
Abstract
This study was designed to examine the effect of adrenomedullin deficiency on cerebral infarction and the relationship between adrenomedullin and cyclic AMP–protein kinase A pathway in regulating reactive oxygen species (ROS). Adrenomedullin heterozygous and wild-type mice were subjected to 60-mins focal ischemia. We used adrenomedullin heterozygous mice because adrenomedullin homozygotes die in utero. Infarct volume, neurologic deficit scores, and immunohistochemical analyses were evaluated at several time points after ischemia. The infarct volume and neurologic deficit scores were significantly worse in adrenomedullin heterozygous mice. Significant accumulation of inducible nitric oxide, oxidative DNA damage, and lipid peroxidation was noted after reperfusion in adrenomedullin heterozygous mice. Treatment of wild-type mice with H89, a protein kinase A inhibitor, resulted in increased infarct size, and worsening of neurologic deficit score and other parameters to levels comparable to those of adrenomedullin heterozygous mice. In contrast, cilostazol, which increases cyclic AMP, rescued neurologic deficit and ROS accumulation in adrenomedullin heterozygous mice. This study showed that adrenomedullin downregulation results in increase in ROS after transient focal ischemia in mice. The results also indicated that adrenomedullin has an important function against ischemic injury through the cyclic AMP–protein kinase A pathway.
Introduction
Adrenomedullin (AM) was originally isolated from pheochromocytoma cells, but it is also produced in and secreted by vascular endothelial cells (Kitamura et al, 1993). The vasodilator action of AM, which causes hypotension, is evident from its ability to elevate intracellular cyclic AMP (cAMP) in vascular smooth muscle cells (Jougasaki and Burnett, 2000; Samson, 1999).
Adrenomedullin is secreted in various organs including the heart, lung, kidney, adipose tissues, and central nervous system (Eto, 2001). Moreover, marked expression of AM was reported under ischemic conditions through the activation of hypoxia-responsive elements in the AM gene through transcription factor hypoxia-inducible factor-1 (Eto, 2001). In the central nervous system, in which AM is mainly expressed in neurons and the endothelium (Serrano et al, 2002), it is reported that transient ischemia boosts AM expression for >15 days after ischemia (Wang et al, 1995). The physiologic role of AM in ischemic brain is controversial; in some studies, it is reported that AM can reduce infarct size after transient ischemia (Dogan et al, 1997; Watanabe et al, 2001), whereas one study detected exacerbation of infarction as a result of AM infusion (Wang et al, 1995).
Reactive oxygen species (ROS) increase AM production in the vascular endothelial and smooth muscle cells (Ando et al, 1998; Chun et al, 2000). On the other hand, AM suppresses the generation of ROS in cultured mesangial cells and macrophages (Chini et al, 1997). Moreover, AM deficiency results in higher levels of ROS with resultant vascular damage (Shimosawa et al, 2002). It was postulated that cAMP–protein kinase A (PKA) pathway has a pivotal function in inhibiting the activation of NADPH oxidase and that AM is a potent intrinsic antioxidant (Liu et al, 2007). We hypothesized that ROS activation could be inhibited by AM, and AM is a protective agent against ischemic brain damage. In this study, we used wild-type (Wt) mice and AM knockout heterozygous (AM(+/−)) mice to examine brain damage and ROS in a transient focal ischemia model.
Materials and methods
Experimental Protocol
All animal procedures were conducted under the approval of the Animal Care Committee of Juntendo University. Eight-week-old male AM(+/−) mice with a disruption in the AM peptide (F10;
Middle cerebral artery occlusion (MCAO) was performed using an intraluminal thread for 60 mins (or 45 mins in only Supplementary Figure 1) as described earlier (Hara et al, 1996). Systolic blood pressure was assessed noninvasively in conscious mice using the tail-cuff system (Softron BP-98A NIBP, Softron Inc, Tokyo). Regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry before, during, and after MCAO, as well as before sacrifice. Neurologic function was assessed using a standard scoring system (Atochin et al, 2003). At the end of the study, the animals of each group were anesthetized by intraperitoneal injection of 50 mg/kg of pentobarbital followed by transcardial perfusion. The area of cerebral infarction was measured using microtubule-associated protein 2, as reported earlier (Lukic-Panin et al, 2007). Brain swelling was calculated as edema index (%) according to the following formula: edema index=(ipsilateral hemispheric volume−contralateral hemispheric volume)/contralateral hemispheric volume × 100 (Maier et al, 1998).
Intraventricular Infusion of Protein Kinase A Inhibitor
An osmotic minipump (model 1002, Alzet; Durect Corporation, Cupertino, CA, USA) was filled with 100
Administration of Phosphodiesterase Type III and IV Inhibitor
From 7 days before MCAO, AM(+/−) mice were provided with laboratory food mixed with 0.1% cilostazol (a potent inhibitor of phosphodiesterase type III, Otsuka Pharmaceutical, Tokyo, Japan) at 50 mg/kg/day or vehicle (normal laboratory food) (AM+PDE3I group and AM+vehicle group, respectively, each group;
Cyclic AMP and Protein Kinase A Assay
Each brain sample was rapidly removed at several time points after MCAO. The samples were lysed in CelLytic reagent (Sigma Chemical Co, St Louis, MO, USA) with protease inhibitor (Calbiochem, La Jolla, CA, USA). The protein concentration in the tissue lysate was determined using the BCA protein assay kit (Pierce, Rockford, IL, USA). Cell debris were removed by centrifugation at 15,000
Immunohistochemistry
Immunohistochemistry was performed on 20
Double Immunofluorescence Histochemistry
Double immunofluorescence staining was performed by simultaneous incubation of the sections overnight at 4°C with primary antibodies: anti-ADM-R (dilution, 1:50), rat anti-CD31 antibody (dilution, 1:100, BD Transduction Laboratories), mouse anti-neuronal nuclei marker (NeuN, a neuronal maker, dilution, 1:100 Chemicon International, Inc, Temecula, CA, USA), anti-iNOs (dilution, 1:100), or anti-Iba-1 (dilution, 1:500) antibody. For double labeling, the primary antibodies were detected with Cy3-conjugated secondary antibody (dilution, 1:500; Jackson Immunoresearch Laboratories, West Grove, PA, USA) and fluorescein isothiocyanate-conjugated secondary antibody (dilution, 1:500; Jackson Immunoresearch Laboratories) after incubation for 1 h at room temperature. The sections were washed with PBS and mounted on microslide glass with Vectashield Mounting Medium (Vector Laboratories).
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis and Immunoblotting
Mice of each group were decapitated at before and 3, 12, or 24 h after reperfusion (
Cell Count and Statistical Analysis
In the immunohistochemical analysis, positively stained cells at the ischemic boundary zone were counted using three sections per animal (0.25 mm2) by an investigator blinded to the experimental groups, using Axio-Vision software (Zeiss, Jena, Germany). Values presented in this study are expressed as mean±s.e.m. One-way analysis of variance followed by
Results
Effects of Adrenomedullin on Infarct Volume and Neurologic Deficit
Figure 1A shows Kaplan–Meier curve of survival rate after reperfusion. The survival rate was significantly worse in AM(+/−) mice than Wt mice (
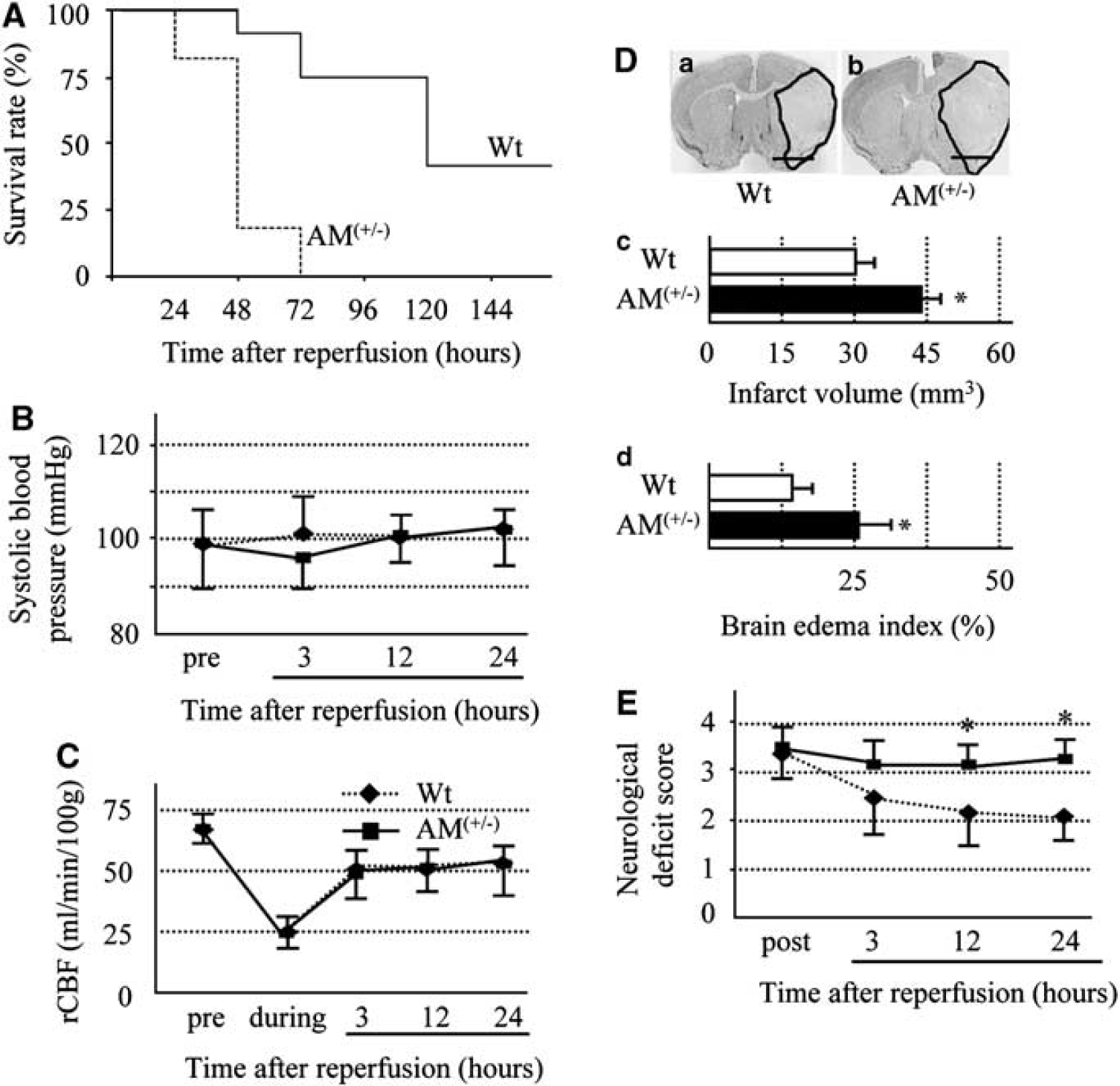
(
Temporal Profile of ADM-R Expression
Next, we assessed the difference in ADM-R expression between Wt and AM(+/−) mice. In both Wt and AM(+/−) mice, ADM-R was expressed in neurons and endothelial cells in nonischemic areas (Figure 2A and 2B). However, in the ischemic boundary zone, the expression of ADM-R was found only in endothelial cells (Figure 2Bc), and it was less in AM(+/−) than Wt mice (Figure 2Bd).
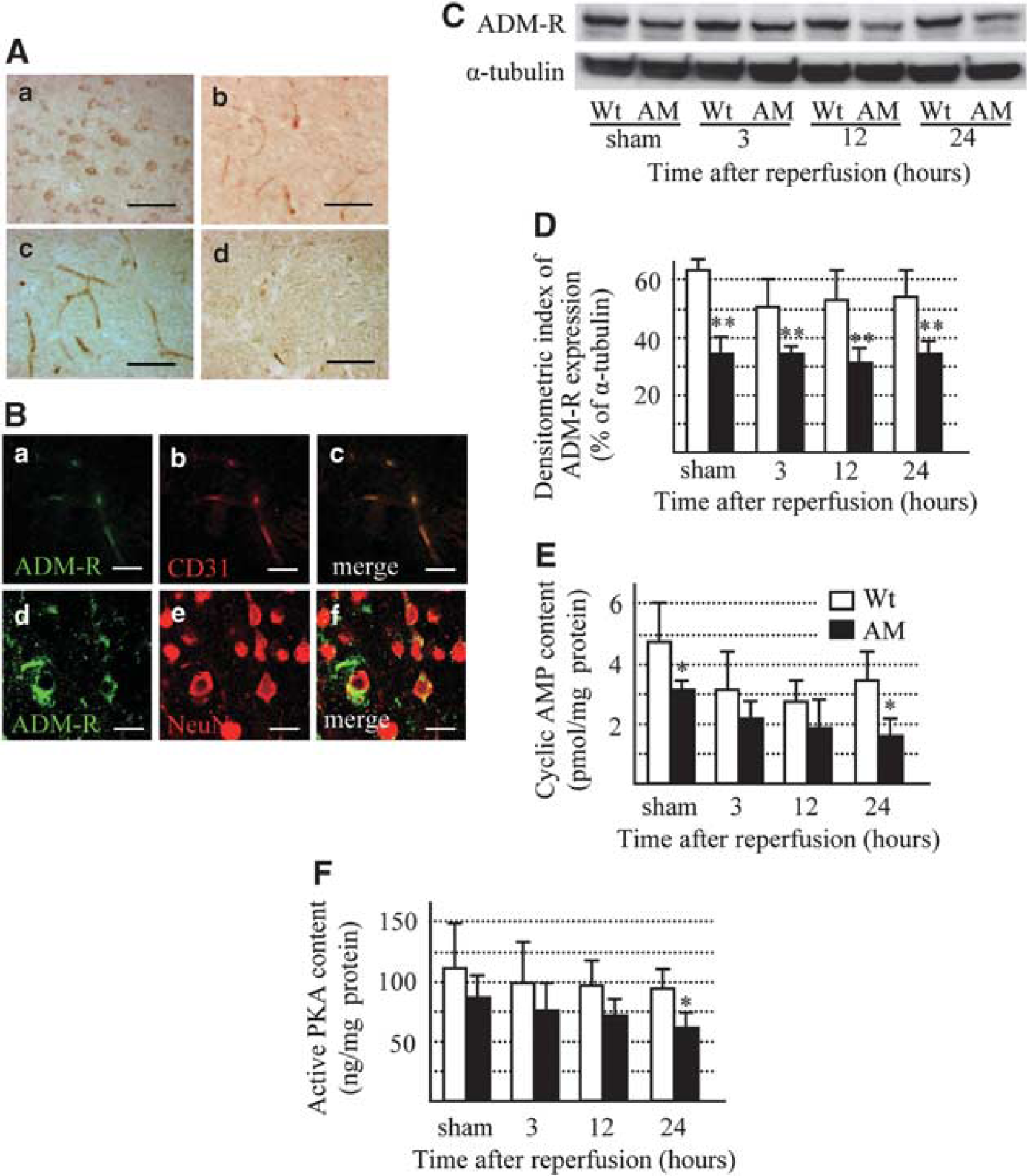
(
ADM-R was detected by immunoblotting as bands of 39 kDa (Figure 2C). In Wt mice, no significant change in ADM-R expression was noted up to 24 h after reperfusion. ADM-R expression in AM(+/−) mice was about half of that in Wt mice and its level did not change throughout the experimental period (Figure 2D). It has been reported that activation of ADM-R increases intracellular cAMP intensity as well as activation of the cAMP/PKA pathway (Chini et al, 1997; Kuwasako et al, 2004; McLatchie et al, 1998). Accordingly, we checked cAMP and PKA activities in the ischemic region. cAMP and PKA level was significantly lower in AM than in Wt mice at sham and 24 h after reperfusion (Figure 2E and 2F).
Effects of Adrenomedullin on Microglial Activation and Inducible Nitric Oxides Formation
Next, we determined the relationship between AM and ROS by examining microglial activation and iNOs formation. In the Wt mice, ramified Iba-1-positive microglia were first observed at 3 h after reperfusion and their number increased till 24 h. On the other hand, in AM(+/−) mice, the number of microglia significantly increased at 3 h of reperfusion compared with Wt mice, but there were no significant differences in localization and time course after 12 and 24 h of reperfusion between Wt and AM(+/−) mice (Figure 3A and 3B). In Wt mice, iNOs immunostaining was observed only in endothelial cells at 3 and 12 h after reperfusion, but also appeared in microglia at 24 h after reperfusion. In AM(+/−) mice, iNOs immunoreactivity was observed only in endothelial cells at 3 h after reperfusion. However, at 12 and 24 h after reperfusion, iNOs staining was observed abundantly in microglia and few endothelial cells in the same animals (Figure 3A, 3C, and 3D). The results of histologic analysis were confirmed by western blotting against Iba-1 and iNOs. The Iba-1-positive band (17 kDa) increased in a time-dependent manner in both groups of mice, and the increase was quantitatively similar in the two groups. However, the iNOs protein level (band, 130 kDa) increased in a time-dependent manner in both groups of mice, but the amount was significantly larger in AM(+/−) than Wt mice (Figure 3E and 3F).
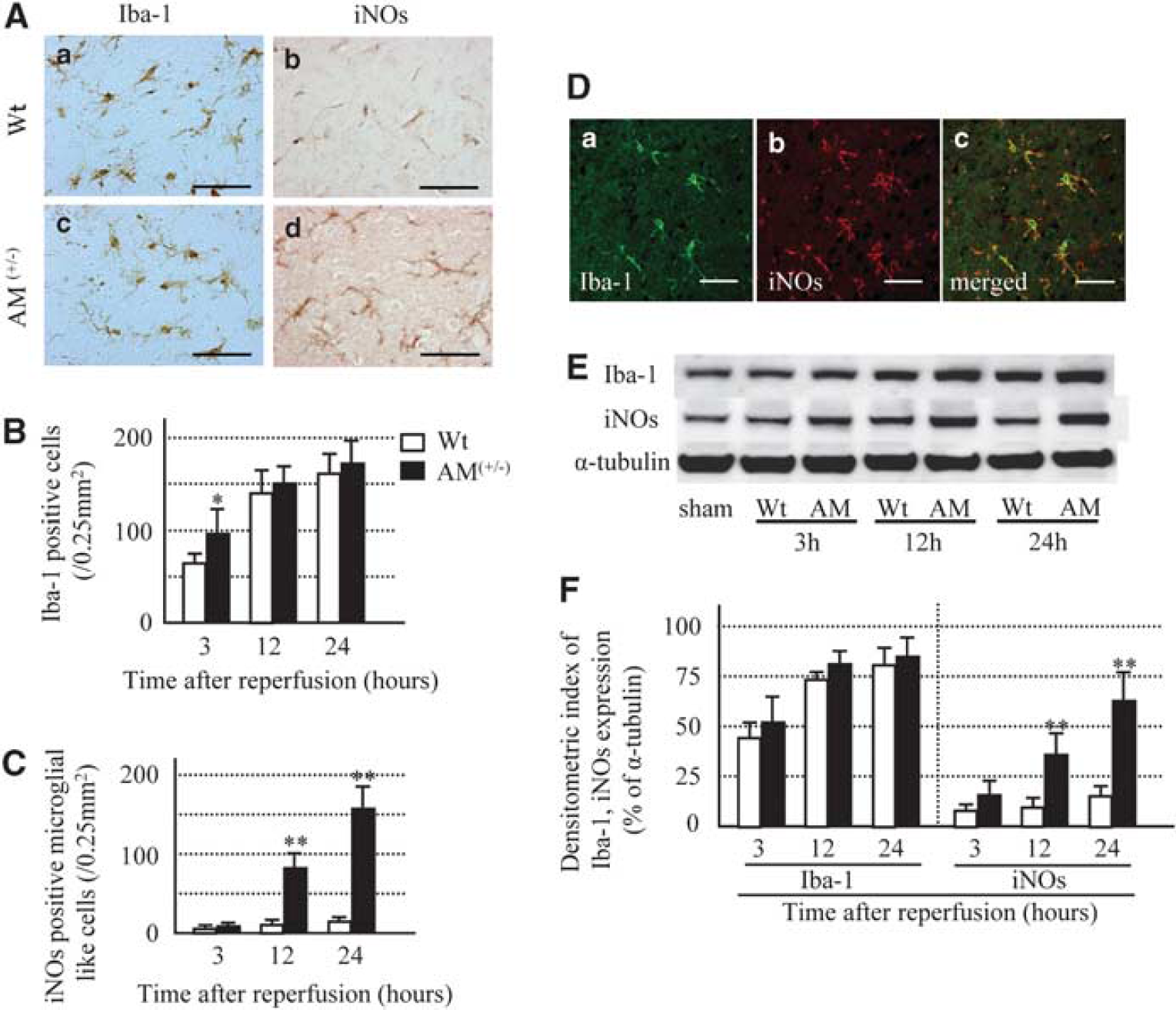
(
Effects of Adrenomedullin on Oxidative Stress
Next, we evaluated oxidative stress by using 8-OHdG (oxidative DNA damage) and HNE (lipid peroxidation). After 3 h of reperfusion, strong 8-OHdG immunoreactivity was evident in the nuclei of neurons in Wt mice, and increased in a time-dependent manner (Figure 4A and 4B). The number of 8-OHdG-positive cells and intensity of immunoreactivity for 8-OHdG at 24 h after reperfusion were significantly higher in AM(+/−) than Wt mice (Figure 4A and 4B). On the other hand, immunoreactivity for HNE was detected as early as 3 h after reperfusion and was present in the cell bodies of neurons (Figure 4A and 4B). The number of HNE-positive cells increased in a time-dependent manner in Wt mice (Figure 4A and 4B). The number of HNE-positive cells and intensity of immunoreactivity for HNE at 24 h after reperfusion were significantly higher in AM(+/−) than Wt mice (Figure 4A and 4B). The results of histologic analysis were confirmed by western blotting against HNE. The protein level of HNE–positive band (34 kDa) increased in a time-dependent manner in both groups of mice, but the amount was larger in AM(+/−) than Wt mice (Figure 4C and 4D).
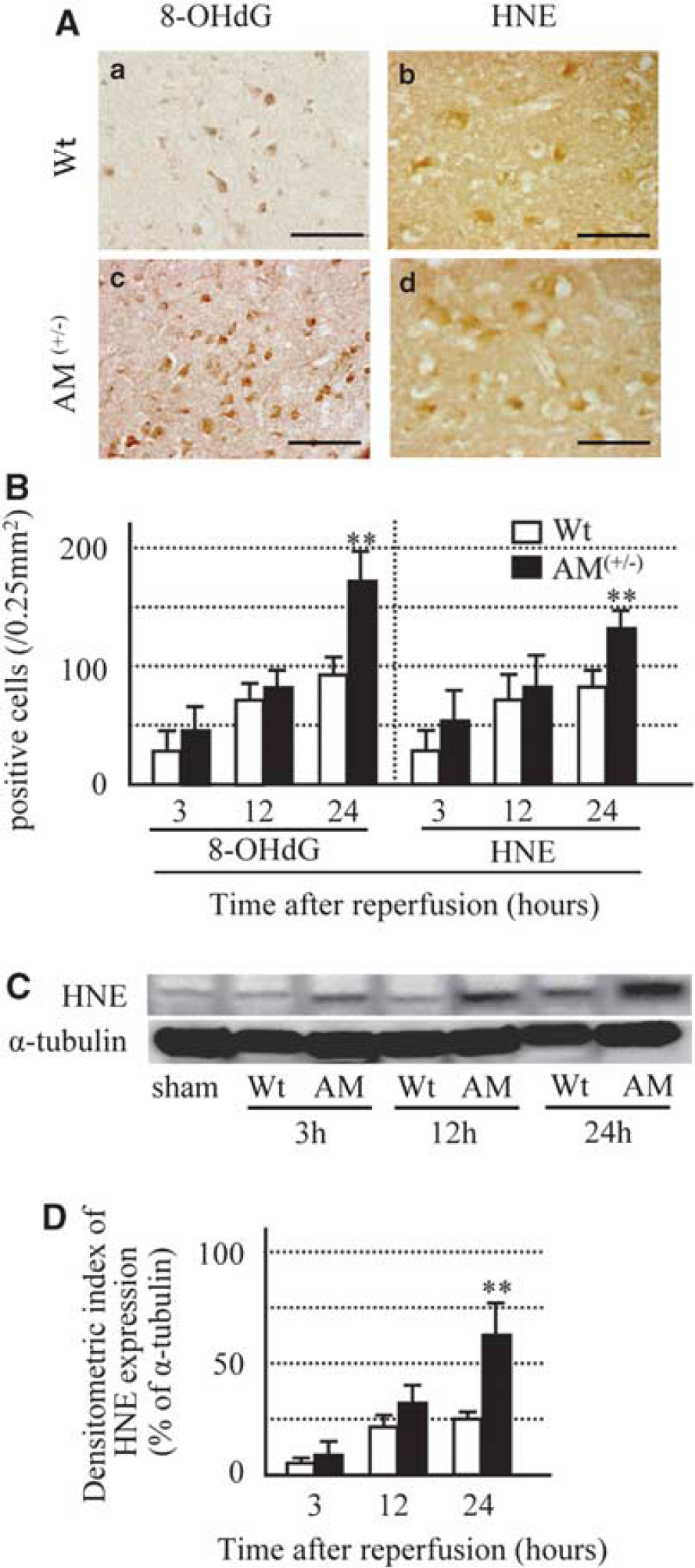
(
Effects of Protein Kinase A Inhibition on Cerebral Ischemia
The above findings suggest that AM blocks ROS formation and oxidative stress through ADM-R, which is expressed on endothelial cells and neurons independent of microglial activation. Activation of ADM-R results in an increase in intracellular cAMP intensity and activation of PKA (Kuwasako et al, 2004; McLatchie et al, 1998). Next, we investigated whether the above effects are dependent on the cAMP–PKA system. Treatment with H89 (a PKA inhibitor) resulted in a comparable increase in the brain infarct volume and edema index both in Wt and AM(+/−) mice (Figure 5A and 5B). The density of Iba-1-positive microglia at 24 h after H89 treatment was not different among the groups, whereas intracerebroventricular infusion of H89 significantly increased the densities of microglial-stained iNOs, 8-OHdG, and HNE, compared with the vehicle (Figure 5C and 5D). With regard to neurologic deficit score, H89 treatment tended to increase the neurologic score (Figure 5E). H89 aggravated those parameters in Wt mice to the same extent as AM(+/−) mice, suggesting that cAMP–PKA-dependent AM effect has an important function in ischemic injury.
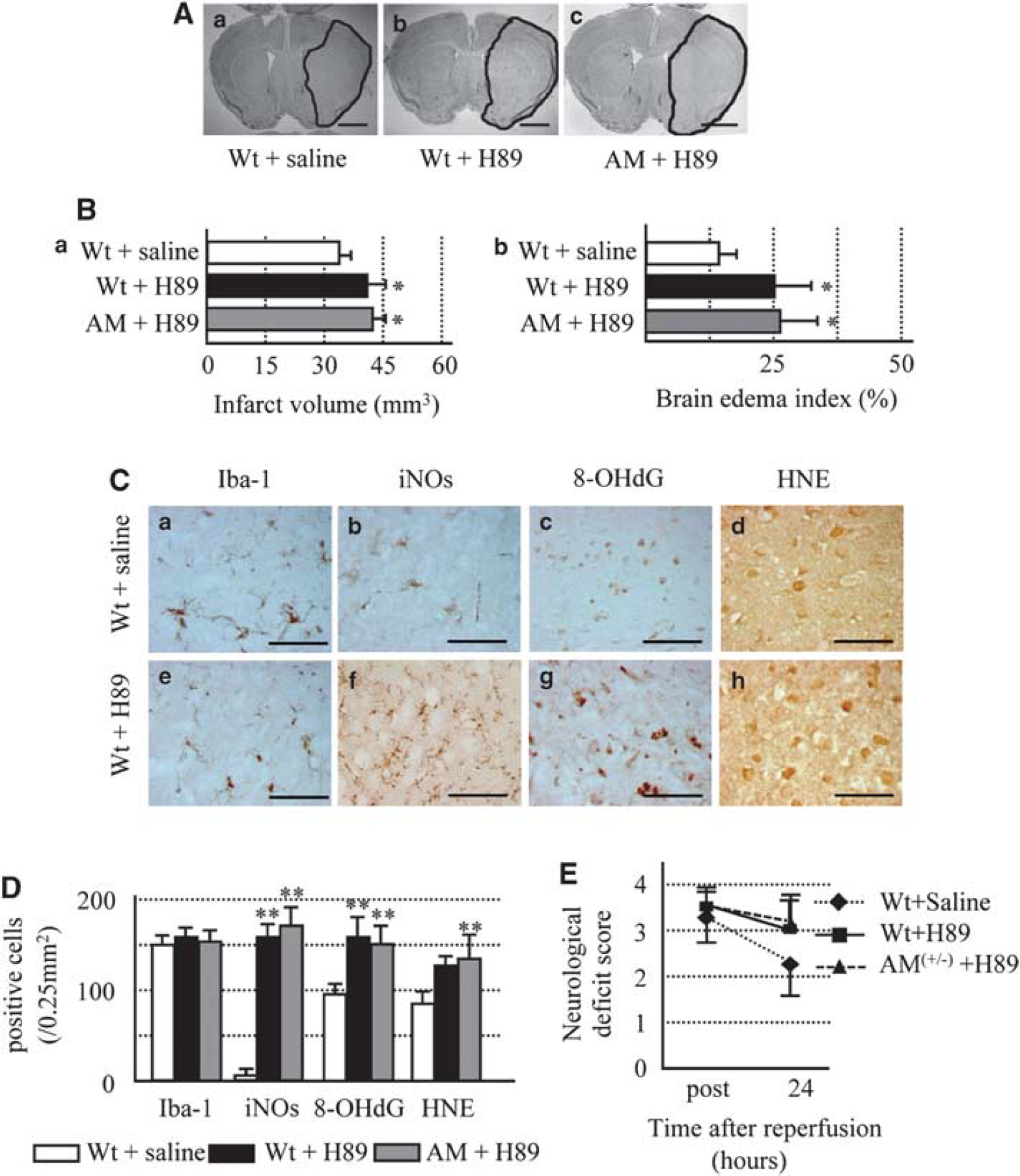
(
Rescue Effects of Protein Kinase A Activation in AM(+/−) Mice
On the basis of the finding of low cAMP level in AM(+/−) mice and that low cAMP levels have an important function in brain damage, we hypothesized that increased PKA level by PDE III inhibitor can reverse brain damage in AM(+/−) mice. PDE III inhibition is modulated by pharmacological and physiologic activators of cAMP–PKA (Suttorp et al, 1993). To confirm the role of this pathway, AM(+/−) mice were treated with cilostazol, a PDE III inhibitor from 7 days before MCAO operation (AM+ PDE3I group), then killed at 24 h after reperfusion. On the basis of the information of stable plasma cilostazol concentrations over a 1-week period, as provided by the supplier, the cilostazol treatment was started before 7 days from operation. There were no significant differences in rCBF and systolic blood pressure between AM+PDE3I and AM+vehicle groups (data not shown). On the other hand, the brain infarct volume and edema index at 24 h after reperfusion were significantly smaller in AM+PDE3I group than in the vehicle group (Figure 6A and 6B). Furthermore, the neurologic deficit score tended to improve after PDE3I treatment (Figure 6E). The density of Iba-1-positive microglia at 24 h was not different between the two groups (Figure 6C and 6D). However, the densities of microglial stained for iNOs, 8-OHdG, and HNE were significantly lower in the AM+PDE3I group than in AM+vehicle group (Figure 6C and 6D). Furthermore, PDE3I increased cAMP content and PKA activity in the ischemic area relative to that seen in Wt mice (Figure 6F and 6G). Further, we repeated the pharmacological studies using 45 mins of MCAO model (MCAO(45)). However, the results were almost similar to those of this study. Specifically, the brain infarct volume and edema index at 24 h after reperfusion was significantly larger in the AM group than in the vehicle and AM+PDE3I group (Supplementary Figure 1A and 1B). Furthermore, the neurologic deficit score tended to be higher for AM mice, but improved after treatment with PDE3I (Supplementary Figure 1C). The density of Iba-1-positive microglia at 24 h was not different between the three groups (Supplementary Figure 1D). However, the densities of microglial stained for iNOs, 8-OHdG, and HNE were significantly higher in AM mice than in Wt and AM+PDE3I group (Supplementary Figure 1D).
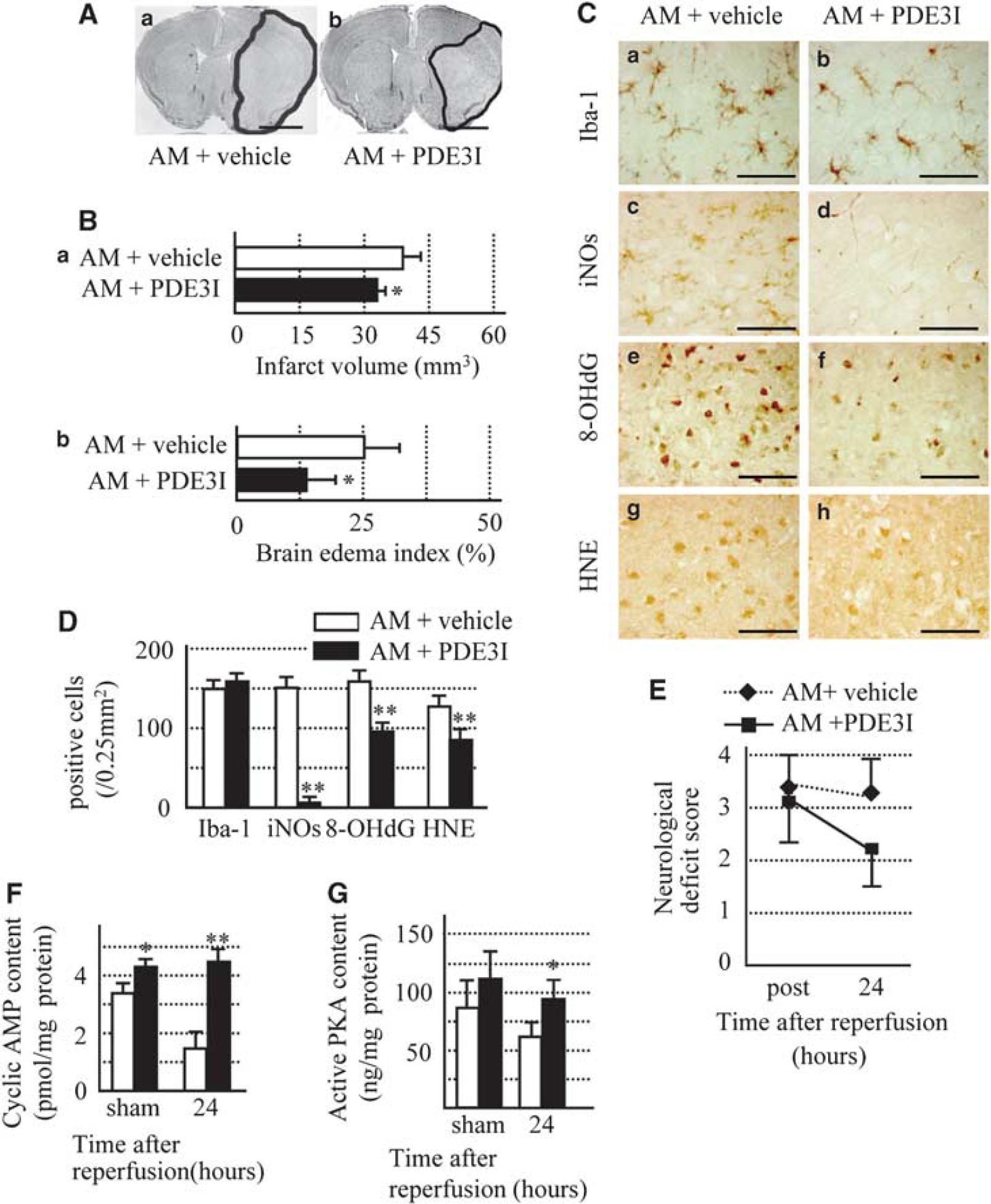
(
Moreover, to confirm the rescue effect of the cAMP/PKA system, we conducted a series of pharmacological studies using rolipram, a PDE4 inhibitor (PDE4I). In these experiments, AM(+/−) mice were treated with rolipram from day 7 before MCAO (AM+PDE4I group). As expected, the results were almost similar to those obtained with PDE3I (Supplementary Figure 2).
Discussion
In this study, we evaluated the
Accumulating evidence strongly supports the hypothesis that AM possesses significant protective properties against end-organ damage under a number of pathophysiological conditions through inhibition of oxidative stress (Kawai et al, 2004; Matsui et al, 2004; Shimosawa et al, 2002). The mechanisms underlying this activity vary depending on cell types and experimental conditions. In mesangial cells, AM suppresses ROS production through the cAMP–PKA pathway (Chini et al, 1997), whereas in the rat ventricle, increased oxidative stress caused by ischemia-reperfusion injury can be attenuated by AM-mediated inhibition of NADPH oxidase through the nitric oxide-cGMP signaling pathway (Kato et al, 2003). In contrast, enhanced hypoxia-induced generation of ROS in human alveolar epithelial cells is attenuated by a marked AM-stimulated increase in glutathione, a potent ROS scavenger (Kim et al, 2006).
Interestingly, our data showed alteration of microglial iNOs expression in AM+/− mice and H89 inhibited Wt mice after MCAO. Two earlier studies reported that hypoxia-induced AM expression in various cell types of the brain including neurons, astrocytes, and endothelial cells (Bernaudin et al, 2002; Ladoux and Frelin, 2000). Microglia are also reported to produce AM in response to hypoxia (Tixier et al, 2008). Moreover, the hypoxic-induced increase in AM mRNA expression was greater in microglia than in endothelial cells and astrocytes (Tixier et al, 2008). Furthermore, supernatants of cultured endothelial cells and microglia exposed to oxygen/glucose deprivation (OGD) significantly reduced OGD-induced neuronal death, and such neuroprotection was inhibited by an AM receptor antagonist (Tixier et al, 2008). These findings suggest that microglial cells are an important source of AM during hypoxic/ischemic stress in the brain. In contrast, accumulation of ROS in microglia is due to underexpression of AM.
In light of the aforementioned reports, the significant findings of this study are as follows. First, the data presented here clearly support the hypothesis that downregulation of AM increases ROS formation under cerebral ischemic condition. Second, in Wt mice, we confirmed that the intracerebroventricular infusion of PKA inhibitor is the source of the markedly increased ROS after MCAO. Third, the results showed for the first time that maintenance of cAMP level in AM(+/−) mice by using PDE3I also provides protection against cerebral ischemia.
Endogenous AM is upregulated by hypoxia in the ischemic brain (Wang et al, 1995). Adrenomedullin acts through at least two subtypes of G protein-coupled receptors, the calcitonin receptor-like receptor (CRLR) and ADM-R (McLatchie et al, 1998). CRLR functions as a calcitonin gene-related peptide receptor or an AM receptor depending on the expression of specific receptor activity-modifying proteins (RAMPs). In combination with RAMP1, CRLR has the highest affinity for calcitonin gene-related peptides, and in combination with RAMP2 or RAMP3, it prefers AM (Kuwasako et al, 2004). The levels of various ADM-R vary according to the cell type. For example, in human adrenal cortex and aldosteronomas, ADM-R mRNA is present at high levels compared with CRLR and the three RAMPs (Albertin et al, 2001).
In this study, we showed increased brain edema in AM mice, in agreement with the finding of an earlier report (Hippenstiel et al, 2002). Moreover, diffuse edema is the hallmark of embryonic lethality in AM−/− mice (Shindo et al, 2001). Earlier studies showed improvement of endothelial barrier function after activation of adenyl cyclase and/or PDE inhibition (Lum et al, 1999; Stevens et al, 2000; Suttorp et al, 1993). Other reports described significant reduction of brain edema in mice with overexpression of AM through reduction of vascular permeability after 2-h MCAO (Miyashita et al, 2006). The result suggests that AM has a significant therapeutic effect on brain edema after ischemia. The therapeutic potential of AM against post-stroke brain edema should be further investigated in the future.
The fact that increased cyclic AMP suppresses the generation of superoxide and hydrogen peroxide (Takei et al, 1998) suggests the synergistic effect of cyclic AMP and oxygen radical scavengers. Moreover, cAMP/PKA system manages the various neurotrophic factors through cAMP response element-binding protein (CREB). A number of studies have shown that the CREB/cAMP response element (CRE) transcriptional pathway regulates the expression of both Bcl-2 and BDNF (Tao et al, 1998; Walton et al, 1996). With respect to the role of the CREB/CRE transcriptional pathway in neuroprotection, CRE-mediated gene expression is necessary for the induction of ischemic tolerance (Hara et al, 2003). Likewise, the neuroprotective effects of an ischemic preconditioning stimulus involve CREB-dependent transcription (Mabuchi et al, 2001). These observations suggest that a signaling cassette formed by the cAMP/PKA/CREB transcriptional pathway contributes to neuroprotection.
In conclusion, this study showed that downregulation of AM results in high levels of ROS and enhanced vascular permeability after transient focal ischemia in mice. Our results should be interpreted cautiously because our study has several limitations. First, the large infarction volume (even at 60 mins of ischemia/reperfusion injury) produced from this study. Second, an alternate strategy needs to be used (i.e., less severity and duration of ischemic injury) when the AM+/− mice are used for the pharmacological studies. Although further studies are required to determine the most appropriate dose of AM in acute ischemia, because of excavated infarct volume using high dose AM i.c.v. (Wang et al, 1995), our results suggest that AM is potentially useful therapeutically in patients with stroke based on a mechanism mediated through cAMP–PKA pathway.
Footnotes
Acknowledgements
This study was supported in part by a High Technology Research Center grant and a Grant-in-Aid for exploratory research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
None of the authors, their institutions or employers has financial or personal relationships that could inappropriately influence or bias the authors’ decisions, work, or manuscript. All authors certify that all their affiliations with and financial involvement, within the past 5 years and foreseeable future (e.g., employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, royalties) with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript are completely disclosed. Finally, the authors have no financial interest related to the material in the manuscript.