
Abstract
Gp91-phox is an integral component of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex that generates reactive oxygen species (ROS) in activated circulating phagocytes. The authors previously demonstrated that gp91-phox knockout (KO) mice show significant protection from neuronal injury after cerebral ischemia–reperfusion injury, suggesting a pivotal role for this enzyme. Moreover, results from chimeric mice suggested that elimination of gp91-phox from both circulating phagocytes and a putative central nervous system (CNS) source were required to confer neuroprotection. In the current study, the authors demonstrated gp91-phox–specific immunostaining of perivascular cells in the CNS of control rats. However, after transient cerebral ischemia, gp91-phox–positive phagocytes were observed within the core ischemic region and activated microglial cells were positive in the penumbra. Such activated microglial cells were also gp91-phox–positive in the CNS of a chimpanzee with mild meningitis. Finally, in humans, both normal adult CNS tissues and isolated fetal microglial cells expressed gp91-phox mRNA. These microglia also expressed mRNA for the five other known components that comprise the NADPH oxidase complex. These data strongly suggest that microglial cells may contain a functionally active NADPH oxidase capable of generating ROS during CNS inflammation.
Keywords
Gp91-phox is a membrane bound glycoprotein component of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a multicomponent redox enzyme found in professional phagocytes that transfers electrons from intracellular NADPH to molecular oxygen, forming superoxide (O2-) (Jesaitis, 1995). This 91 kD protein heterodimerizes with a 22 kD protein (p22-phox) to form cytochrome b588, which together with the cytosolic components p47-phox, p67-phox, p40-phox, and rac 2 constitute the catalytic form of the enzyme (Heyworth et al., 1998; Jesaitis, 1995). Mutations in the p22-phox, gp91-phox, p47-phox, or p67-phox protein results in a dysfunctional NADPH oxidase and leads to a rare congenital disorder characterized by life-threatening microbial infections and inflammatory granuloma called chronic granulomatous disease (CGD) (Curnutte, 1993). The most common form of this disease is inherited in an X-linked manner (X-CGD) and these patients lack the gp91-phox protein. Whereas the ability of professional phagocytes to generate oxygen radicals constitutes an integral part of our innate host immunity, these same molecules contribute to tissue destruction during a variety of inflammatory diseases.
Free radicals have been reported to play a significant role in the pathogenesis of cerebral ischemia–reperfusion injury. They can be measured in the central nervous system (CNS) after transient ischemia using a range of techniques including chemiluminescence (Dirnagl et al., 1995; Yamaguchi et al., 1998), cytochemical detection (Kontos et al., 1992), nitroblue tetrazolium reduction (Kontos et al., 1985), electron spin resonance spectroscopy (Kumura et al., 1996), and salicylate trapping (Cao et al., 1988). Studies using free radical scavengers such as copper-zinc superoxide dismutase (SOD–1) or catalase conjugated to polyethylene glycol showed reduced neuronal injury after ischemia and reperfusion (Baker et al., 1998; He et al., 1993; Imaizumi et al., 1990; Liu et al., 1989; Truelove et al., 1994). Neuronal protection has more recently been observed in transgenic mice overex-pressing intracellular superoxide scavengers such as SOD-1 (Murakami et al., 1997; Yang et al., 1994) or glutathione peroxidase (Weisbrot-Lefkowitz et al., 1998). In addition to these studies, the authors have shown that neuroprotection is afforded in mice deficient in gp91-phox (Walder et al., 1997). Some reduction of brain injury associated with transient ischemia can also be shown by depletion of neutrophils from the circulation using anti-neutrophil antibodies (Matsuo et al., 1995) or by interfering with cellular adhesion (Connolly et al., 1996; Soriano et al., 1996; Zhang et al., 1994). There is evidence that neutrophils infiltrate into the vessels and parenchyma of the ischemic brain after reperfusion (del Zoppo et al., 1991; Zhang et al., 1994). However, the authors' previous results using chimeric mice generated by bone marrow transfer suggest that neuroprotection is only achieved by eliminating gp91-phox, and thus NADPH oxidase activity, from both infiltrating phagocytes and endogenous CNS cells (Walder et al., 1997).
Many cells have shown evidence for expression of gp91-phox mRNA or protein—for example, cultured human podocytes (Greiber et al., 1998), chondrocytes (Moulton et al., 1998), pulmonary neuroepithelial bodies (Youngson et al., 1993), endothelial cells (Jones et al., 1996), type I cells of carotid body (Kummer and Acker, 1995), glomerular mesangial cells (Jones et al., 1995), and microglial cells (Sankarapandi et al., 1998). In addition to its role in NADPH oxidase, gp91-phox has been proposed to act as an oxygen sensor (Youngson et al., 1993), although evidence for this has not been firmly established (Wenger et al., 1996).
Low level superoxide generation by nonprofessional phagocytes may contribute to microbial killing and play a role in signal transduction (Finkel, 1998, 1999) or gene regulation (Dalton et al., 1999). Microglial cells are the endogenous phagocytes of the CNS and appear to play a major role during the inflammatory process (Gonzalez-Scarano and Baltuch, 1999). The inflammatory response in the CNS is different from that in other organs of the body because of the presence of the blood-brain barrier, the absence of mast cells and classical antigen presenting cells, and the reduction of lymphoid and neutrophil cell migration. This inflammatory response in the CNS involves the activation of the microglial cells, which usually results in a distinct morphologic change consisting of a larger soma and shorter, more numerous, processes. Activated microglia appear to respond rapidly to ischemic injury, often within the first 24 hours, by releasing cytokines such as TGF-β and IL-1β and up-regulating a variety of cell surface components including CR3 and MHC class II (Gehrmann et al., 1992a,1992b). In vitro, activated microglia have the ability to kill cocultured embryonic neurons, which can be blocked by the addition of catalase suggesting a role for ROS (Thery et al., 1991). Also in vitro, reoxygenation of hypoxic murine microglial cells (Spranger et al., 1998) or activation of sheep microglial cells with PMA (Sankarapandi et al., 1998) resulted in the production of ROS as measured by chemiluminescence.
The objective of the current study was to investigate the in situ expression of gp91-phox in normal and inflamed CNS tissues in multiple species. Here, the authors demonstrated localization of gp91-phox in specific cells within the CNS and implicated the microglia as the cell type that has the potential to contribute to free radical-mediated injury during an inflammatory response.
MATERIALS AND METHODS
All animal experiments were performed in compliance with the standards established in the Guide for Care and Use of Laboratory Animals (NIH, Publication Number 85–23) and internal IACUC guidelines at Genentech, Inc., South San Francisco, CA, U.S.A.
Development of 18C7 monoclonal antibody
Eleven male CGD knockout mice (Pollock et al., 1995) (Charles River Laboratories, Wilmington, DE, U.S.A.) were hyperimmunized with purified cytochrome b (Cross et al., 1999b) in Freund's or Ribi adjuvant (Ribi Immunochem Research, Hamilton, MO, U.S.A.). Splenocytes from three mice demonstrating high anti-cytochrome b antibody titers were fused with mouse myeloma cells (×63.Ag8.653; American Type Culture Collection, Rockville, MD, U.S.A.) using a modified protocol analogous to one previously described (Hongo et al., 1995; Kohler and Milstein, 1975). After 10 to 14 days, the supernatants were harvested and screened for antibody production by direct enzyme-linked immunosorbent assay (ELISA). Five positive clones, showing the highest immunobinding after the second round of subcloning by limiting dilution, were injected into Pristane-primed mice (Freund and Blair, 1982) for in vivo production of monoclonal antibodies. The ascites fluids were pooled and purified by Protein A affinity chromatography (Pharmacia fast protein liquid chromatography (FPLC); Amersham Pharmacia Biotech AB, Uppsala, Sweden) as previously described (Hongo et al., 1995). The purified antibody preparations were sterile filtered (0.2 μm in pore size; Nalgene, Rochester, NY, U.S.A.) and stored at 4†C in phosphate-buffered saline (PBS).
Preparation of human neutrophils
Human neutrophils from normal volunteers or patients with X-CGD were isolated from whole blood obtained by venipuncture with appropriate consent and purified by dextran sedimentation, hypotonic cells lysis to remove residual erythrocytes and density gradient centrifugation (Badwey et al., 1982). Cells were resuspended in PBS at 2 × 107/mL and stored on ice for a maximum period of 15 minutes before use. All reagents used were endotoxin-free as determined by the limulus amebocyte lysate assay (Biowhittaker, Walkersville, MD, U.S.A.) in which the minimum detectable levels are <0.03 EU/mL.
Isolation of murine and rat bone marrow-derived leukocytes
Murine or rat leukocytes were collected from the bone marrow of the tibia and femur. Erythrocytes were lysed by incubation with 0.15 mol/L NH4Cl, 10 mmol/L KHCO3, and 10 nmol/L Na2 EDTA pH 7.3 for 5 minutes on ice. Cells were resuspended either in PBS for preparation of cell pellets for immunohistochemistry and flow cytometry or prepared for Western blot as described.
Preparation of cell pellets for immunohistochemistry
Purified human neutrophils and rat and mouse bone marrow samples were suspended in PBS buffer and centrifuged for 5 minutes at 500 g. The supernatant was removed and pellets embedded in OCT Tissue Tek compound (VWR), frozen in isopentane cooled in liquid nitrogen, and stored at −80†C.
Western blot
Human neutrophils or bone marrow-derived murine leukocytes (107/mL) were lysed in 100 mmol/L NaCl containing 10 mmol/L Tris-HCl, 0.5% (w/v) deoxycholate (Sigma, St. Louis, MO, U.S.A.), 1% Triton X–100 (Sigma), 10 mmol/L MgCl2, pH 7.5, and Complete protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN, U.S.A.) for 30 minutes on ice. Cellular debris and nuclei were removed by centrifugation (20,000 g, 4†C, 5 minutes) and postnuclear cell lysates mixed with an equal volume of Laemmli sample buffer and heated for 3 minutes at 95†C.
Rat bone marrow-derived leukocytes were resuspended in 10 mmol/L PIPES, 100 mmol/L KCl, 3.5 mmol/L MgCl2, 3 mmol/L NaCl, pH 7.3 containing Complete protease inhibitor cocktail (Boehringer Mannheim) at 1.4 × 107/mL and sonicated as previously described (Curnutte, 1985). Samples were solubilized with an equal volume of Laemmli sample buffer and heated for 3 minutes at 95†C. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% to 20% gradient) and electrophoretic transfer, the nitrocellulose filters were blocked by incubation with 10 mmol/L HEPES, 0.5 mol/L NaCl, 3% bovine serum albumin (BSA), 10% goat serum, pH 7.4 for 1 hour at 22†C. Gp91-phox was detected by incubation with 10 mmol/L HEPES, 0.5 mol/L NaCl, 0.2% Tween-20, 1% BSA, 3% goat serum, pH 7.4 containing 100 ng/mL 18C7 monoclonal antibody for 12 hours at 22†C. Filters were incubated with a goat anti-mouse IgG monoclonal antibody conjugated to horseradish peroxidase (Sigma) for 1 hour at 22†C. Gp91-phox was visualized using a chemiluminescence detection system (Boehringer Mannheim) performed as recommended by the manufacturer.
Flow cytometric analysis of gp91-phox
For flow cytometric analysis, human leukocytes were isolated from 1 mL whole blood. Erythrocytes were lysed by incubation with 0.15 mol/L NH4Cl, 10 mmol/L KHCO3, and 10 nmol/L Na2 EDTA pH 7.3 for 5 minutes at 22†C. Murine and rat bone marrow-derived leukocytes were isolated as described above. Cells were washed with PBS, incubated in 10% formalin-buffered saline for 10 minutes at 22†C, washed again, and resuspended in undiluted acetone for 10 minutes at 22†C. Leukocytes were resuspended in blocking buffer (PBS containing 0.5% BSA, 10% donkey serum, and 0.02% sodium azide) for 1 hour at 22†C, washed, adjusted to 107/mL, and incubated with 18C7 monoclonal antibody or isotype control at 1 μg/mL, 45 minutes, 22†C. After incubation with primary antibody, a donkey anti-mouse antibody conjugated to fluoroscein isothiocyanate (FITC) (Jackson, West Grove, PA, U.S.A.) was the secondary antibody used. Gp91-phox–stained leukocytes were analyzed on a FACScan using Cell-Quest software (Becton Dickenson, Franklin Lakes, NJ, U.S.A.).
Immunohistochemistry
Seven-micrometer sections of the cell pellets and coronal sections of the rat CNS were cut serially using a cryostat (Leica 3000; Leica, Deerfield, IL, U.S.A.) onto Plus slides (VWR) and left to air dry overnight. Sections were fixed in acetone/ethanol for 5 minutes, washed with PBS, and blocked for endogenous biotin using an avidin-biotin blocking kit (Vector Laboratories, Burlingame, CA, U.S.A.). To prevent nonspecific binding, the sections were incubated with 10% horse serum (Gibco, Rockville, MD, U.S.A.), diluted in 0.1% BSA/PBS (Sigma) for 1 hour before treatment with primary antibodies diluted in 0.1% BSA/PBS for 1 hour at room temperature. The following monoclonal antibodies were used: 18C7, anti-human gp91-phox (Genentech, South San Francisco, CA, U.S.A., 1:100); OX-6, anti-rat MHC class II (Serotec, Raleigh, NC, U.S.A. 1:50); OX-42, anti-rat CD11b/c (Serotec, 1:50); MHM23, antihuman CD18 (Dako, Carpinteria, CA, U.S.A.; 1:500); CR3/43, anti-human MHC class II (Dako, 1:50); and a negative mouse IgG isotype control antibody (Zymed, South San Francisco, CA, U.S.A.; undiluted). After several PBS washes, sections were incubated with biotinylated horse anti-mouse secondary antibody (Vector) diluted 1:200 for 30 minutes, washed again in PBS, and then incubated in an avidin-biotin horseradish peroxidase complex solution (Vectastain ABC kit, Vector) for 30 minutes. The chromagen was developed using metal enhanced diaminobenzidine solution (Pierce, Rockford, IL, U.S.A.).
Rat embolic stroke protocol
Male Sprague-Dawley rats weighing in excess of 500 g were anesthetized with a gaseous mixture of halothane, oxygen, and nitrous oxide. One milliliter of whole blood was drawn through cardiac puncture into PE-90 tubing coated with bovine thrombin, and the ends of the tubing were plugged. Tubes were incubated at 37†C for 18 hours and continuously rotated to maintain uniform cross-sectional consistency of the clot. The tubing was cut into 5-mm lengths; the clots were extruded and then were introduced into a cannula made with polyethylene PE-60 tubing (Clay Adams, Becton Dickinson, Franklin Lakes, NJ, U.S.A.). Ten male Sprague-Dawley rats weighing 270 to 300 g were anesthetized as before and a midline incision of the neck exposed the right common carotid, external carotid, and its branches and internal carotid arteries. The right common carotid artery was isolated, ligated proximal to the site of cannulation, and loose ligatures were placed distally. The artery then was cannulated with the PE-60 tubing containing the 5-mm preformed whole blood clot and its tip advanced to the origin of the internal carotid artery. Care was taken to ligate all accessible branches of the internal and external carotid arteries. The clot then was flushed into the cerebral circulation with 0.3 mL sterile saline. The cannula was removed, the common carotid artery ligated distal to the site of cannulation to prevent retrograde flow, and the surgical incision closed. At 6 (n = 5) and 16 hours (n = 5) postsurgery, rats were anesthetized with Nembutal and decapitated; the brains were removed and sliced into 4-mm coronal sections. Each slice was embedded in OCT Tissue Tek (VWR), frozen in isopentane cooled in liquid nitrogen, and stored at −80†C.
Chimpanzee
The chimpanzee cortex was obtained from a 2-year-old female. The chimpanzee had suffered from a progressive neurologic disease for 1 year that had resulted in her inability to feed herself. Pathologic postmortem analysis revealed extensive cerebellar hypocellularity (cerebellar cortical abiotrophy, presumptive) and a mild inflammation of the meninges.
Northern blot
Human brain multiple tissue northern blots, II and III (Clontech, Palo Alto, CA, U.S.A.) containing 2 μg poly A+ RNA per lane isolated from different brain sections, were hybridized with a radiolabeled 121 bp (bp 127–248) gp91-phox probe (1.4 × 105 cpm/ng) or a random primed β-actin probe (1.3 × 106 cpm/ng) for 16 hours at 42†C using Express Hybridization Solution (Clontech). Prehybridization, hybridization, and washing procedures were performed as recommended by the manufacturer. gp91-phox mRNA was visualized using a Fujix BAS2000 Phosphoimager (Fuji Medical Systems, Stamford, CT, U.S.A.). The gp91-phox and β-actin blots were exposed to the phosphoimager for 16 hours and 1 hour, respectively.
Reverse transcription-polymerase chain reaction
Human fetal microglial cells were cultured from fetal brain tissue as previously described (Chao et al., 1995). Reverse transcription (RT) of 1 μg total RNA was performed using an oligo (dT) 12–18 primer (Pharmacia). Briefly, 1 μg RNA was incubated with 1 μL of 0.5 μg/μL oligo (dT) 12–18 primer for 10 minutes at 70†C. The RT reaction was performed in 50 mmol/L Tris-HCl (pH 8.3), 75 mmol/L KCl, 3 mmol/L MgCl2, 10 mmol/L DTT, 0.5 mmol/L dNTPs (Pharmacia) containing 200 units SuperScript II RT (Gibco BRL). A control reaction for RT had the SuperScript II RT enzyme omitted. The reaction mixture was incubated at 42†C for 1 hour followed by termination at 95†C for 5 minutes in a programmable Tempcycler (Coy, Ann Arbor, MI, U.S.A.). The cDNA was stored at −80†C before amplification.
Amplification of NADPH oxidase component's cDNA was performed in 50 mmol/L KCl, 10 mmol/L Tris-HCl (pH 9.0 at 25†C), 0.1% Triton X-100, 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 2 U Taq DNA polymerase (Promega), 0.5 μmol/L of each (sense and antisense) primer, and 2 μL cDNA. The following sense (S) and antisense (AS) primers, shown in 5′ to 3′ were used: gp91-phox, (S) TTC ACT CTG CGA TTC ACA CCA TTG C and (AS) CCT CAG GGG CGG ATG TCA GTG TAA; p22-phox, (S) ATT GTG GCG GGC GTG TTT GTG T and (AS) GTT GCT GGG CGG CTG CTT GAT G; p47-phox, (S) CTT CGT ACC CAG CCA GCA CTA TGT and (AS) CAT GGA CGG AAA GTA GCC TGT GAC; p67-phox, (S) GCT GGC AGC GGA CAA GAA GGA CT and (AS) GGC ACA AAC CCA AAT AGC ACA CG; p40-phox, (S) CCA GCC ACT TTG TTT TCG TCA TCG and (AS) GCC CTC CAG CCA GTC TTT GTT G; rac, (S) CCC CCT ATC CTA TCC GCA AAC A and (AS) CTT GTC GTC CCG CAG GTC CAG (primers amplify both rac-1 and rac-2). Control reaction for PCR contained no cDNA. The mixture was subjected to 30 amplification cycles, with each cycle as follows: 94†C for 45 seconds, 58†C for 45 seconds, and 72†C for 90 seconds. Samples were analyzed by 2% agarose gel electrophoresis and amplified DNA fragments visualized with ethidium bromide stain under ultraviolet light.
RESULTS
Monoclonal antibodies against purified human cytochrome b558 generated in gp91-phox KO mice were analyzed by Western blot. This analysis identified a specific antibody, 18C7, that recognized human gp91-phox in normal, but not X-CGD, neutrophils (Fig. 1A). The antibody labeled only permeabilized, but not intact, normal human neutrophils as demonstrated by flow cytometry (Fig. 1B), indicating recognition of an intracellular epitope. Negligible staining was observed in neutrophils from patients with X-CGD (Fig. 1C). Immunohistochemical staining of gp91-phox was observed in normal, but not X-CGD, human neutrophil pellets confirming the specificity of 18C7 (Fig. 1D and 1E).
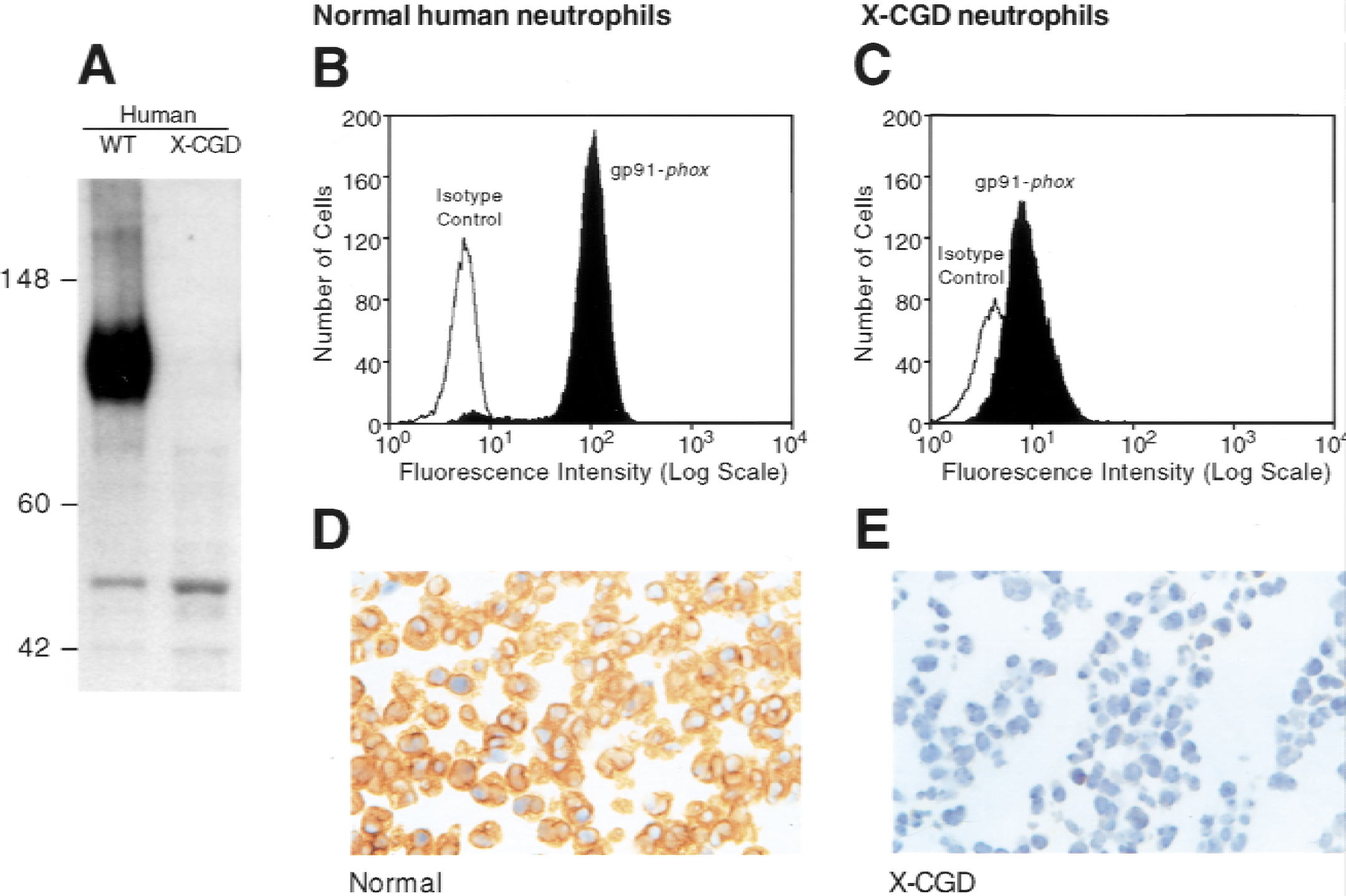
Specificity of the anti-gp91-phox monoclonal antibody 18C7 in normal and X-linked chronic granulomatous disease (X-CGD) human neutrophils. (
The ability of 18C7 to recognize both murine and rat gp91-phox in bone marrow–derived neutrophils was demonstrated by Western blot analysis, flow cytometry, and immunohistochemical staining (Fig. 2). This crossreactivity is not surprising because human and murine gp91-phox share 93% amino acid identity. Because of differences in glycosylation, rodent gp91-phox has an apparent molecular weight of 62 kD (Fig. 2A), compared with 91 kD in humans (Fig. 1A). 18C7 also appears to weakly recognize a band at approximately 50 kD by Western blot in human and rodent neutrophils (Figs. 1A and 2A). This band appears to be unrelated to gp91-phox as it can be seen in both human and mouse X-CGD extracts. Importantly, such reactivity was not observed by immunohistochemical analysis. Gp91-phox was not detected by Western blot in bone marrow–derived neutrophils isolated from gp91-phox KO mice (Fig. 2A). Flow cytometric analysis indicated that 18C7 specifically labeled gp91-phox positive cells in normal murine and rat bone marrow but not those from gp91-phox KO mice (Fig. 2B to 2D). Although immunohistochemical staining of murine bone marrow pellets was not possible for technical reasons (18C7 is a murine antibody), 18C7 specifically stained phagocytes in rat bone marrow but not erythroid or lymphoid cells (Fig. 2E). These data indicate that 18C7 recognizes gp91-phox and demonstrates crossreactivity with rodent species by Western blot, flow cytometry, and immunohistochemistry.
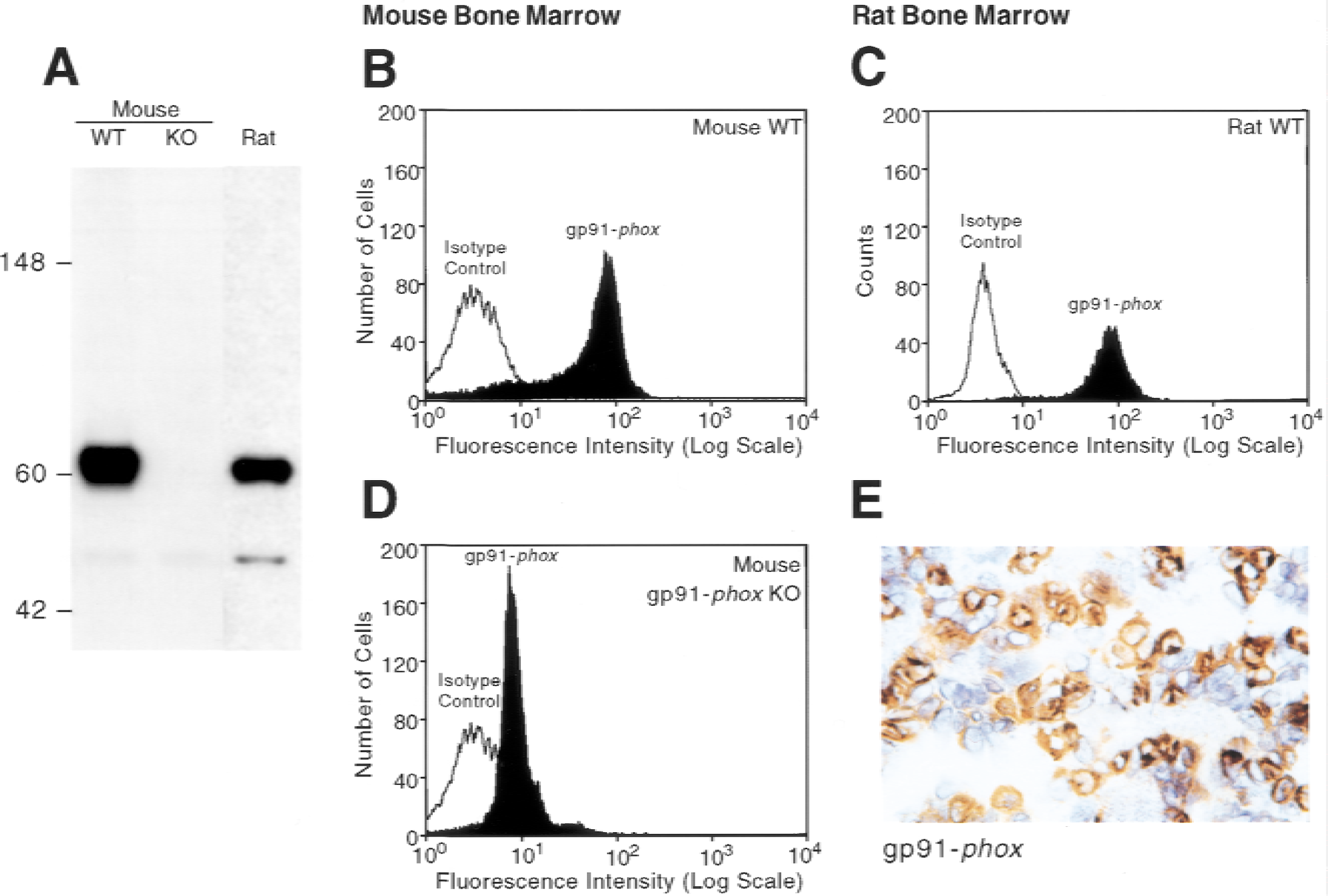
Crossreactivity of the anti-gp91-phox monoclonal antibody 18C7 for phagocytic precursors in mouse and rat bone marrow. (
Using 18C7, the authors evaluated gp91-phox expression in a rat embolic ischemia–reperfusion stroke model. This model is described here for the first time in Materials and Methods. This rat embolic stroke model leads to a visible infarct core in the caudate putamen region of the CNS with a variable surrounding area of lesser damage, which the authors will define as the penumbra. Immunostaining of normal rat CNS with 18C7 identified gp91-phox only in perivascular cells (Fig. 3B). These cells are morphologically identical to ED2-positive perivascular cells (Graeber et al., 1989) (unpublished observations) and are phenotypically different from the microglia seen in a serial section using the anti-CD11b/c antibody (Fig. 3A). As previously demonstrated, this CD11b/c monoclonal antibody stained resting parenchymal microglial cells throughout the brain (Mato et al., 1996). These perivascular cells and parenchyma microglial cells failed to stain for MHC class II using the authors' standard immunohistochemical techniques (Fig. 3C).
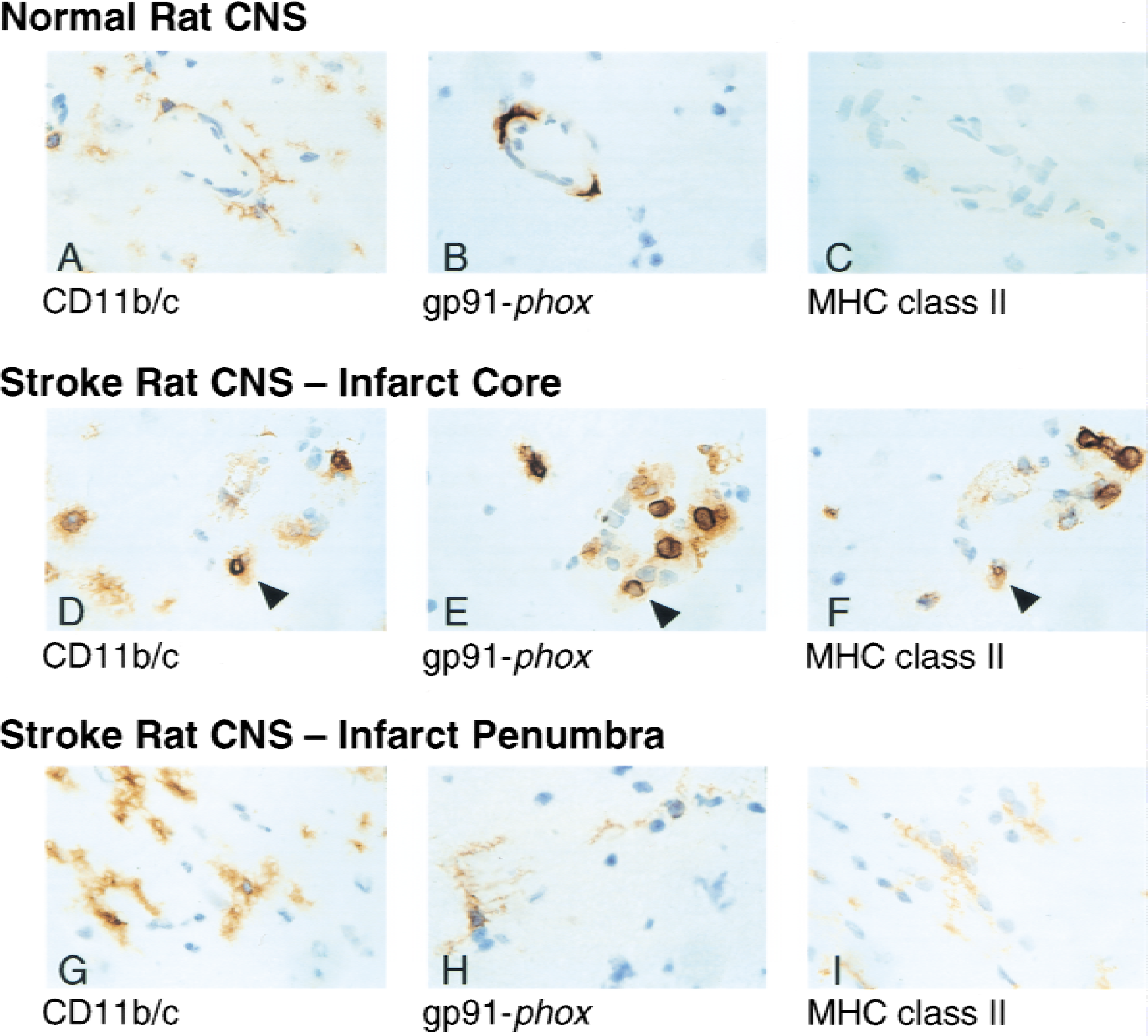
Detection of gp91-phox in rat central nervous system (CNS) after cerebral ischemia–reperfusion injury. Immunohistochemistry of rat CNS tissue from normal animals or animals 16 hours postembolic stroke. Serial sections from normal rat CNS (top panels), infarct core (middle panels), or infarct penumbra (bottom panels) were stained with anti-CD11b/c (
At 6 hours postischemia, only a small infarct was observed. Gp91-phox--positive expression appeared very similar to its expression in the normal CNS. However, by 16 hours postembolic stroke, analysis of the rat CNS revealed intense inflammation within the large ischemic core region. In contrast with the normal rat CNS, gp91-phox–positive phagocytes were observed in abundance in both the parenchyma and around blood vessels (Fig. 3E). Serial sections revealed identical cells positive for gp91-phox as well as CD11b/c and MHC class II, indicating that these cells are either infiltrating activated monocytes or endogenous macrophages (Fig. 3D and 3F). Because these cells are MHC class II positive, by definition they can not be neutrophils. Immediately outside the infarct core region, in an area defined as the penumbra, gp91-phox was clearly upregulated in parenchymal microglial cells 16 hours postembolic stroke (Fig. 3H). Enhanced expression of CD11b/c and MHC class II was also observed in these cells, indicating their immunologic activation (Fig. 3G and 3I).
To determine whether gp91-phox was upregulated in primate microglia during neuroinflammation, the authors performed immunohistochemistry on the CNS of a chimpanzee with mild meningitis (Fig. 4). Focal areas with large, activated parenchymal microglial cells were observed within the cortex. These cells were immunopositive for gp91-phox (Fig. 4B and 4E). Analysis of serial sections indicated that the identical cells also expressed CD18 and MHC class II (Fig. 4A, 4C, 4D, and 4F). Although microglial expression of CD18 was evident in noninflamed regions of the CNS, no staining of parenchymal microglial cells for gp91-phox or MHC class II was observed. Similar to the rat CNS, gp91-phox also stained perivascular cells throughout the brain in the chimpanzee (data not shown).
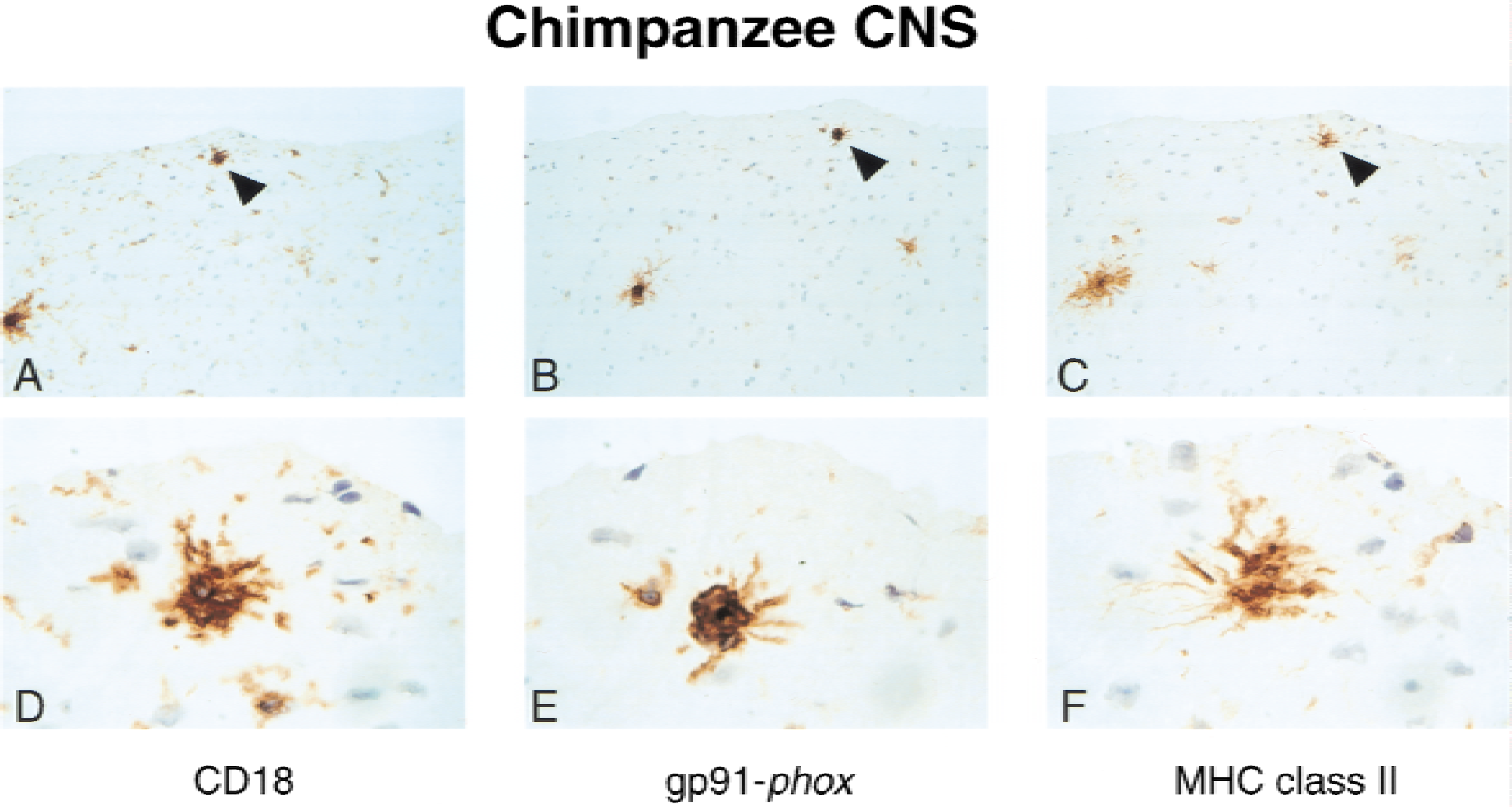
Induction of gp91-phox in brain parenchymal microglial cells from a chimpanzee with meningitis. Serial sections from a chimpanzee diagnosed at autopsy with mild meningitis stained with anti-CD18 (
To evaluate if gp91-phox is expressed in normal human brain, the authors analyzed mRNA levels in 16 different brain sections by Northern blot analysis (Fig. 5). They were able to consistently detect expression of the 4.7 kb gp91-phox mRNA transcript in spinal cord and medulla (Fig. 5). Expression was also observed in other regions of the CNS. However, these Northern blots are not quantitative as the number of donors for each tissue type is variable on a given blot as well as between each blot lot. Similar levels of β-actin mRNA transcript were detected in all lanes suggesting equivalent loading of poly A+ RNA.
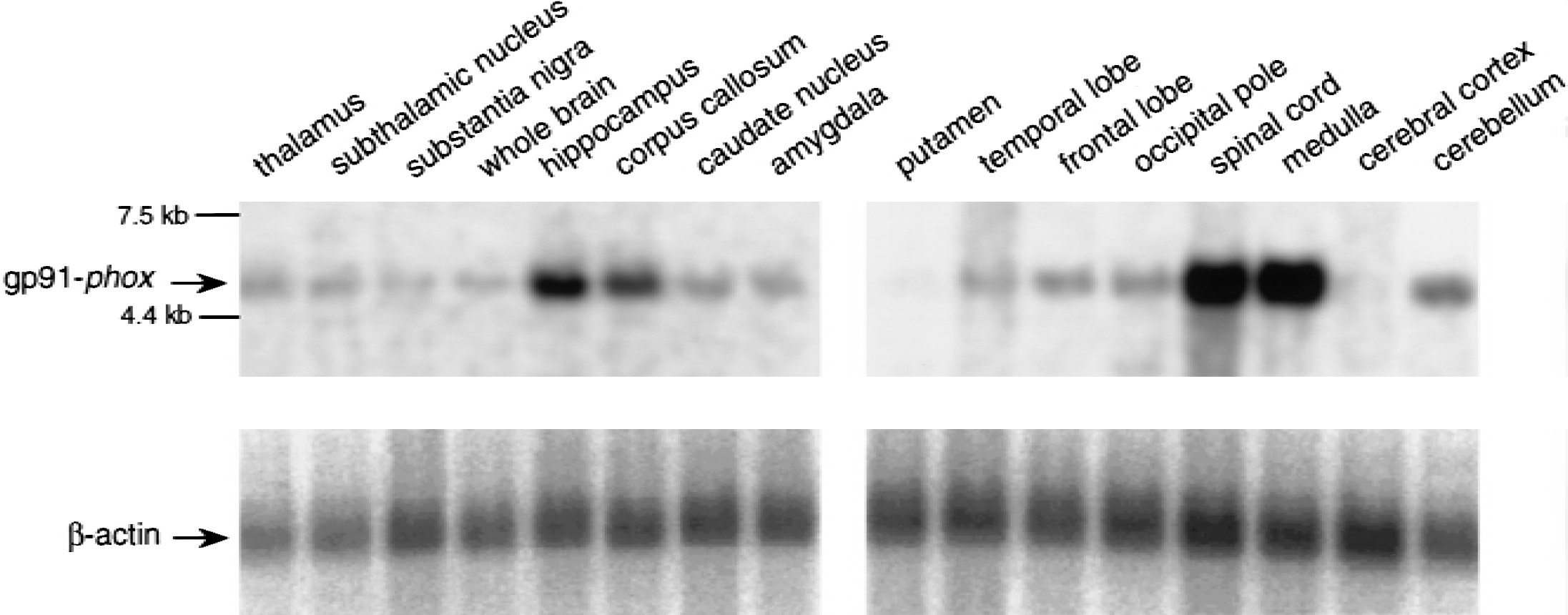
Expression gp91-phox mRNA in normal human central nervous system (CNS) tissue. Human multiple brain tissue northern blots (Clontech) show gp91-phox mRNA as a 4.7 kb band (top panels). The bottom panels show consistent β-actin expression in all lanes. The gp91-phox and β-actin blots were exposed to the phosphoimager for 16 hours and 1 hour, respectively.
Human microglial cells have been reported to generate superoxide (Chao et al., 1995; Colton and Gilbert, 1987), although it is unknown if these cells express all the functional components of NADPH oxidase and whether this enzyme is responsible for the production of ROS. Analysis of human fetal microglial cells by reverse transcription-polymerase chain reaction shows the presence of all the unique functional components of this enzyme complex, including gp91-phox, p22-phox, p47-phox, p67-phox, p40-phox, and rac (Fig. 6). These results suggest cultured human fetal microglial cells have the capacity to produce superoxide through NADPH oxidase.
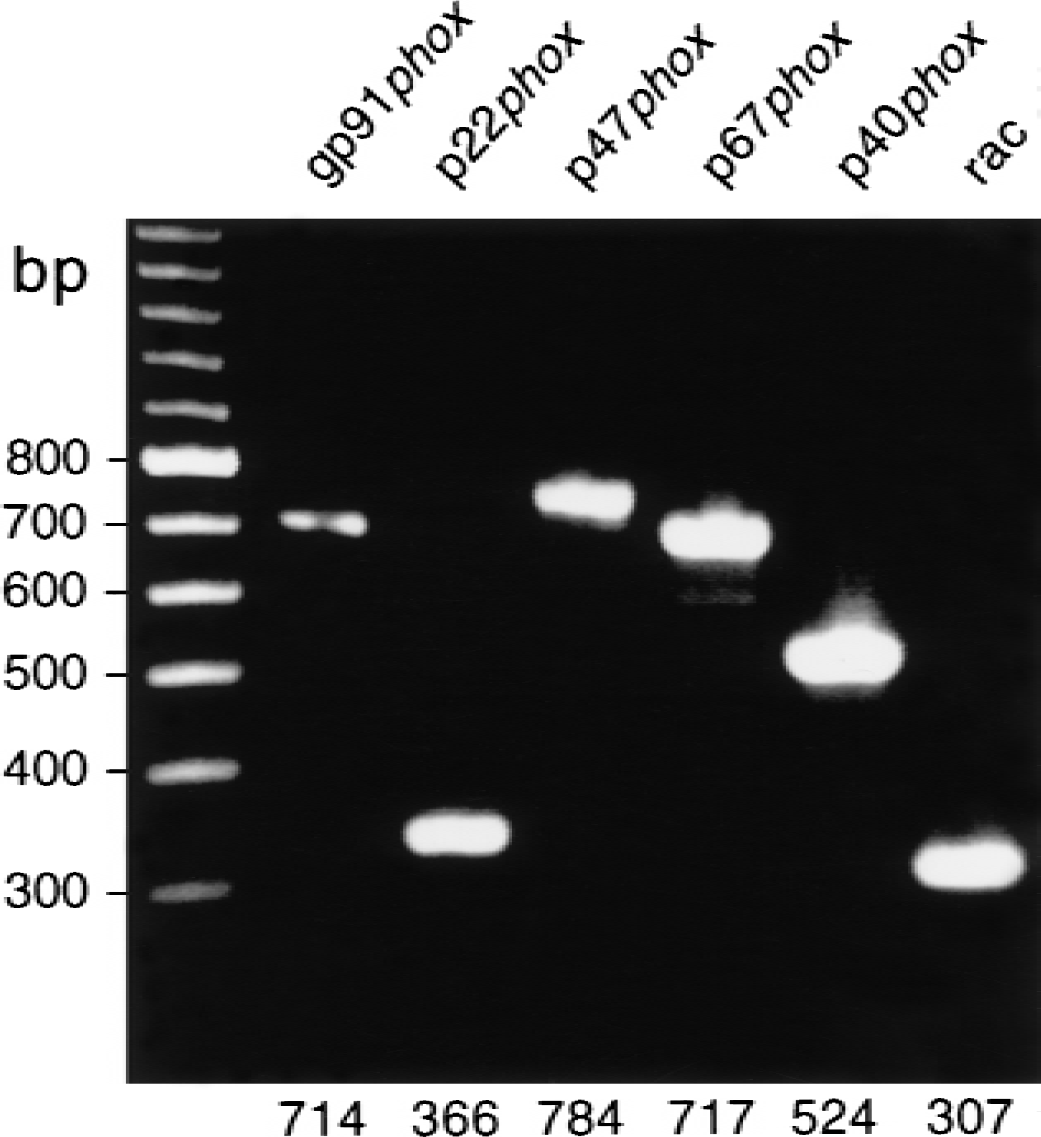
Detection of gp91-phox in human fetal microglial cells by reverse transcription-polymerase chain reaction (RT-PCR). RT-PCR analysis of gp91-phox, p22-phox, p47-phox, p67-phox, p40-phox, and rac from cultured human fetal microglial cells. Predicted sizes for the correct RT-PCR products are shown.
DISCUSSION
Despite the technical difficulties in measuring oxygen radical generation in vivo, numerous reports have implicated these molecules in the pathogenesis of cerebral ischemia–reperfusion injury. Transgenic mice with increased ROS scavenging activities, such as glutathione peroxidase (Weisbrot-Lefkowitz et al., 1998) or copper-zinc superoxide dismutase (Murakami et al., 1997; Yang et al., 1994), confer neuroprotection. These observations are supported by the use of chemical inhibitors and antioxidants designed to scavenge oxygen radicals (Baker et al., 1998; He et al., 1993; Imaizumi et al., 1990; Liu et al., 1989; Park and Hall 1994; Truelove et al., 1994) that are known to modify a range of macromolecules including DNA, proteins, and lipids. In vitro, it has been shown that there are many sources for free radical generation, including nitric oxide synthase, xanthine oxidase, mitochondria, and NADPH oxidase. However, the cell types responsible for generating free radicals during neuroinflammation have not been determined. Genetic deficiency in the neuronal form of nitric oxide synthase affords some neuroprotection after cerebral ischemia (Huang et al., 1994), suggesting neurons as a possible in vivo source for free radicals. Indeed, a recent paper has indicated that NADPH oxidase components are present in cultured sympathetic neurons and may contribute to neuronal apoptosis (Tammariello et al., 2000). Alternatively, reactive microglia and activated phagocytes are frequently observed within the inflamed CNS (Gonzalez-Scarano and Baltuch, 1999) suggesting these cells may contribute to free radical generation. It is likely that the cell types, sources, and mechanisms by which oxygen radicals contribute to neuronal injury after transient cerebral ischemia are complex and may involve direct injury to neurons, oligodendrocytes, and endothelial cells.
The authors previously have reported that mice lacking the gp91-phox component of NADPH oxidase (gp91-phox KO mice) were significantly protected from neuronal injury after transient cerebral ischemia (Walder et al., 1997). Moreover, generation of chimeric animals by bone marrow transfer indicated that elimination of NADPH oxidase from both circulating phagocytes and from an endogenous CNS source was required to confer neuroprotection (Walder et al., 1997). These results suggested that a cell intrinsic to the CNS contains NADPH oxidase and that this enzyme may be functionally active during cerebral ischemia–reperfusion injury.
To evaluate the expression of gp91-phox in the CNS of rats during ischemia–reperfusion injury, the authors generated a gp91-phox–specific monoclonal antibody, 18C7. In the normal rat CNS, gp91-phox expression was observed only in perivascular cells. These cells appear morphologically similar to cells that stain positive for ED2 (unpublished data), an antibody that specifically labels perivascular macrophages (Graeber et al., 1989). The constitutive expression of gp91-phox in these cells is consistent with their possible role in host defense against microorganisms invading the CNS (Gonzalez-Scarano and Baltuch, 1999). After transient embolic ischemia, abundant gp91-phox positive cells were observed within the core region of the infarct. These cells also expressed CD11b/c and MHC class II indicating they were endogenous or infiltrating activated macrophages. Within the penumbra of the stroke, gp91-phox was upregulated in activated parenchymal microglial cells that also stained positive for CD11b/c and MHC class II. These results indicate that there is an increased expression of gp91-phox during the neuroinflammatory process, implicating NADPH oxidase as an important mediator of neuronal injury. Interestingly, CNS neurons were not gp91-phox positive in this rat model of cerebral ischemia.
The observation that gp91-phox is expressed in activated microglia in the chimpanzee with mild meningitis further supports the notion that NADPH oxidase is involved in neuroinflammation. As observed in the rat, gp91-phox protein expression was upregulated in CD18/ MHC class II immunopositive-activated microglial cells in the inflamed chimpanzee CNS. The activation status of these cells is confirmed morphologically by the large amoeboid shape and ramified processes. These microglial cells are known to be phagocytic, to have increased MHC class II expression, and to contribute to the inflammatory response by releasing cytokines such as TNF-α (Gonzalez-Scarano and Baltuch, 1999).
The expression of gp91-phox in parenchymal microglial and perivascular cells indicates that these cells have the potential to generate oxygen radicals through NADPH oxidase. The gp91-phox component is the catalytic core of the enzyme as it contains both the FAD and redox centers that are responsible for superoxide generation (Yu et al., 1998). Detailed studies on patients with CGD and NADPH oxidase cell free activation systems have established that 4 other proteins—p22-phox, p47-phox, p67-phox, and rac 2—are necessary, along with gp91-phox, for superoxide generating activity (Curnutte, 1993). The function of p40-phox, which is not required for cell-free oxidase activity, is still under investigation (Cross et al., 1999a). Examination of these other NADPH oxidase components in microglial cells in situ is currently ongoing. In vitro, cultured human fetal microglial cells express mRNA for all six of the oxidase components (Fig. 6) and are known to generate superoxide (Chao et al., 1995; Colton and Gilbert, 1987). These results are supported by the published observation that cultured microglial cells contain p22-phox and gp91-phox protein and can undergo an oxidative burst when stimulated (Sankarapandi et al., 1998; Spranger et al., 1998).
Preclinical studies have identified many factors that contribute to ischemia–reperfusion injury including glutamate-triggered neuronal excitotoxicity, calcium and zinc neurotoxicity, adenosine triphosphate depletion, hypoxia-induced neuronal apoptosis, ion shifts leading to acidosis, phagocyte infiltration, and generation of nitrogen and reactive oxygen intermediates (Lee et al., 1999). Uniformly, activated microglial cells are observed after transient ischemic insult and appear to play a major role in coordination of the neuroinflammatory response (Zielasek and Hartung, 1996). In injured CNS tissue, these cells phagocytose, release proinflammatory cytokines, chemotax, present antigen, and generate oxygen free radicals (Gehrmann et al., 1995; Zielasek and Hartung, 1996). The observation that gp91-phox is upregulated in parenchymal microglia in the penumbra after ischemia–reperfusion is consistent with the known activation state of these cells during neuroinflammation. It is likely that NADPH oxidase-derived free radicals are generated by infiltrating and endogenous phagocytes in the core region of the infarct and by perivascular microglial cells in the penumbra. The authors' previous observations using gp91-phox KO mice suggest that elimination of both circulating and CNS sources of NADPH oxidase are required for neuroprotection (Walder et al., 1997). The capability of resident microglial cells, as well as infiltrating phagocytes, to produce oxygen radicals through the NADPH oxidase pathway suggests that these cells may play an important role in the neurologic damage observed in CNS inflammatory disease.
Footnotes
Acknowledgments:
The authors thank Drs. Chun Chao and Wen Sheng, Minneapolis Medical Research Foundation, for determining expression of NADPH oxidase components in human fetal microglial cells by reverse transcription-polymerase chain reaction. The authors also thank Denise Harrison for her assistance in the generation and purification of 18C7 monoclonal antibody and Walter Darbonne for independent Northern blot analysis in human tissues. Finally, they would like to thank Alison Bruce for her invaluable graphic support.