures
Experimental procedure was performed following five criteria derived from the Stroke Therapy Academic Industry Roundtable (STAIR) group guidelines for preclinical evaluation of stroke therapeutics (Philip et al, 2009): (1) cerebral blood flow was measured to confirm the vascular occlusion, as an index of the reliability of the ischemic model; (2) animals were randomly assigned to treatment groups of the study; (3) researchers were blinded to treatment administration; (4) researchers were blinded to treatments during outcome assessment; and (5) temperature was controlled during the ischemic period.
Experimental Groups
Two experimental groups were designed: MCAO control group (
In both experimental groups, glutamate blood levels, infarct volumes, and edema development analysis were performed. Furthermore, somatosensory tests were performed on day 7. A sham group was also performed to confirm that the surgery did not affect the analyzed variables.
To study the effect of oxalacetate on blood glutamate and aspartate levels in health animals, two doses of oxaloacetate were tested, 1.5 mg/100 g (intravenously) (
Endothelial Cells Culture
Previous studies have established that the close proximity of astrocytes is crucial to the development of endothelial cells in culture models (Cohen-Kashi Malina et al, 2009), therefore, an
In brief, initial primary astrocyte cultures were prepared from neonatal (P0) Sprague-Dawley rat cortex, as previously described (McCarthy and de Vellis, 1980). For the noncontact coculture, 105 astrocytes from the primary culture were seeded on the bottom of poly-D-lysine (PDL) coated six-well plates for 5 to 6 days in Dulbecco's modified Eagle growth medium supplemented with 10% horse serum, 100 U/mL penicillin, and 100 µg/mL streptomycin (all from Gibco-Invitrogen Ltd, Paisley, UK). Then, Dulbecco's modified Eagle medium was removed and 7 × 104 rat endothelial cells (R840-05, Cell Applications Inc, San Diego, CA, USA) were seeded on rat tail collagen precoated (R35442, BD Biosciences, Madrid, Spain) transwell inserts in the six-well plate. Astrocytes and endothelial cells were grown to confluence for 4 days in EGM MV Microvascular Endothelial Cell Growth Medium (Lonza, Basel, Switzerland).
Immunocytochemistry
To characterize the endothelial cells used, the specific cell marker von Willebrand factor was used (Jaffe, 1977). For this, 104 endothelial cells were grown onto culture slides (R354632, BD Biosciences) for 4 days as previously described. Then, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 minutes and subsequently permeabilized with 0.3% Triton X-100 for 5 minutes. After washing with Tris-buffered saline, they were blocked with 5% bovine serum albumin in phosphate-buffered saline for 30 minutes. Cells were subsequently incubated with a polyclonal antibody raised in rabbit against von Willebrand factor for 1 hour (1:400, Abcam, Cambridge, UK), washed in Tris-buffered saline, and incubated with an anti-rabbit tetramethylrhodamine isothiocyanate (TRITC) conjugated for 1 hour (1:50, Invitrogen). Nuclei were counterstained by incubating with Hoescht 33342 dye (2 µg/mL, Molecular Probes-Invitrogen, Paisley, UK) for 5 minutes and then washed in Tris-buffered saline. All incubations were made at room temperature. After mounting, slides were observed under a fluorescence microscope (Olympus, Hamburg, Germany) and images were acquired with a CCD camera (Nikon, Nikon Precision Europe, Langen, Germany) using the NIS Elements software.
Immunoblotting
Expression of tight junction proteins (Zonula occludens protein-1 (ZO-1) and occludin) and glutamate transporters (EAAT1, EAAT2, and EAAT3) were detected in the endothelial cells by immunoblotting technique. At the end of the culture period, endothelial cells grown on transwell insert were rinsed with phosphate-buffered saline and collected in ice-cold lysis buffer (Invitrogen) with 1% antiprotease cocktail (Sigma-Aldrich Chemie GMBh, Taufkirchen, Germany). A sample of brain cortex (200 mg) from an adult health rat was also lysed to be used as control. The different samples were sonicated for 15 seconds, boiled for 5 minutes, and cooled immediately on ice. Sample extracts were centrifuged at 14,000 g for 5 minutes at 4°C and supernatant protein concentration was quantified using the Bradford method (Bio-Rad Protein Assay Kit, Bio-Rad Laboratories Ltd, Herts, UK). Forty micrograms of protein extract was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis on 8% polyacrylamide gels. Gels were run at 20 mA for 90 minutes. Separated proteins were electroblotted onto polyvinylidene fluoride (PVDF) membranes at a constant current intensity of 15 mA for 15 hours (semidry blotting). Membranes were blocked for 60 minutes at room temperature in blocking buffer containing 5% nonfat dry milk and then the blots were incubated overnight at 4°C with the primary antibody against the different proteins studied. Primary antibodies used were rabbit polyclonal antibody against ZO-1 (1:250, Invitrogen), mouse monoclonal antibody against occludine (1:500, Invitrogen), rabbit polyclonal antibody against EAAT1 (1:1000, Abcam), mouse monoclonal antibody against EAAT2 (1:500, Abcam), mouse monoclonal antibody against EAAT3 (1:500, Abcam), and mouse monoclonal antibody against actin (1:1000, Sigma). Actin was used as loading control. All antibodies used were diluted in Tris-buffered saline-Tween-bovine serum albumin (BSA) buffer (20 mmol/L Tris base, 140 mmol/L NaCl, 0.1% Tween-20). After washing, the blots were incubated for 1hour at room temperature with the following secondary antibodies: goat anti-mouse Cy3 (1:3000, GE Life Sciences, GE Healthcare Europe GmbH, Munich, Germany) and goat anti-rabbit Cy5 (1:3000, GE Life Sciences), according to the primary antibody used. Western image was acquired with fluorescence scam (Molecular Imager PharosFX Plus System, Bio-Rad).
Serum Glutamate and Aspartate Analysis
Glutamate and aspartate serum levels were determined by high performance liquid chromatography analysis, following a previously described method (White et al, 1986). The experiments with oxalacetate administration with health animals, glutamate and aspartate levels were measured before the treatment (basal) and 1, 2, and 3 hours after the treatment injection. In experiments with animal model of ischemia, the blood samples were acquired before surgery (basal sample) and 90 minutes (before treatment injection), 4 hours, 24 hours, 3 days, and 7 days after the occlusion.
Magnetic Resonance Imaging Protocol
Infarct size and edema extension were assessed by means of magnetic resonance imaging. Magnetic resonance imaging studies were conducted on a 9.4-T horizontal bore magnet (Bruker BioSpin, Ettligen, Germany) with 20 cm wide actively shielded gradient coils (440 mT/m). Radiofrequency transmission was achieved with a birdcage volume redsonator; signal was detected using a fourelement surface coil, positioned over the head of the animal, which was fixed with a teeth bar, earplugs, and adhesive tape. Transmission and reception coils were actively decoupled from each other. Gradient-echo pilot scans were performed at the beginning of each imaging session for accurate positioning of the animal inside the magnet bore. Apparent diffusion coefficient maps were acquired using a spin-echo echo-planar imaging sequence with the following acquisition parameters: field-of-view 19.2 × 19.2 mm2, image matrix 128 × 128 (in-plane resolution 0.15 mm/pixel × 0.15 mm/pixel), 14 consecutive slices of 1 mm thickness, repetition time = 4 seconds, four segments, echo time = 30 ms, spectral bandwidth 2,50,000 Hz, and seven diffusion
In vivo Magnetic Resonance Spectroscopy
Magnetic resonance spectra were acquired as previously described (Higuchi et al, 1997; Tkac et al, 1999). Local shimming was performed by manual adjustment of first-and second-order shim coil currents using a Press-waterline sequence. The field homogeneity in a 3-mm3 voxel typically resulted in signal line widths of 11 to 20 Hz for water. Water signal was suppressed by variable power RF pulses with optimized relaxation delays.
Image Analysis
All images were processed using ImageJ (Rasband WS, ImageJ, NIH, http://rsb.info.nih.gov/ij). Infarct volumes were determined from quantitative diffusion and T2 maps by manually selected areas of elevated apparent diffusion coefficient/T2 values by a researcher blinded to animal handling. Edema was estimated by measuring the volumes of the affected (
Somatosensory Test
Sensorimotor deficits were assessed as previously described (Reglodi et al, 2003). All sensorimotor tests were performed during the light cycle of animal housing, with environmental conditions consistently maintained across examinations. Briefly, the rodent's walking pattern was observed during a 1-minute period by a researcher blinded to the animal grouping. A sensorimotor score on a scale of 0 to 3 was assigned to rats on the basis of their gait behavior. 0, Straight walking; 1, walking toward the contralateral side; 2, alternate circling and walking straight; 3, circling and/or other gait disturbance (backing, crawling, walking on digits).
Statistical Analysis
Results are expressed as percentages for categorical variables and as mean (s.d.) or median (quartiles) for the continuous variables, depending on the normal or not normal distribution of data. Proportions were compared using the χ2 test, and the Student's
Results
Characterization of Excitatory Amino-Acid Transporters on Endothelial Cells
A noncontact coculture of rat endothelial cells with rat brain astrocytes was chosen to check the expression of EAATs. This model allows the removal of endothelial cells from the culture without contamination from astrocytes (Figure 1A). Our cells were positive for von Willebrand factor, a specific marker for endothelial cells (Figure 1B). To check that cultured endothelial cells had functional properties close to those observed
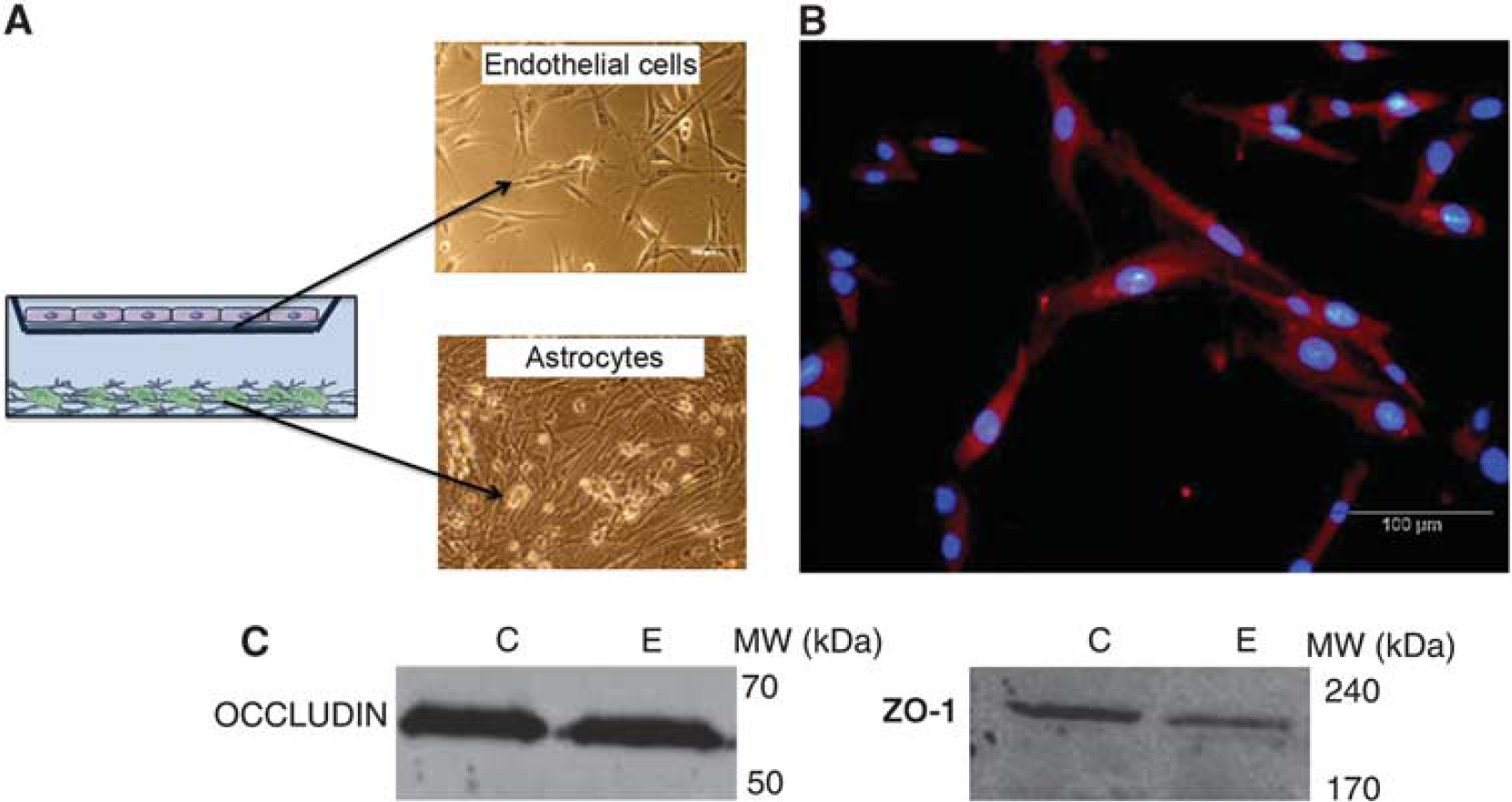
Coculture of astrocytes and endothelial cells used in the study. (
Once endothelial cells culture was characterized, EAATs expression was analyzed. Figure 2 shows the expression of EAAT1 and EAAT3 in endothelial cells. We did not detect the presence of EAAT2 under our experimental conditions. The expression of EAAT1, EAAT2, and EAAT3 in a brain tissue sample, used as positive control, confirmed that bands observed in the Western blots effectively corresponded to the analyzed proteins.
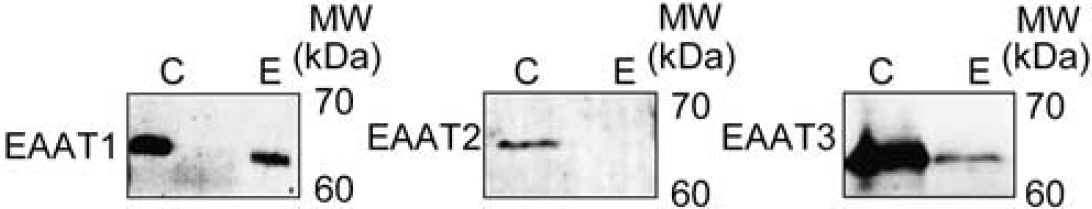
Excitatory amino-acid transporters (EAATs) protein expresion in rat endothelial cells. Western blot analysis shows the expression of EAAT1 and EAAT3, but not EAAT2, in endothelial cells (E). Tissue from the brain cortex (C) was used as positive control of EAATS.
Effect of Oxaloacetate-Mediated Glutamate Oxaloacetate Transaminase Activation on Glutamate Blood Levels
A 1.5-mg/100 g dose of oxaloacetate was administered by intravenous injection to healthy animals, observing no changes on blood glutamate levels within the first 3 hours after administration (Figure 3A). As GOT metabolizes glutamate to 2-ketoglutarate and aspartate, we also analyzed aspartate levels on blood to confirm that the dose of oxaloacetate used was not enough to activate GOT. No changes on aspartate levels were observed (Figure 3B). When a higher dose of oxaloacetate of 3.5 mg/100 g (intravenously) was used, a significant decrease of glutamate levels was observed at 1 hour after treatment, and maintained within the first 3 hours after administration. The reduction of glutamate was about 60% with respect to basal levels (
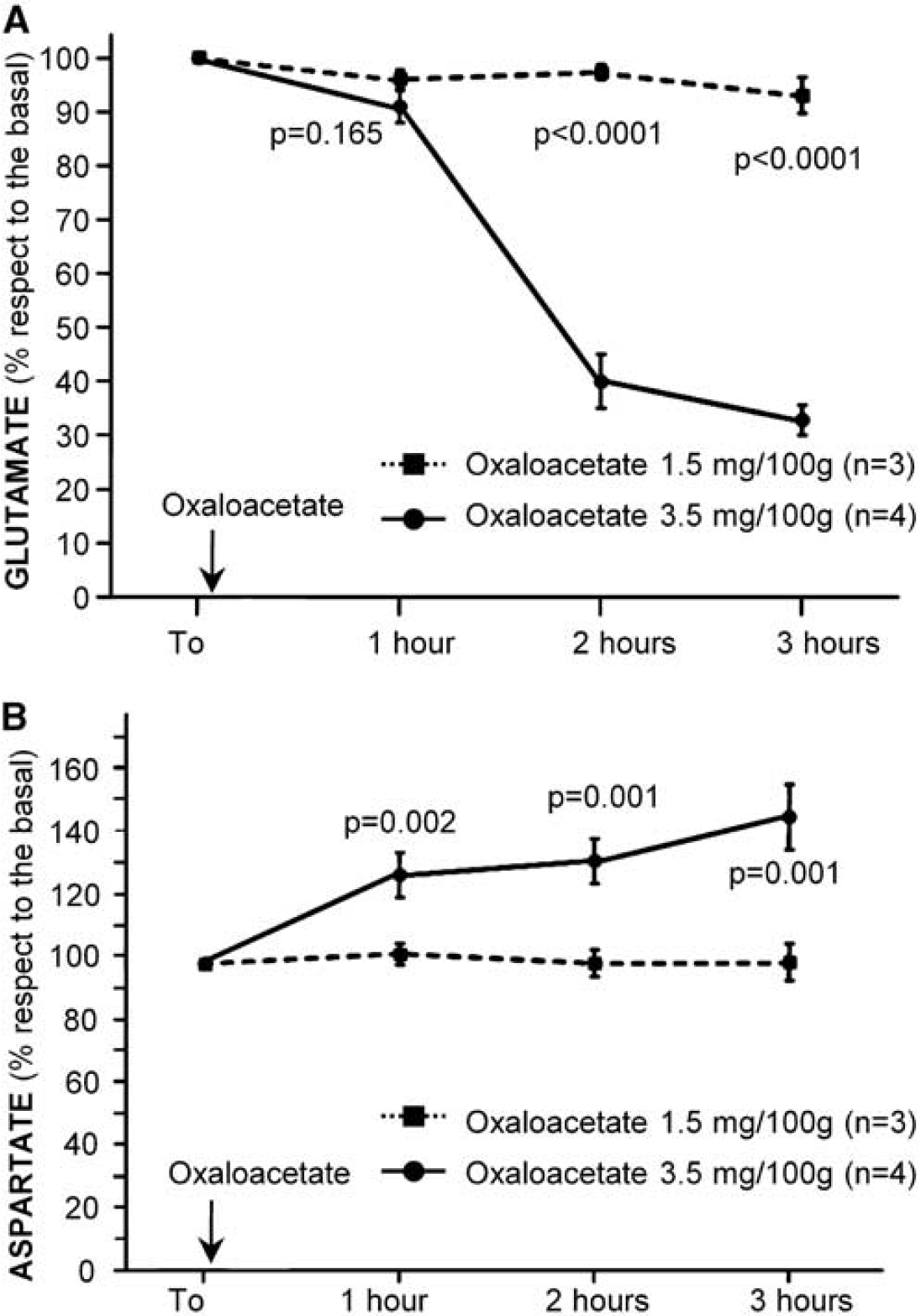
Doses-reponse effect of oxaloacetate on blood glutamate (
Neuroprotective Effect of Oxaloacetate-Mediated Glutamate Oxaloacetate Transaminase Activation on Middle Cerebral Arterial Occlusion Animal Models
Middle cerebral arterial occlusion induced an increase of 40% on blood glutamate levels at 4 hours after ischemia onset (Figure 4), which returned to normal levels 24 hours later. The increasing effect of cerebral ischemia on glutamate levels was inhibited when oxaloacetate-mediated GOT activation (3.5 mg/100 g) was induced (
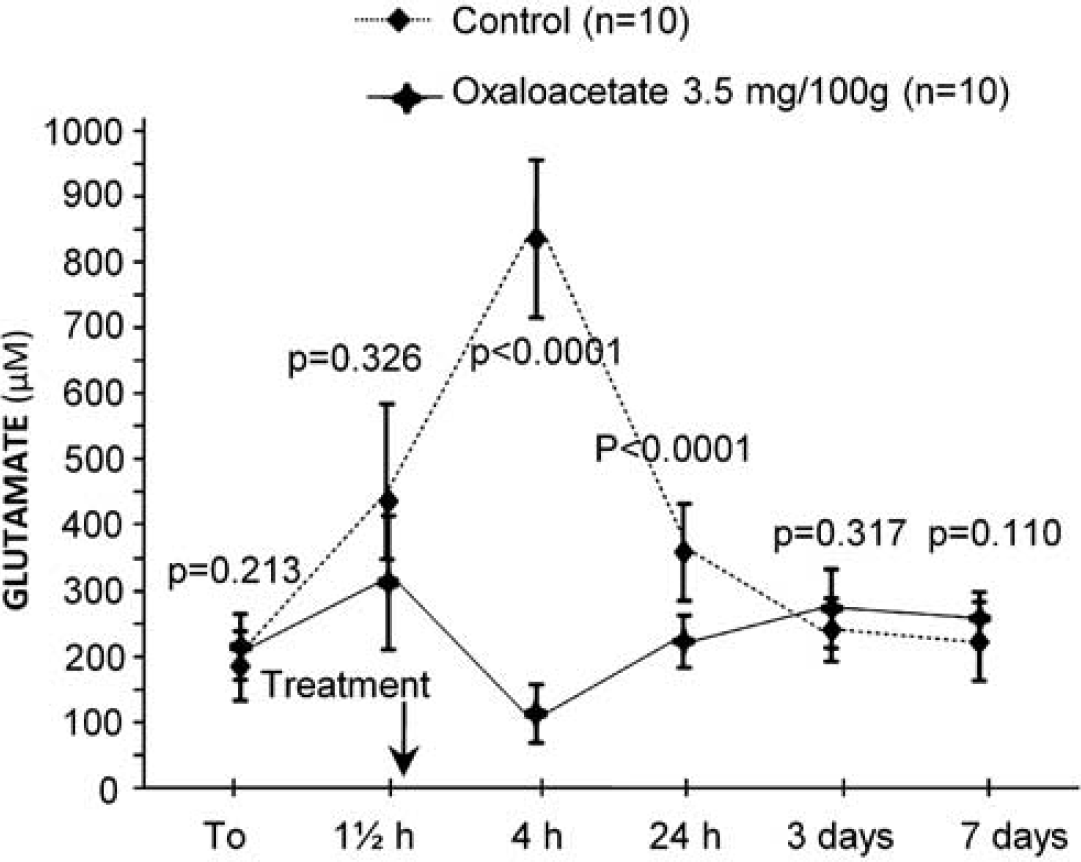
Time course of blood glutamate levels in control (
To demonstrate that the reduction on blood glutamate levels observed after GOT activation led to a neuroprotective effect on our animal model of cerebral ischemia, the infarct volumes of treated versus control animals were compared. As shown in Figures 5A and 5B, oxaloacetate-mediated GOT activation decreased lesion sizes about 80%. At day 7, infarct volumes of treated group were significantly lower than those for control group (
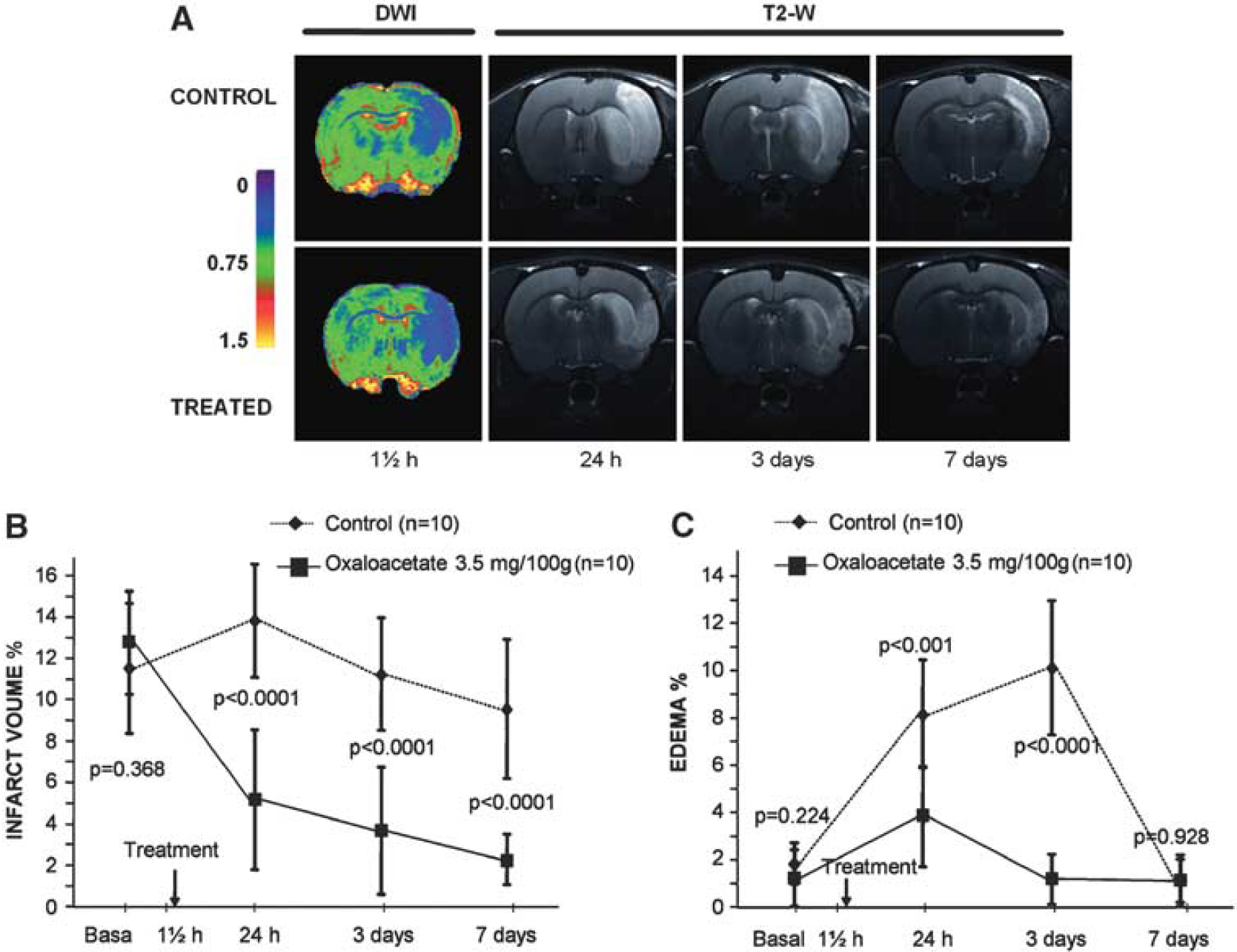
Infarct size and edema development were assessed by means of magnetic resonance image (MRI). Diffusion-weighted images (DWI) were acquired immediately after removing the suture from the animal (90 minutes after the occlusion); T2-weighted (T2-W) images were acquired at 24 hours, 3 days, and 7 days after ischemia onset (
On the other hand, a lower sensorimotor deficit was observed in treated animals compared with controls (1 [1,2] versus 2 [2,3];
Magnetic Resonance Spectroscopic Analysis of Cerebral Glutamate Levels in Middle Cerebral Arterial Occlusion Animal Models
Magnetic resonance spectroscopic (Figures 6A and 6B) analysis was used to study the time course of cerebral glutamate levels in ischemic animals treated with (3.5 mg/100 g) oxaloacetate and with saline. Quantitative analysis of magnetic resonance spectra revealed a persistent increase of glutamate levels after the occlusion onset for control group. Four hours after occlusion, the increase of extracellular glutamate levels was about 300% with respect to basal levels. A significant decrease of glutamate levels in the brain parenchyma was observed in the treated group with respect to the control animals (
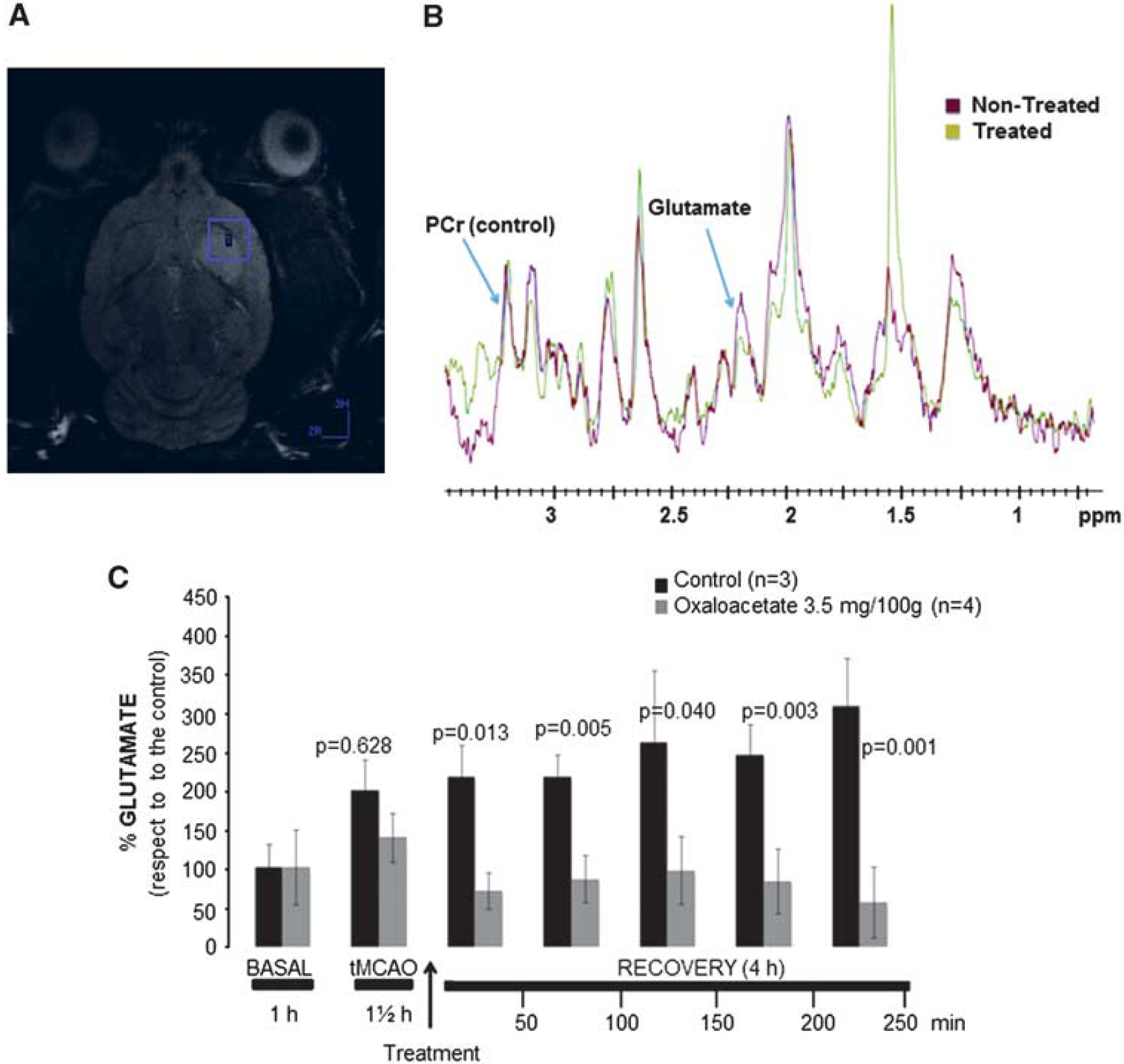
Magnetic resonance spectroscopy (MRS) studies. (
Discussion
The brain is an organ with a high content of glutamate, the most important excitatory neurotransmitter. Although its presence is essential for the correct function of neuronal tissue, its excitatory properties become dangerous for neurons when its concentration in the extracellular fluid rises under pathological conditions like ischemic stroke.
During ischemic stroke, glutamate is highly released to the extracellular space, mediating a marked increase in intracellular calcium, followed by the activation of intracellular enzymes, which provoke neuronal death. The central role glutamate in the ischemic cascade, make this neurotransmitter a good target in the search for neuroprotective agents in ischemic stroke.
In agreement with the previous findings (Zlotnik et al, 2007, 2008), in the present manuscript, we demonstrated that in ischemic stroke, a decrease in blood glutamate levels by means GOT, provides a neuroprotective mechanism to remove this neurotransmitter from the brain tissue to the blood through the blood-brain barrier.
The expression of EAATs on brain capillary endothelial cells has been described to be essential for the diffusion of glutamate from the brain parenchyma to blood. Prior to study the effects of GOT and given that we used rats in our experiments, the first step of our research was to demonstrate the presence of EAATs on rat endothelial cells, where, to the best of our knowledge, they have never been described before. We focused our studies on EAAT1, EAAT2, and EAAT3 due to their ubiquitous location and because they have been previously described in porcine and bovine brain endothelial cells (O'Kane et al, 1999; Teichberg et al, 2009). We detected EAAT1 and EAAT3, but not EAAT2 expression in rat endothelial cells. The expression of at least two types of EAATs on this cell type in rats seems to demonstrate the permeability for glutamate from the brain parenchyma into the blood torrent through the blood-brain barrier.
A high number of neuroprotective agents found in experimental research have failed in stroke clinical trials, most of the cases due to the narrow therapeutic window (Castellanos et al, 2006). Another important reason has been the low quality and adequacy of animal testing for neuroprotective agents for stroke (Philip et al, 2009). For these reasons, oxaloacetate was administered to demonstrate the properties of GOT activation as a neuroprotective tool within a wide therapeutic window and following the STAIR guidelines for preclinical evaluation of stroke therapeutics (Philip et al, 2009). Besides, our primary end point in the experimental study was infarct volume. The infarct volume analysis represents an objective and quantitative tool to estimate ischemic brain injury and it is commonly used to determine the efficacy of neuroprotective agents in preclinical studies (Lin et al, 1993).
It has been described that GOT activation with oxaloacetate 1.2 mg/100 g (intravenously) reduces blood glutamate levels (Gottlieb et al, 2003; Nagy et al, 2009). In disagreement with these results, we observed no effects with a dose of 1.5 mg/100 g dose within the first 4 hours after treatment under our experimental conditions. However, when oxaloacetate 3.5 mg/100 g (intravenously) was tested, a significantly maintained decrease on blood glutamate levels was observed within the first hours. Therefore, this last dose was chosen to test the neuroprotective effect of oxaloacetate-mediated GOT activation.
The increase of blood glutamate levels observed after MCAO disappeared when GOT was activated in the treated group with oxaloacetate. This effect on the reduction of blood glutamate levels was associated with a decrease of infarct sizes and edema development, as well as to a reduction on motor deficits.
Preclinical testing criteria for neuroprotective agents establishes that they have to reduce the ischemic damage at least by 50% or even better to 80% before being considered for clinical trials (Fisher et al, 2007; Rother, 2008). Considering that GOT activation reduces the ischemic damage about 80%, an optimal neuroprotective capacity is expected for this enzyme.
High levels of glutamate in brain parenchyma cause an overstimulation of AMPA and NMDA receptors. Overstimulation of both receptors induces an influx of calcium and sodium, leading to an electrolytic disturbance in brain tissue and the subsequent formation of cytotoxic edema. The increase in tissue water content and brain swelling that contribute to the ischemic injury worsen (Lin et al, 1993; Moldes et al, 2008). Therefore, the development of brain edema represents another good marker to determine the neuroprotective efficacy of GOT activation. In line with this, our results show that oxaloacetate-mediated GOT activation reduced the edema development.
It could be argued that observed effects could be actually caused by neuroprotective properties of oxaloacetate and not to a real effect of GOT on brain glutamate levels. To clarify this point, we performed a magnetic resonance spectroscopy analysis of glutamate levels in brain parenchyma. In agreement with previous
As it is shown in Figures 4 and 6, the decrease of blood and brain glutamate levels takes place simultaneously after oxaloacetate treatment, reflecting a rapid diffusion of glutamate from the brain into the blood torrent. In this regard, kinetic studies by means of radiolabeled glutamate injection into rat lateral ventricles have also shown a fast (within 1 minute) diffusion of glutamate from the brain parenchyma to blood (Gottlieb et al, 2003). Therefore, the efficacy of GOT to remove brain glutamate depends on its ability to metabolize blood glutamate to increase the concentration gradient between the brain parenchyma and blood torrent.
Although clinical studies are necessary to perform and validate these results, in conclusion, the capacity of the GOT to remove glutamate from the brain by means of blood glutamate degradation show that this enzyme could be considered as an efficient and novel neuroprotective tool against ischemic stroke.
Conflict of interest
The authors declare no conflict of interest.