
Abstract
Effect of tissue-type plasminogen activator (tPA) on oxygen–glucose deprivation (OGD) was studied in cultured cortical neurons prepared from tPA gene knockout (tPA-KO) and wild-type (Wt) mice. Three hours of OGD induced 45% and 23% of neuronal death in Wt and tPA-KO mice, respectively. Neuronal death in tPA-KO mice was increased to 42% by additional tPA. Six hours of OGD induced 80% and 40% of neuronal death in Wt and tPA-KO mice, respectively, whereas the addition of tPA increased to 62% in tPA-KO mice. These results suggest that tPA is directly involved in the process of neuronal death induced by ischemia-mimic stress without involving vascular or circulatory components.
Tissue-type plasminogen activator (tPA), a secretory serine protease, changes plasminogen into plasmin, which then degrades insoluble fibrin and is used for thrombolytic therapy. In patients with ischemic stroke, tPA treatment within 3 hours after the onset of symptoms restored reperfusion and reduced morbidity and mortality (NINDS rt-PA stroke study group, 1995). In experimental animals, tPA limited cerebral infarct size (Vanderschueren et al., 1997). These observations suggest that the treatment with tPA reduces the ischemic neuronal damage by recanalizing occluded vessels.
Recent reports, however, have suggested that tPA mediates excitotoxicity in the hippocampus through the activation of plasmin and degradation of laminin (Tsirka et al., 1995; Chen and Strickland 1997). These observations suggest that tPA may also deteriorate cerebral infarction, because glutamate excitotoxicity may exacerbate ischemic neuronal death. However, reports for the effects of tPA on cerebral ischemic injury are controversial (Wang et al., 1998; Nagai et al., 1999a, 1999b; Tabrizi et al., 1999; Kilic et al., 1999). The discrepancy among these results seems to be explained by the opposing roles of tPA: one as a mediator of neurotoxicity and the other as a thrombolytic agent to recover cerebral blood flow.
In this study, the effect of tPA on neuronal death induced by oxygen-glucose deprivation (OGD) that mimics ischemic insults was investigated. Cortical neurons obtained from tPA-KO and Wt mice were used in culture to exclude the possible effects of tPA on cerebral blood flow and vessels through thrombolytic activity.
MATERIALS AND METHODS
Primary culture of cortical neuron
Cortical neurons were prepared from one-day-old mice. After removal of the brain under brief anesthesia, the cerebral cortex was dissected and triturated repeatedly with Pasteur pipettes. Dissociated cells were placed on a collagen-coated, glass-bottomed petri dish in a culture medium as previously reported (Yamamoto et al., 1996). The dish then was incubated for a week at 37°C under 95% air and 5% CO2, and was subjected for the experiment.
Induction of oxygen–glucose deprivation
After changing the culture medium to a glucose-free artificial cerebrospinal fluid (aCSF), cells were submitted to hypoxia in a humidified chamber containing oxygen absorbing agent (Powdertech, Tokyo, Japan) under 95% N2 and 5% CO2 for 3 hours or 6 hours at 37°C. Artificial CSF contained (in mmol/L) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 2.6 NaHCO3, and 10 glucose. In another set of experiments, neurons prepared from tPA-KO mice were treated with culture medium containing 10 μg/mL human recombinant tPA (hr-tPA; Sumitomo Pharmaceuticals, Osaka, Japan) for 1 hour, and were exposed to hypoxia after application of glucose-free aCSF containing 10 μg/mL hr-tPA. To analyze the effect of N-methyl-d-aspartate (NMDA) receptor antagonist on the OGD-induced neuronal death, neurons prepared from wild-type (Wt) mice were treated with 20 μmol/L MK-801 (Sigma, St. Louis, MO, U.S.A.) for 20 minutes, and were exposed to OGD for 3 hours with coincubation of MK-801 (20 μmol/L). Control culture dishes were maintained under 95% air and 5% CO2 in a glucose-containing aCSF.
Assessment of neuronal death
To assess neuronal death, cells were stained with 0.4% trypan blue added to aCSF for 15 minutes (Yamamoto et al., 1996) and were observed under an inverted differential interference contrast microscope (Axiovert 10; Zeiss, Oberkochen, Germany) equipped with a video contrast-enhancement system. The number of living and dead neurons, respectively, were counted in three different observation fields of each dish, and then the mean death rate was calculated.
Statistical analysis
Data were expressed as mean ± SD and were evaluated using one-way analysis of variance. Two group comparisons were made using Student's t-test. Differences were considered significant for P < 0.05.
RESULTS
In control neurons (exposed to normoxia and normoglycemia), few neurons were stained with trypan blue after 6 hours (Fig. 1A). After 6-hour exposure to OGD in the culture prepared from Wt mice, most neurons were stained with trypan blue, and even in trypan blue–negative neurons, the cell bodies were swollen (Fig. 1B). In contrast, in the culture from tPA-KO mice, trypan blue–positive neurons and swollen neurons were less frequent (Fig. 1C). This reduction in the trypan blue–positive neurons was reversed to the same level as observed in Wt mice by the treatment of 10 μg/mL of hr-tPA (Fig. 1D).
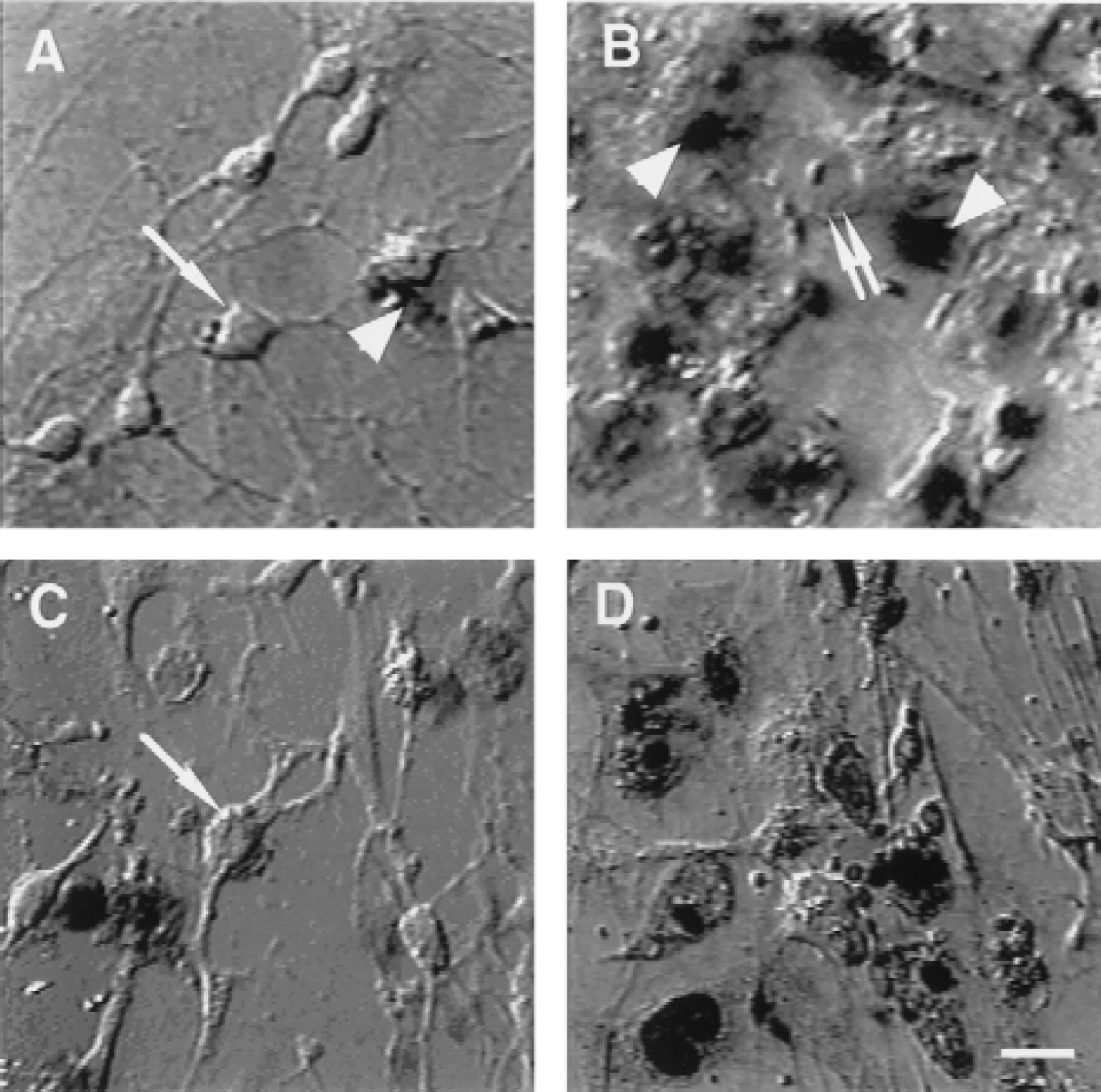
Differential interference contrast images of cultured cortical neurons stained with trypan blue.
In control culture, the death rates of neurons after 3 hours or 6 hours were between 6.8% and 12% in Wt, tPA-KO, or tPA-KO in the presence of hr-tPA. There was no statistical difference among these values. After 3-hour OGD, 46% ± 9.2% of the neurons died in Wt cultures (n = 7), whereas 23% ± 18% died in tPA-KO mice, which was significantly less than that of Wt mice (P < 0.05) (Fig. 2). This reduction was completely reversed (to 42% ± 11%) by hr-tPA (n = 7) (Fig. 2). The treatment with MK-801 (20 μmol/L) decreased neuronal death induced by 3-hour OGD in Wt culture (to 20% ± 11%;P < 0.05, n = 7). After 6-hour OGD, the rate of the neuronal death was 80% ± 11% in Wt mice (n = 7) and 40% ± 17% in tPA-KO mice (n = 7) (Fig. 2). Neuronal death significantly decreased (P < 0.05) in tPA-KO, which was partially reversed (to 62% ± 16%) by hr-tPA (n = 7) (Fig. 2).
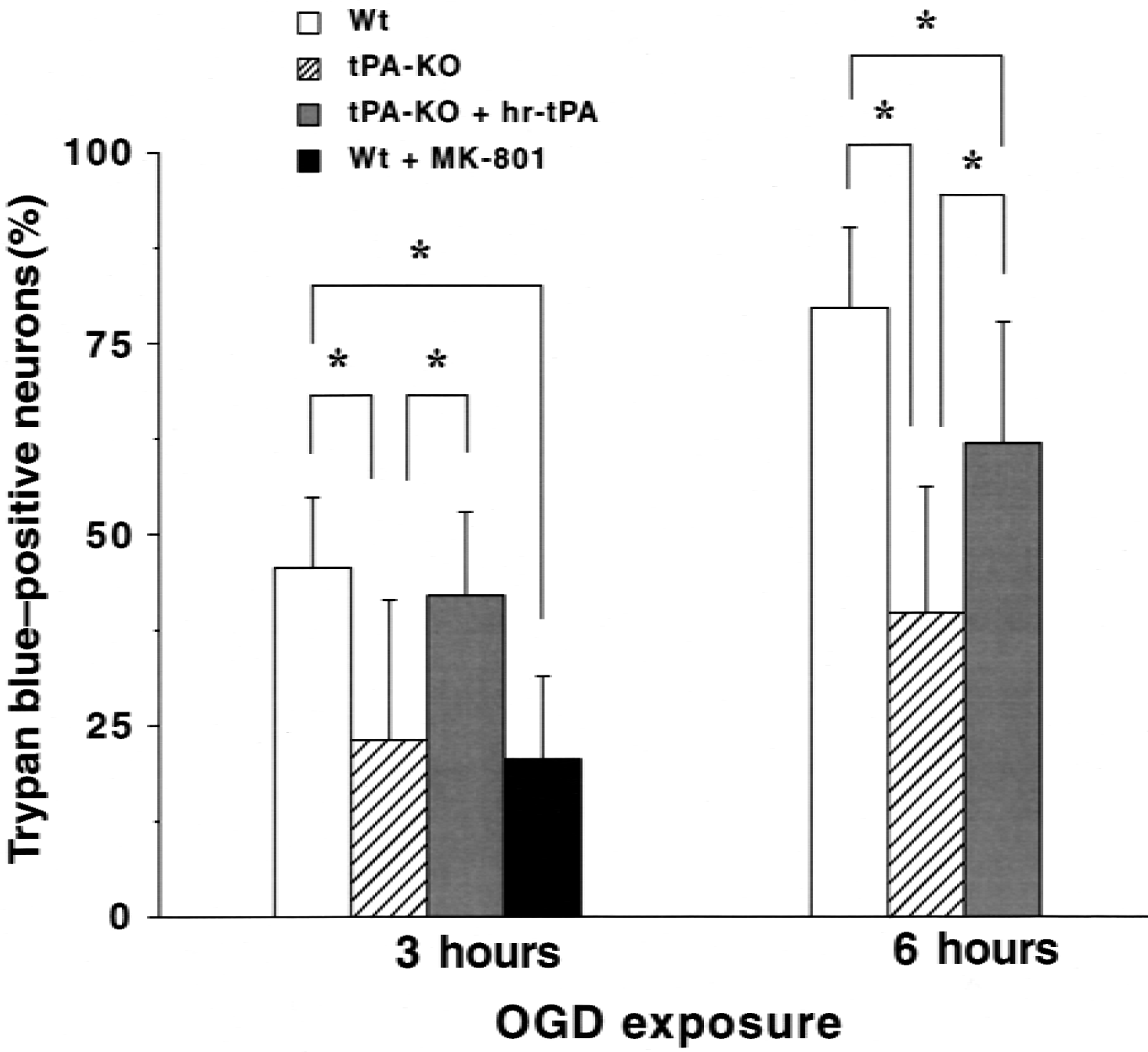
Effect of tissue-type plasminogen activator deficiency (tPA-KO) and administration of human recombinant tPA (hr-tPA) on neuronal cell death after oxygen–glucose deprivation (OGD). In control culture (exposed to normoxia and normoglycemia), the rates of trypan blue–positive neurons after 3 or 6 hours were between 6.8% and 12% in wild-type (Wt), tPA-KO, or tPA-KO in the presence of hr-tPA. Cultured neurons from tPA-KO mice and Wt mice were exposed to OGD. OGD significantly increased the neuronal death. MK-801 (20 μmol/L) significantly reduced the neuronal death induced by 3 hours of OGD. Note that tPA-KO significantly decreased neuronal death, which was restored completely in 3 hours and partially in 6 hours by additional hr-tPA. Neuronal death was determined by trypan blue staining. Rates of trypan blue–positive neurons were expressed as percentage of the total number of neurons counted. A mean percentage was calculated for three fields of each culture dish, and seven dishes were measured in each experiment. Data are expressed as mean ± SD. * P < 0.05.
DISCUSSION
Neuronal death induced by OGD in culture was suppressed in tPA-KO mice, which was reversed by the addition of hr-tPA. These results suggest that endogenous or exogenous tPA is directly involved in the process of the ischemic neuronal death without any effects on systemic circulation or blood vessels.
This direct neurotoxic effect of tPA is consistent with the authors' previous observations that tPA expands cerebral infarction induced by permanent middle cerebral artery occlusion in mice and hamsters in the absence of recanalization (Nagai et al., 1999a, 1999b). However, tPA neurotoxicity apparently contradicts the result that early treatment with tPA reduced cerebral infarct size in thrombus model (Vanderschueren et al., 1997). This reduction may be because of recanalization by thrombolytic action of tPA. Therefore, it appeared that the effects of tPA on the ischemic brain are multifactorial, which seems to explain the contradictory results in tPA action in in vivo experiments for cerebral infarction.
The mechanisms by which the ischemic damage was reduced in tPA-KO mice are currently unclear. The difference of sensitivity to OGD between Wt and tPA-KO cortical neurons may explain the result; it is not likely, however, because additional tPA restored neuronal death in tPA-KO culture. Because in this study, MK-801, NMDA receptor antagonist, reduced neuronal death induced by OGD, tPA must be directly involved in the excitotoxic pathway. Tsirka et al. (1995) suggested that tPA can amplify the glutamate-mediated ischemic neuronal death through generation of plasmin. However, this also is unlikely because plasminogen gene deficient mice always have a larger infarct (Nagai et al., 1999b). Another possibility is that tPA exerts its effect through receptor activation. Zhuo et al. (2000) reported that low-density lipoprotein receptor-related protein acts as a receptor for tPA on neurons. Other tPA receptors might be present in neurons and astrocytes.
In conclusion, this study demonstrates a direct involvement of tPA in the process of ischemic neuronal death. The precise mechanism by which tPA enhances the ischemic damage requires further investigation.