
Abstract
Selective neuronal cell death in the CA, pyramidal cells of the hippocampus and neurons of the dorsolateral striatum as a consequence of brain ischemia/reperfusion (IR) can be ameliorated with brain hypothermia. Since postischemic injury is mediated partially by chemical production of reactive oxygen species (ROS), decreased ROS production may be one of the mechanisms responsible for cerebral protection by hypothermia. To determine if ischemic brain temperature alters ROS production, reversible IR was produced in rats by occlusion of both carotid arteries with hemorrhagic hypotension. After 15 min of ischemia, circulation was restored for 60 min. Brain temperature was maintained during ischemia at either 30, 36, or 39°C and kept at 36–37°C after reperfusion. Using cerebral microdialysis, we measured nonenzymatic hydroxylation of salicylate by HPLC with electrochemical detection in the hippocampus. CBF was also compared among the groups during IR. The results were that normothermic animals during reperfusion had persistently increased levels of the salicylate hydroxylation product, 2,3-dihydroxybenzoic acid (2,3-DHBA), reaching 251% of control at 60 min. This increase in 2,3-DHBA production was potentiated after 60 min of reperfusion (406% of control) with ischemic hyperthermia. In hypothermic ischemia, 2,3-DHBA production at 60 min was attenuated to 160% of control. CBF decreased to ∼5% of baseline value during ischemia, but increased three- to four-fold relative to control in all three groups. Therefore, the effects of ischemic brain temperature on 2,3-DHBA production did not correlate with changes in CBF during IR. We conclude that brain-temperature-related changes in OH · production are readily detected in the rat and decreased ROS generation may contribute to cerebral protection afforded by hypothermia during brain ischemia.
After 10 min of ischemia, most of the neurons die in vulnerable brain regions such as CA1 pyramidal cells of the hippocampus and the small- to medium-sized neurons of the dorsolateral striatum (Ginsberg et al., 1985). Moderate to deep brain hypothermia significantly increases neuronal tolerance to ischemia in both two- and four-vessel occlusion models, leading to decreased neuronal death (Churn et al., 1990; Minamisawa et al., 1990; Green et al., 1992) and behavioral deficits (Green et al., 1992). In contrast, brain hyperthermia interferes with blood–brain barrier integrity (Dietrich et al., 1990), accentuates loss of CA1 neurons, and extends neuronal loss to CA3 (Churn et al., 1990). Hyperthermia may also transform selective neuronal injury into infarction (Ginsberg et al., 1992).
The mechanisms by which temperature modulates outcome after brain ischemia are uncertain. Brain hypothermia seems to reduce intracranial pressure after intracranial hemorrhage (Howell et al., 1956), preserve blood–brain barrier function (Dietrich et al., 1991), improve glucose utilization, and facilitate restoration of CBF after ischemia (Ginsberg et al., 1992). Other potential protective mechanisms of hypothermia include inhibition of hypoxic brain depolarization (Chen et al., 1993), leading to decreased release of neurotransmitters during ischemia, e.g., excitatory amino acids (EAAs) and catecholamines (Globus et al., 1988), attenuation of N-methyl-D-aspartate (NMDA) activity (Zeevalk and Nicklas 1993), depression of brain metabolism, thereby retarding the rate of high-energy phosphate depletion (Kramer et al., 1968), and inhibition of protease activation (Taft et al., 1993).
Recently, hypothermia has been reported to decrease lipid peroxidation after brain ischemia (Baiping et al., 1994), suggesting that decreased production of reactive oxygen species (ROS) may be involved in cerebral protection by hypothermia. ROS are formed by incomplete reduction of O2 to superoxide (· O2−) or hydrogen peroxide (H2O2) (Freeman and Crapo, 1982). H2O2 can lead to production of highly reactive hydroxyl radical (OH ·) in the presence of transition metal ions such as iron. Cellular iron release has been implicated after ischemia and may contribute to the OH · generation (Patt et al., 1990). Hydroxyl radicals oxidize essential cellular lipids, proteins, and nucleic acids, leading to cell damage and ultimately to cell death (Oliver et al., 1990; Floyd and Carney, 1992). The brain is particularly vulnerable to oxidative damage because it is rich in the unsaturated fatty acids and iron, but relatively poor in antioxidant defenses (Floyd and Carney, 1992).
Forebrain ischemia increases ROS production and lipid peroxidation in the hippocampus of the gerbil relative to the cerebral cortex (Carney and Floyd, 1992; Hall et al., 1993). In cerebral ischemia/reperfusion (IR), brain damage can be attenuated by exogenous superoxide dismutase (Kitagawa et al., 1990), inhibition of xanthine oxidase (Martz et al., 1989), and ROS scavenging by spin-trapping agents (Carney and Floyd, 1991). ROS production also appears to be related to increased synaptic EAA (Oh and Betz, 1991) and catecholamine accumulation during ischemia (Simonson et al., 1993). Hence, hypothermia may protect the brain after ischemia by decreasing ROS production. In this study, we investigated whether altering ischemic brain temperature altered the rate of nonenzymatic hydroxylation in rat hippocampus and cortex during global brain IR. The effects of the brain temperature protocols on CBF also were evaluated using laser Doppler flowmetry to determine whether or not temperature-dependent differences in blood flow could contribute to differences in OH · generation.
MATERIALS AND METHODS
Animal preparation
Male Sprague-Dawley rats weighing 280–320 g were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and ventilated with room air using a small animal ventilator (Edco ventilator, Chapel Hill, NC, U.S.A.). Both femoral arteries and one femoral vein were cannulated with PE-50 tubing. Common carotid arteries were dissected from the vagus nerve. Arterial blood pressure was monitored continuously (Gould model 481) through a calibrated pressure transducer, and the signals were integrated to display the MABP. Arterial blood gases were measured periodically (IL model 1304 blood gas pH analyzer) to maintain pHa, Pao2, and Paco2 at normal ranges. Brain temperature was estimated using a deep nasopharyngeal temperature probe (YSI-400) and kept at the desired temperature.
IR protocol
Five minutes before inducing brain ischemia, 50 IU of heparin was injected intravenously. To induce hypotension, the rats were bled over 2–3 min from a femoral artery to 35 mm Hg. Immediately after reaching 35 mm Hg, both carotid arteries were occluded using atraumatic aneurysm clamps. An MABP of 35 mm Hg was maintained for 15 min by withdrawing or infusing blood into a 10-ml heparinized syringe during ischemia. Brain circulation was then restored for 60 min by unclamping the carotid arteries and rapidly reinfusing the blood into the rat.
Modulation of brain temperature
Rats were divided into three experimental groups: ischemic brain hypothermia (30°C, n = 8), ischemic normothermia (36°C, n = 6), and ischemic hyperthermia (39°C, n = 7). The brain was cooled with a fan and crushed ice or heated with a lamp placed over the head. Brain temperature could be adjusted from 36 to either 30 or 39°C over ∼3 min before ischemia. Brain temperature was restored to 36–37°C immediately after brain ischemia. This process was completed within 3 or 4 min. During the experiments, body core temperature was maintained at 36–37°C in all animals.
Brain microdialysis
The microdialysis probe was implanted as described previously (Benveniste et al., 1984). A CMA/12 microdialysis probe (3-mm dialysis membrane, OD 500 μm; Carnegie Medicine, Stockholm, Sweden) was placed into the left hippocampus using coordinates of AP — 3.4 mm, ML + 2.2 mm, and DV — 3.0 mm. The probe was perfused continuously at 2 μl/min with artificial cerebrospinal fluid (NaCl 117 mM, KCl 3 mM, MgSO4 1.2 mM, KH2PO4 0.4 mM, NaHCO3 25 mM, CaCl2 1.2 mM) containing sodium salicylate (5 mM) (Chiueh et al., 1992) through a microinjection pump (CMA/microdialysis; Carnegie Medicine AB). After equilibration for 2 h, perfusate was collected every 15 min using a CMA/40 microfraction collector (Carnegie Medicine AB). All samples were frozen immediately and stored at −80°C until analysis. This technique collects interstitial molecules primarily from the hippocampus with contributions from corpus callosum and the cortex. This procedure produces hippocampal concentrations of salicylate in the 400–700 μM range.
Measurement of 2,3-dihydroxybenzoic acid
Salicylic acid reacts with hydroxyl radical (OH ·) to produce 2,3- or 2,5-dihydroxybenzoic acid (2,3- or 2,5-DHBA) and a small amount of catechol. We measured only 2,3-DHBA as an indicator of OH · production because, unlike 2,5-DHBA, which can be generated by mixed function oxidases, it can be generated only by nonenzymatic hydroxylation (Halliwell et al., 1991). The 2,3-DHBA was detected by injecting 25 μl of dialysate directly into the HPLC electrochemical detection system (ESA, Bedford, MA, U.S.A.). The elution buffer consisted of 30 mM sodium citrate/1.8% acetic acid/1.5% methanol. The ECD system was set at −0.40 and +0.40 VDC (Zhang and Piantadosi, 1992). This system is capable of detecting hydroxylation products of salicylate to low picomolar concentrations.
Measurement of CBF
CBF was measured in separate animals to determine whether hyperemic responses after ischemia were affected by brain temperature. A 0.8-mm-diameter fiberoptic probe, coupled to a BMP2 laser Doppler flow perfusion monitor (Vasamedics, St. Paul, MN, U.S.A.), was placed onto the surface of the brain. Large pial vessels were avoided and the probe was sheltered from direct light sources by covering it with foil. After 60 min of equilibrium, CBF measurements were performed in animals in all three experimental groups during IR (n = 4 for each group). Laser Doppler flowmetry correlates well with flow measured by other techniques such as hydrogen clearance (Skarphedinsson et al., 1988). Most of the CBF signal measured by this probe is derived from the first 2 mm of tissue beneath the device since it does not detect flowing red blood cells in vitro beneath slices of rat brain thicker than 2 mm (data not shown). The microdialysis probe sampled a tissue volume from just beneath the brain surface to ∼3 mm deep.
Chemicals
Sodium chloride, potassium chloride, magnesium sulfate, sodium bicarbonate, and potassium dihydrogen phosphate were purchased from Mallinckrodt (Paris, KY, U.S.A.). Calcium chloride and salicylate were purchased from Sigma (St. Louis, MO, U.S.A.).
Statistical analysis
Statistical analysis were performed using a Statview 512+ (Brain Power, Calabasas, CA, U.S.A.) computer software package. The data were entered into a computer spreadsheet and evaluated by repeated measures analysis of variance and the Scheffé F test. A p value of <0.05 was considered significant.
RESULTS
Physiologic variables
Table 1 shows the effects of the IR protocol and intraischemic changes in brain temperature on MABP, Pao2, Paco2, and pHa in the rats. Significant differences were not observed before or after ischemia or between any groups. The measured values of Pao2 were above 85 mm Hg, whereas the values of Paco2, and pHa were in the physiologically acceptable range for the rat. MABP was restored to >100 mm Hg within 1 min of reperfusion, and it gradually rose to nearly preischemic values.
Physiological measurements in brain ischemia/reperfusion
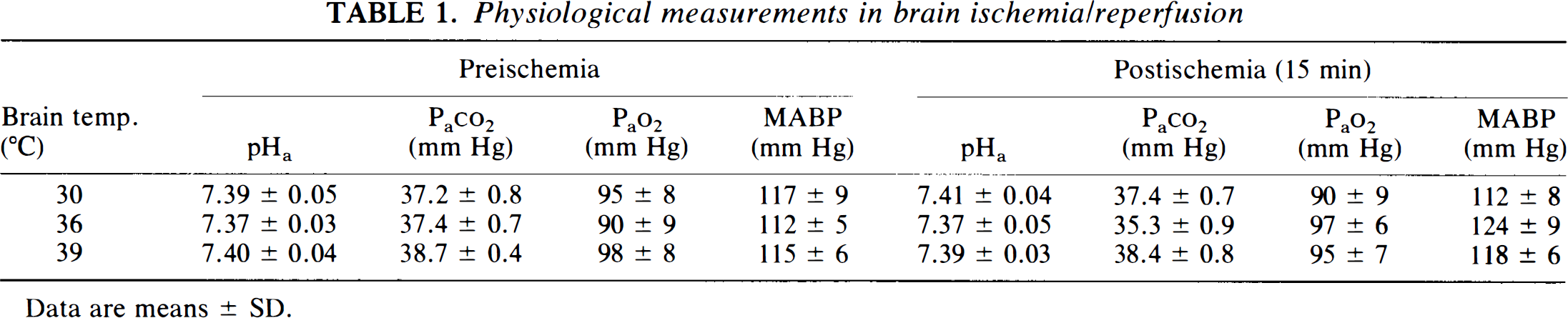
Data are means ± SD.
Effect of IR on hydroxyl radical production
In control experiments, the 2,3-DHBA level in the dialysate tended to decrease in animals that did not undergo IR (Fig. 1), possibly due to recovery from the process of inserting the probe. The rats experiencing IR, however, showed significant increases in 2,3-DHBA concentrations during reperfusion (p < 0.05). The mean values for 2,3-DHBA concentration were 1.32 ± 0.21 and 3.33 ± 0.37 pmol/25 μl dialysate for preischemia and 60 min after reperfusion, respectively. Of note, the efficiency for OH · trapping by salicylate is <10%. In addition, the recovery rate of the microdialysis probe for 2,3-DHBA in vitro is −23%.
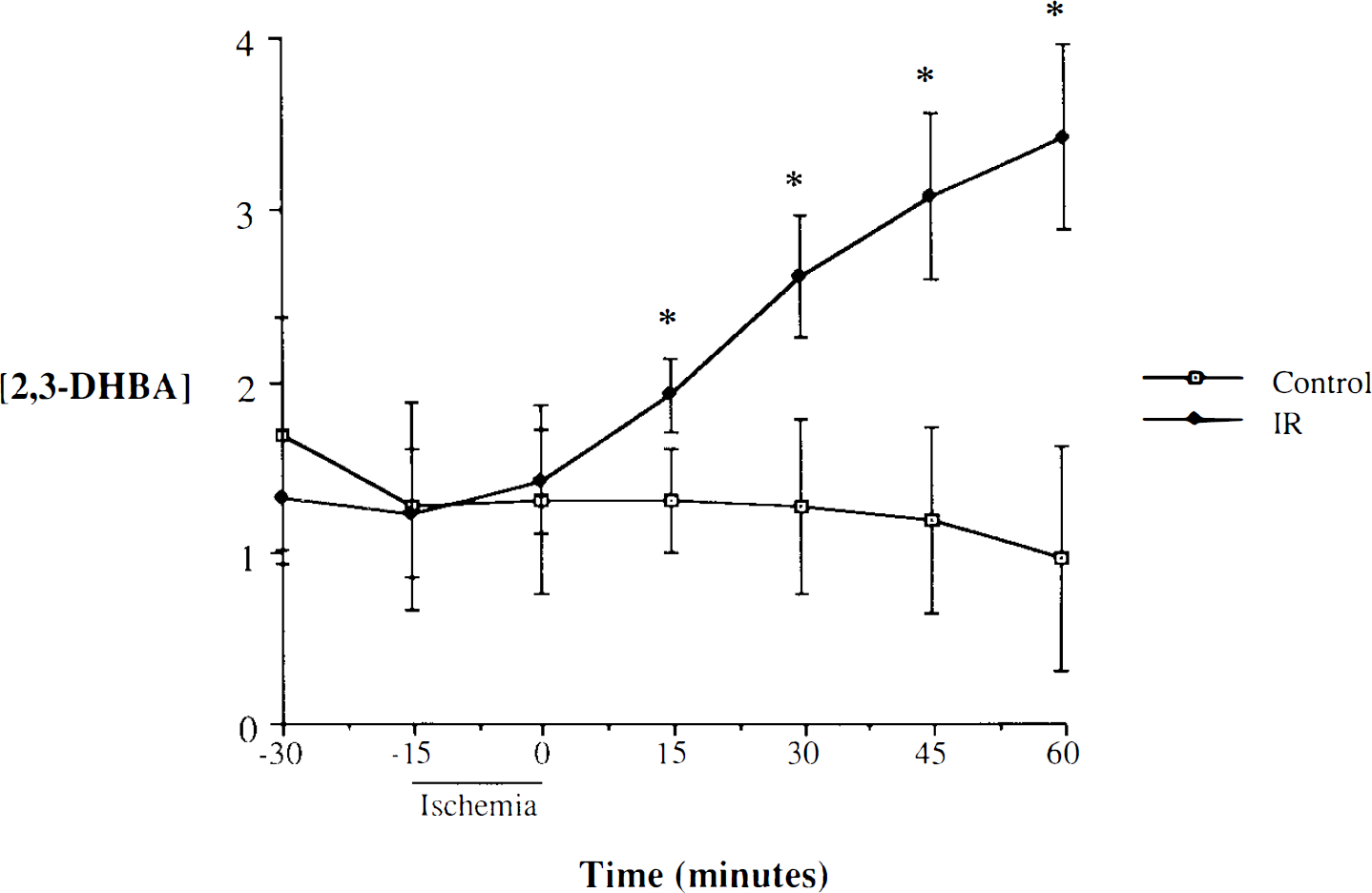
Effects of brain ischemia/reperfusion (IR) on hydroxyl radical production. Values are expressed as means ± SD (pmol/25 μl dialysate). *p < 0.05 for IR (n = 6) vs. control (n = 5). 2,3-DHBA, 2,3-dihydroxybenzoic acid.
Ischemic brain temperature and hydroxyl radical production
The effects of modulating ischemic brain temperature on OH · production are summarized in Fig. 2. In control experiments, changing brain temperature between 30 and 39°C without IR did not alter the temporal profile of 2,3-DHBA production (data not shown). Increases in ischemic brain temperature, however, significantly elevated both the rate of formation and the concentration of 2,3-DHBA during IR compared with IR control. The increase in 2,3-DHBA concentration with time was significant by analysis of variance at 0, 15, and 45 min of reperfusion. In contrast, when the same experiments were carried out in animals treated with ischemic hypothermia at 30°C, the rate of appearance and concentration of 2,3-DHBA in the dialysate were attenuated substantially, although the values were still greater than in nonischemic control animals.
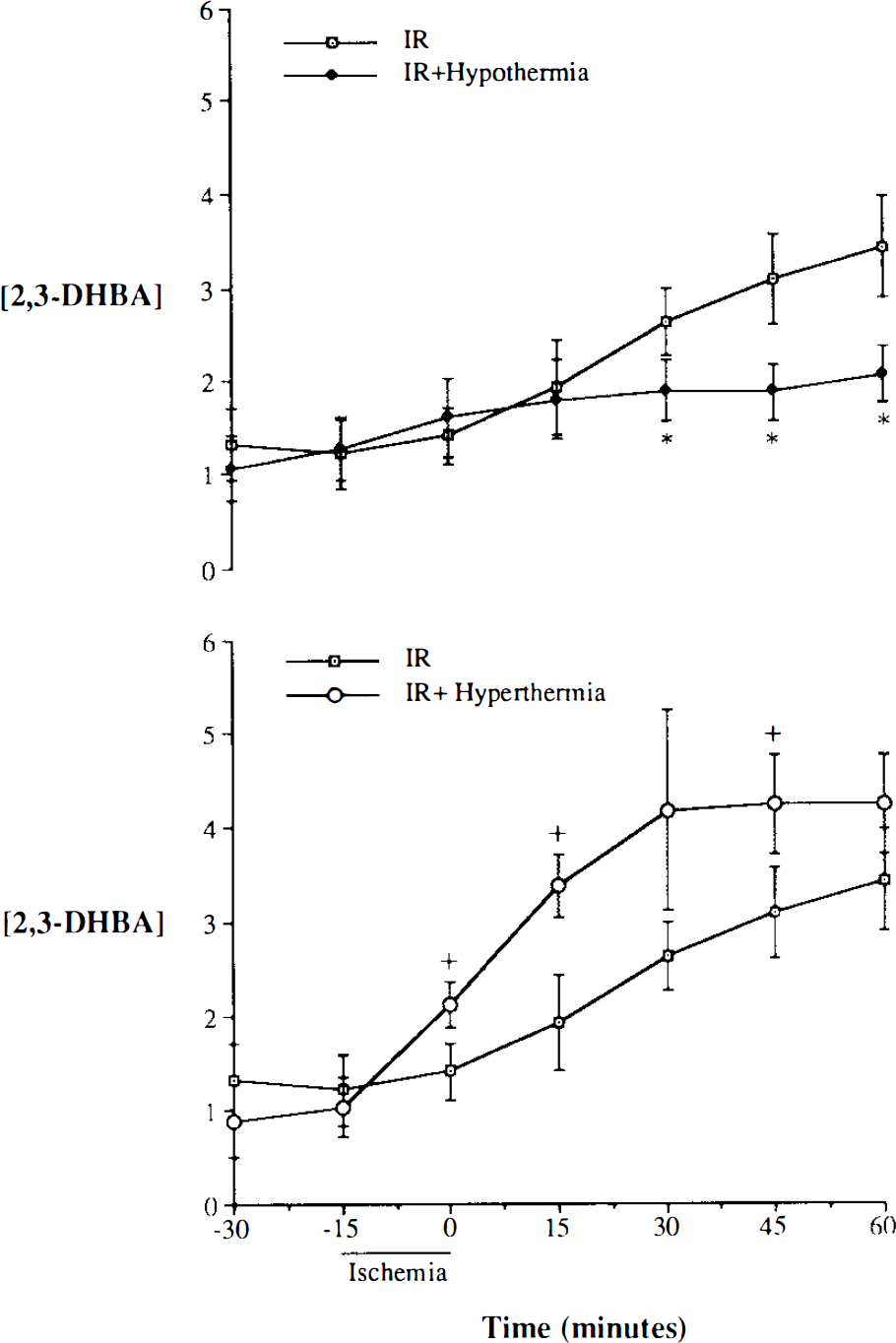
Effects of ischemic brain temperature during ischemia on the temporal profile of hydroxyl radical production. Values are expressed as means ± SD (pmol/25 μl dialysate). *p < 0.05 for ischemia/reperfusion (IR) + hypothermia (n = 8) vs IR; +p < 0.05 for IR + hyperthermia (n = 7) vs. IR. 2,3-DHBA, 2,3-dihydroxybenzoic acid.
Ischemic brain temperature and CBF
The results of CBF measurements are summarized in Fig. 3. In IR experiments at normal brain temperatures, CBF decreased to <5% of control values during ischemia. A moderate hyperemic response was observed after reperfusion, and flow values reached three to four times above the preischemic CBF after 5–15 min. No significant differences in the CBF response were found during ischemia among any of the temperature groups. Post-ischemic hyperemia was not altered significantly by ischemic brain temperature, although the hyperthermic animals tended to have a slightly higher peak response after ischemia.
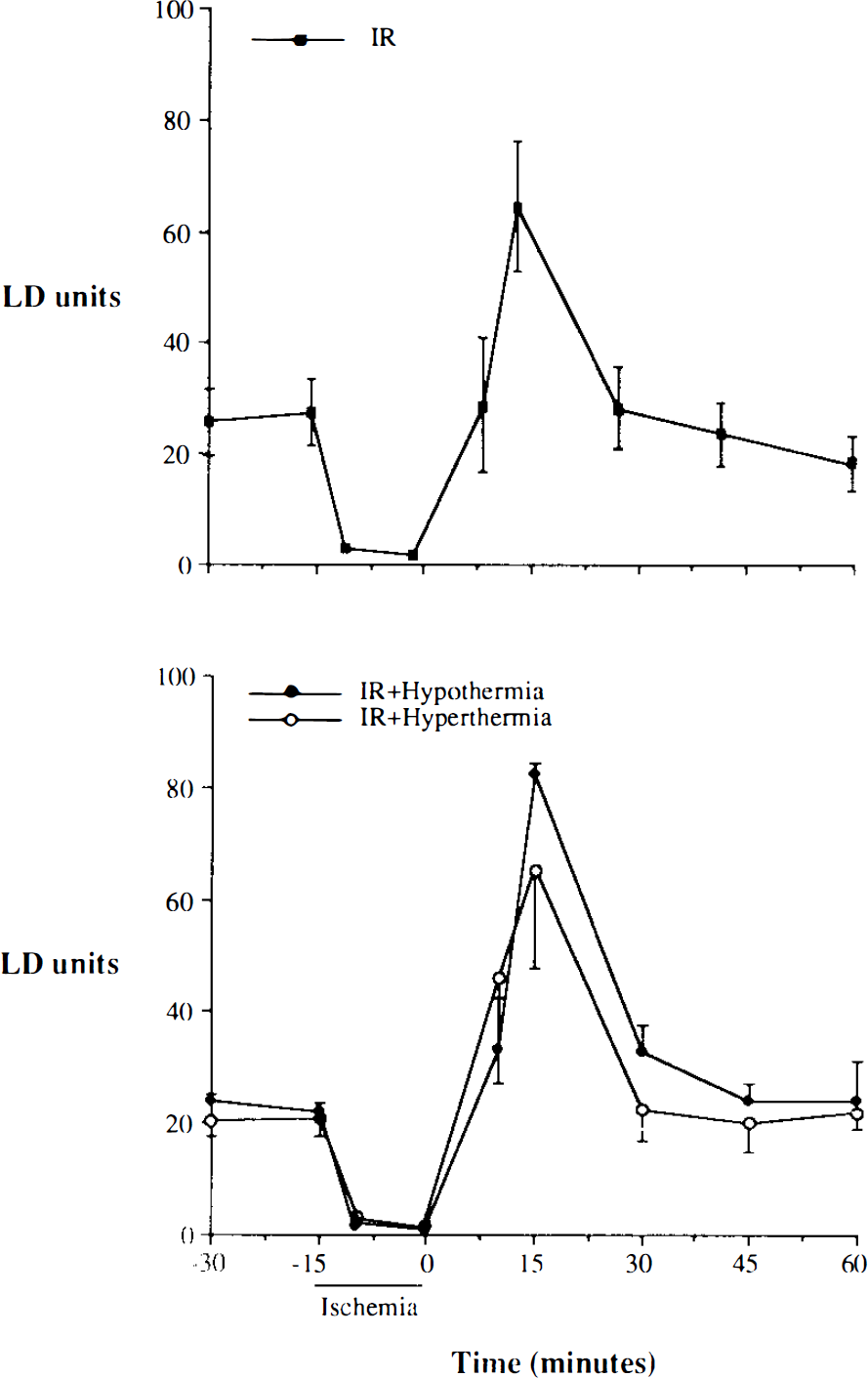
Effects of ischemic brain temperature on CBF. Values are expressed as means ± SD [laser Doppler (LD) units/min, n = 4] for animals in each group. IR, ischemia/reperfusion.
DISCUSSION
The major new finding of this study is that modulation of brain temperature during global transient ischemia significantly altered OH · detected during reperfusion. Production of OH · in rat hippocampus and cortex during reperfusion was attenuated or enhanced by moderate hypothermia (30°C) and mild hyperthermia (39°C), respectively. These changes in OH · production were not seen in control animals at different brain temperatures that did not experience IR and did not correlate to changes in CBF. There was no difference in CBF measured by cortical laser flowmetry during either ischemia or reperfusion related to ischemic brain temperature. These CBF data are in agreement with earlier reports of the effects of both moderate intraischemic hypothermia and hyperthermia (Busto et al., 1989; Morikawa et al., 1992).
The mechanisms by which brain temperature influenced OH · production in this study are unknown. The exact sources of ROS production and iron mobilization during brain IR are unclear (Floyd and Carney, 1992; Hall et al., 1993; Simonson et al., 1993). It has been hypothesized that ROS are produced by IR-induced intracellular events associated with synaptic EAA accumulation. Increased intracellular Ca2+, for instance, induces mitochondrial oxidative stress (Halestrap et al., 1993), stimulates phospholipase A2, and activates lipoxygenase and cyclooxygenase, leading to ROS production (Hall et al., 1993). Calcium is involved in converting xanthine dehydrogenase to xanthine oxidase, which produces · O2− (Carney and Floyd, 1991). EAA-mediated oxidative stress has been suggested from a neuronal cell line study (Murphy et al., 1990) and rat studies showing EAAs enhance OH · production in the brain (Boisvert and Schreiber, 1992). Activation of NMDA or kainic acid receptors produces lipid peroxidation in the hippocampus and cerebellum, respectively (Haba et al., 1991; Sun et al., 1992). It is plausible, therefore, that decreases or increases in synaptic EAA accumulation would lead to a decreased or increased ROS production.
Even small variations in brain temperature can influence synaptic EAA accumulation substantially. At a brain temperature of 30°C during ischemia, EAA accumulation can be totally inhibited (Busto et al., 1989), whereas keeping brain temperature at 39°C enhances EAA accumulation during both ischemia and reperfusion (Sternau et al., 1992). The exact relationship between hypothermia/hyperthermia and synaptic EAA accumulation is not understood, but hypothermia seems to inhibit ischemia-induced depolarization (Chen et al., 1993). Therefore, hypothermia and hyperthermia may modulate ROS production by decreasing or increasing synaptic EAA accumulation and consequently Ca2+ influx (Choi, 1988).
In addition to EAA-mediated oxidative stress, oxidative deamination of catecholamines during reperfusion may also contribute to ROS production (Simonson et al., 1993). The release of catecholamines during ischemia not only increases oxidative stress, but also has detrimental effects on brain tissue. Chemical depletion of catecholamines or nigral lesioning before ischemia appears to be protective (Globus et al., 1987). Brain hypothermia to 30°C decreases catecholamine release by 60% during brain ischemia (Busto et al., 1989). Consequently, hypothermia may decrease · O2 and H2O2 production during reperfusion by this mechanism since autooxidation and oxidative deamination of catecholamines would be decreased. This effect should lead to decreased OH · production.
Hypothermia and hyperthermia also may alter ROS production by altering the cerebral metabolic rate for oxygen. Examination of the present data for the rate of recovery of 2,3-DHBA in the first 30 min of reperfusion indicates that the overall recovery rate increased by a factor of 5.4 between 30 and 39°C. The observed temperature dependence fits an exponential model quite well (r2 = 0.96); however, the rate of 2,3-DHBA recovery is >2.5 times that expected from the Arrhenius relationship. The failure of the data to conform more reasonably to the Arrhenius equation is an indication that the apparent rate constant is a sum or some other more complex function of the rate and equilibrium constants for OH · generation in the in vivo system. The possibility was also considered that changes in intraischemic brain temperature altered the efficiency of salicylate hydroxylation; however, such a chemical effect should be related to the change in absolute temperature and could account for only a 1–2% difference in 2,3-DHBA production. It is also important to note that brain temperature in this study was different only during the period of ischemia (15 min); during reperfusion, brain temperature was essentially the same among all groups of animals.
Hypothermia decreases brain energy demand and consequently decreases the extent of brain acidosis (Ginsberg et al., 1992). Tissue acidosis enhances ROS generation by a variety of mechanisms, e.g., elevated tissue Po2 (Bohr effect), thereby increasing · O2− production (Wolbarsht and Fridovich, 1989), more rapid dismutation of · O2− into H2O2 (Fridovich, 1989), and mobilization of tissue-bound iron. Changes in iron compartmentation have been implicated in ischemic CNS damage (Patt et al., 1990). Finally, hypothermia/hyperthermia-induced changes in OH · production may be associated with changes in blood–brain barrier integrity and neutrophil activation after brain ischemia. In an earlier report, brain IR increased vascular permeability and adhesion and activation of neutrophils after forebrain ischemia at normal or hyperthermic temperatures (Dietrich et al., 1991), but not with hypothermia (Dietrich et al., 1991). Preservation of blood–brain barrier integrity and decrease in neutrophil adhesion/activation afforded by hypothermia (Jurkovich et al., 1988; Dietrich et al., 1990) have been attributed to slow release of lipid mediators, probably due to hypothermia-induced decreased Ca2+ entry combined with a decreased degradation of arachidonic acid (Dempsey et al., 1987).
Excessive ROS production, regardless of the mechanisms, is deleterious to cells. Along with direct toxic effects, ROS enhance EAA toxicity. Glutathione deficiency intensifies NMDA toxicity in cortical cultures (Bridges et al., 1991). ROS increase EAA accumulation in brain slices (Pellegrini-Giampietro et al., 1990), which could be due to ROS-mediated inhibition of EAA reuptake by glial cells (Keyser and Pellmar, 1993) and/or inhibition of glutamine synthetase (Oliver et al., 1990). This effect may be more significant during later reperfusion if the ratio between Ca2+ -permeable and -impermeable AMPA subunits increases following global brain ischemia (Pellegrini-Giampietro et al., 1992). AMPA receptor antagonism may improve neuronal survival in global ischemia even 2 h after brain ischemia (Sheardown et al., 1990).
In conclusion, our results show that OH · production after transient ischemia is attenuated by ischemic hypothermia and enhanced by ischemic hyperthermia. Since hypothermia enhances neuronal tolerance to global ischemia in rat, attenuated OH · production may be a neuroprotective mechanism of hypothermia (Minamisawa et al., 1990). Neuropathological studies will be needed to support the hypothesis that hypothermia protects ischemic brain by decreasing OH · production.