
Abstract
Traumatic brain injury elicits acute inflammation that in turn exacerbates primary brain damage. A crucial part of innate immunity in the immune privileged central nervous system involves production of proinflammatory cytokines mediated by inflammasome signaling. Here, we show that the nucleotide-binding, leucine-rich repeat pyrin domain containing protein 1 (NLRP1) inflammasome consisting of NLRP1, caspase-1, caspase-11, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), the X-linked inhibitor of apoptosis protein, and pannexin 1 is expressed in neurons of the cerebral cortex. Moderate parasagittal fluid-percussion injury (FPI) induced processing of interleukin-1β, activation of caspase-1, cleavage of X-linked inhibitor of apoptosis protein, and promoted assembly of the NLRP1 inflammasome complex. Anti-ASC neutralizing antibodies administered immediately after fluid-percussion injury to injured rats reduced caspase-1 activation, X-linked inhibitor of apoptosis protein cleavage, and processing of interleukin-1β, resulting in a significant decrease in contusion volume. These studies show that the NLRP1 inflammasome constitutes an important component of the innate central nervous system inflammatory response after traumatic brain injury and may be a novel therapeutic target for reducing the damaging effects of posttraumatic brain inflammation.
Introduction
Traumatic brain injury (TBI) is a complex and devastating clinical condition mediated by proinflammatory cytokines that produce neuronal loss, axonal destruction, and demyelination during the secondary injury cascade (Bramlett and Dietrich, 2007; Povlishock, 1992). Increased production of cytokines of the interleukin-1 (IL-1) family, such as IL-1β, is well documented, providing clear evidence for a pivotal role of this cytokine in triggering TBI-induced inflammatory processes (Ciallella et al, 2002; DeKosky et al, 1994; Fan et al, 1995; Fassbender et al, 2000; Fink et al, 1999; Goss et al, 1995; Hutchinson et al, 2007; Kinoshita et al, 2002; Knoblach and Faden, 2000; Morita-Fujimura et al, 1999; Utagawa et al, 2008). IL-1β and IL-18 are potent mediators of inflammation and initiate and/or amplify a wide variety of effects associated with innate immunity, host responses to tissue injury, and microbial invasion (Bhat et al, 1996; Dinarello, 2004; Dinarello, 2005a, b, 2006). Although the majority of studies indicate that inflammatory processes associated with the adaptive immune response contribute to secondary injury after TBI, few, if any, information is available about the innate central nervous system (CNS) immune response after brain trauma.
In the innate immune response, activation and processing of proinflammatory cytokines IL-1β, IL-18, and IL-33 are controlled by inflammatory caspase-1 and caspase-5 in cytoplasmic multiprotein complexes known as inflammasomes (Arend et al, 2008; Li et al, 2008; Martinon et al, 2002). Assembly of inflammasomes depends on the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) (nucleotide-binding domain, leucine-rich repeat containing) family of proteins (Ting et al, 2008). To date, more than 20 NLR proteins have been identified with more than 200 members predicted from recent genomic analysis (Rast et al, 2006). Inflammasomes are composed of three proteins: (1) an NLR family member, (2) the adaptor protein apoptosis speck-like protein with a caspase recruitment domain (ASC), and (3) caspase-1. The exception is the NLRC4 (Ipaf) inflammasome that consists of NLRC4 and caspase-1 (Poyet et al, 2001). The inflammasome regulates caspase-1 processing and activity and, consequently, the levels of the active cytokines IL-1β and IL-18.
Our recent work shows that the NLRP1 inflammasome is present in spinal cord and cortical neurons and plays an important role in the innate CNS inflammatory response after injury and stroke (Abulafia et al, 2009; de Rivero Vaccari et al, 2008). In these studies, the NLRP1 inflammasome was shown to be an important therapeutic target to reduce caspase-1 activation and tissue damage leading to improved functional outcomes. Here, we extend this approach to determine whether TBI would also induce inflammasome activation in vulnerable brain regions. The cell-type-specific distribution of inflammasome proteins was evaluated in control and traumatized brains. To establish the importance of inflammasome activation in the innate CNS immune response in pathophysiology of TBI, we therapeutically neutralized the inflammasome that resulted in reduced caspase-1 and IL-1β activation, leading to significant improvement in tissue sparing.
Methods
Animals and Traumatic Brain Injury
Male Sprague–Dawley rats (250-350 g) were anesthetized using 3% halothane and a gas mixture of 70% N2O and a balance of O2 to achieve deep sedation. Injury was produced using a fluid-percussion injury (FPI) device that consisted of a saline-filled Plexiglas cylindrical reservoir bent at one end with a rubber-covered piston and with the opposite end fitted with a transducer housing and injury screw adapted for the rat's skull as described earlier (Dietrich et al, 1994). The metal screw was firmly connected to the plastic injury tube of the intubated anesthetized rat and the injury was induced by the descent of a metal pendulum that struck the piston. A power laboratory system (CB Sciences, Dover, NH, USA) was used to measure the atmospheric level of the fluid-percussion impact. After TBI, all rats were returned to their cages and allowed to recover from the surgical procedures. Injury was moderate (1.7 to 2.2 atmospheres) and animals were killed at different times after TBI. Sham and naive animals were used as controls. Naive rats were anesthetized and killed. Tissue samples were snap frozen in liquid nitrogen and stored at −80°C until the time of assay. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Miami.
Antibodies
Rabbit anti-Rattus-novegicus ASC and NALP1 antisera were prepared by Bethyl Laboratories as described (de Rivero Vaccari et al, 2008). Other antibodies were purchased from commercial sources, which include: anti-IL-1β (Cell Signaling Technology, Beverly, MA, USA), anti-caspase-1 (Upstate Biologicals, Lake Placid, NY, USA), anti-caspase-1 (Santa Cruz Biologicals, Santa Cruz, CA, USA), anti-caspase-11 (Alexis Biochemicals, Plymouth Meeting, PA, USA), anti-caspase-11 (Santa Cruz Biological, Santa Cruz, CA, USA), anti-XIAP (X-linked inhibitor of apoptosis protein) (BD Transduction Laboratories, San Jose, CA, USA); anti-caspase-3 (Upstate), anti-pannexin-1 (Zymed, South San Francisco, CA, USA), and anti-MAP2 (Chemicon, Billerica, MA, USA).
ASC Neutralization
To dissect the contribution of the NLRP1 inflammasome to TBI-induced inflammation, we blocked the activity of the inflammasome with antibodies against the inflammasome adaptor protein ASC. Antibody treatment was started immediately after trauma. One group of animals received 15 μg of anti-ASC intracerebroventricularly into the right ventricle after injury, whereas control groups received a similar treatment regimen, but using IgG of the same isotype corresponding to anti-ASC. Cortices were removed at 24 h after treatment and lysates were prepared and immunoblotted for caspase-1 and XIAP. For lesion volume analysis, rats were subjected to moderate FPI and then treated with 15 μg of anti-ASC intracerebroventricularly and then injected with 50 μg of the anti-ASC antibody intraperitoneally at 24 and 48 h after injury. Another group of rats was treated with IgG and served as control. At 3 days after TBI, animals were perfusion fixed for quantitative analysis of contusion areas and volumes.
Immunoblotting
A 2-mm section of cortex was homogenized in PTN50 extraction buffer (50 mM NaPi, pH: 7.4, 50 mM NaCl, 1% Triton X-100) with proteases (1 μg/ml pepstatin A, 1 μM aprotinin, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin). Proteins were resolved in 10−20% Tris–HCl criterion precasted gels (Bio-Rad) as described (de Rivero Vaccari et al, 2008).
Coimmunoprecipitation
To assess the protein composition and association of proteins in the inflammasome, 500 μg of cortical lysates from sham and traumatized animals at 4 h were immunoprecipitated with anti-ASC or anti-NALP1 antibodies using TrueBlot anti-rabbit Ig immunoprecipitation beads. Cortical lysates were precleared by adding 50 μl of anti-rabbit TrueBlot beads to 500 μg of lysate in a microcentrifuge tube as described (de Rivero Vaccari et al, 2008).
Immunohistochemistry
Immunostained brain sections of uninjured and injured rats at 4 h were examined with a Zeiss laser scanning confocal microscope (Zeiss, Thornwood, NY, USA). Rats were perfused with 4% paraformaldehyde as described and processed for cryostat sectioning (Leica SM 2000R sliding microtome). Sections (50 μm) were blocked by treatment with normal goat serum (Vector Laboratories, Burlingame, CA, USA). Tissue sections were rinsed with 0.1 M phosphate-buffered saline (pH 7.4) and incubated overnight at 4°C with primary antibodies against caspase-1 (1:500), caspase-11 (1:500), ASC (1:500 dilution), and NALP-1 (1:500). To determine the precise cellular distribution of inflammasome proteins, sections were double stained with the neuronal marker antimicrotubule-asso-ciated protein-2 (MAP2, neurons—Chemicon, Billerica, CA, USA) as described (de Rivero Vaccari et al, 2008).
Contusion Volume Analysis
At 3 days after TBI, rats were killed and perfusion fixed. For calculating the contusion areas and volume TBI, six coronal sections were chosen for morphometric study. Quantification of lesion volume in the injured brain was calculated using computer-assisted microscopy and Neurolucida software (MicroBrightfield, Willeston, VT). After perfusion, brains were removed and fixed in 4% paraformaldehyde (n = 5 animals per group), transverse sectioned at 10 μm, and then stained with hematoxylin and eosin (H&E) for histologic assessment of contusion areas by an individual masked to the experimental groups. Coronal sections spaced at every 780 μm from bregma levels 1.8 to 7.3 were used for contusion area and volume measurements (Zilles, 1985). Volumes were determined by tracing the area of cortical damage that was demarcated in hematoxylin and eosin stained sections. The contused area consisted of pyknotic neurons, reactive astrocytes, and an area of shearing at the gray/white matter interface of the lateral cerebral cortex. The extracellular space also appeared edematous relative to regions outside the contused area. The Neurolucida, Microbrighfield software program for lesion volume calculations, made numerical integration of the successive areas.
Statistical Analysis
Data are expressed as standard error of the mean (± s.e.m.). Statistical comparisons between uninjured and injured groups were made using Student's t-test, one-way analysis of variance, or two-way paired comparisons analysis of variance followed by Tukey's multiple comparison tests. P-values of significance used were *P < 0.05 and #P < 0.10.
Results
Traumatic Brain Injury Induces Processing of Interleukin-iβ in the Cerebral Cortex
To determine whether TBI induces the processing of IL-1β from a precursor into a mature secreted form, we performed quantitative immunoblot analysis on cortical lysates from sham-operated and traumatized animals at 15, 30 mins, 1, 3, 6, and 24h after TBI. As shown in Figure 1, within 15 mins after TBI, pro-IL-1β (35kDa) is rapidly processed to the mature form (17kDa) in cortices of traumatized rats. These results indicate that increased levels of the mature form of IL-1β are induced rapidly within the cortex by moderate TBI and are consistent with the rapid processing and secretion observed by the majority of investigations studying secretion of IL-1β from primary macrophages or cell lines (Dinarello, 2005a).
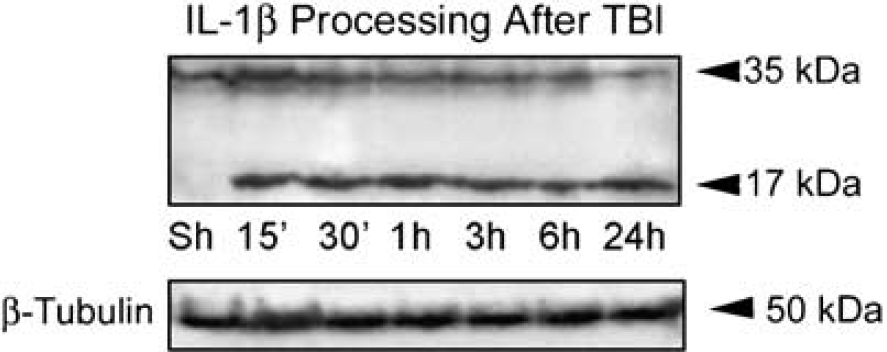
TBI induces interleukin-1β (IL-1β) processing in the injured cortex. Immunoblot analysis of IL-1β in cortical lysates of sham-operated animals (Sh) and traumatized rat cortices at 15, 30 mins, 1, 3, 6, and 24 h after injury, β-tubulin was used as internal standard and control for protein loading. N = 5 per group.
Traumatic Brain Injury Induces Expression of Inflammasome Proteins
Processing of pro-IL-1β and IL-18 involves the activation of a caspase-1-activating platform, termed as the inflammasome (Martinon and Tschopp, 2007; Martinon et al, 2002; Ogura et al, 2006). To provide direct evidence for involvement of the inflammasome in TBI-induced inflammation, we analyzed traumatized cortices for the time course of expression of key inflammatory caspases and inflammasome proteins (Figure 2). Antibody specificity for the reagents used in preliminary studies has been rigorously documented in our earlier manuscript (de Rivero Vaccari et al, 2008). TBI rapidly activated caspase-1 (Figure 2A) and upregulated caspase-11, the rodent ortholog of human caspase-5 (Figure 2B). Proteolytic processing of procaspase-1 was detected at 15 mins after trauma. Accordingly, there were significant increases in the levels of the adaptor protein ASC within 1 h after TBI (Figure 2C), whereas no significant changes in the levels of NLRP1 were observed (Figure 2D). NLRP3 was not detected in lysates from sham and traumatized animals at any time point examined (data not shown) and served as a negative control. These results show that TBI rapidly stimulates expression of NLRP1 inflammasome signaling molecules.
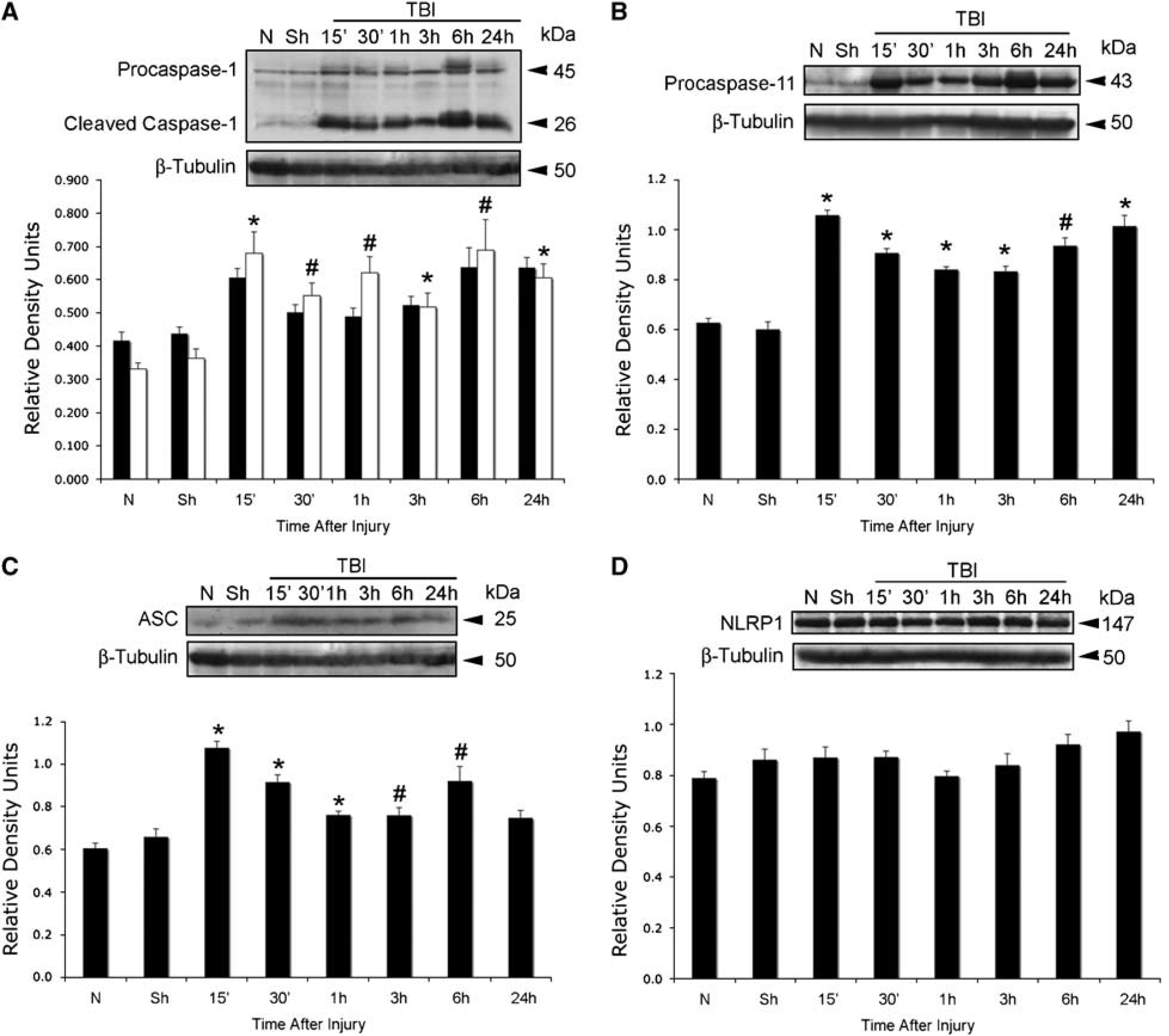
TBI induces activation and processing of caspase-1 and increases levels of ASC and caspase-11, but not NLRP1. Representative immunoblot analysis of (
Traumatic Brain Injury Induces Dramatic Changes in the Composition of the Inflammasome
We next determined whether the increased expression levels of inflammasome proteins lead to formation of an inflammasome complex. To characterize the associations of inflammasome proteins after TBI, coimmunoprecipitations of cortical lysates from injured animals at 4 h after trauma were performed using anti-ASC antibody, anti-NLRP1, and preimmune serum as control (Figure 3). In sham cortices, ASC was immunoprecipitated with anti-ASC, caspase-1, caspase-11, and pannexin-1; however, very low levels of NLRP1 and XIAP were present in this signaling complex (Figure 3). At 4 h after TBI, the composition of the signaling complex changed within the traumatized cerebral cortex. Notably, there was increased association of caspase-1, caspase-11, NLRP1, XIAP, and pannexin 1 with ASC. However, the levels of full-length 53-kDa XIAP protein was cleaved to generate a 25-kDa fragment. Anti-ASC did not immunoprecipitate caspase-3, whereas preimmune serum did not immunoprecipitate the inflammasome-associated proteins, demonstrating antibody specificity, and thus, serving as negative controls. In reciprocal coimmunoprecipitation experiments, anti-NLRP1 immunoprecipitated ASC, caspase-1, caspase-11, pannexin-1, as well as XIAP, but it did not immunoprecipitate caspase-3; thus, providing additional evidence for formation of the inflammasome complex after TBI. Thus, although the levels of NLRP1 did not change at different time points after TBI (Figure 3B), the proportion of NLRP1 recruited into the inflammasome complex increased. These findings indicate that TBI activates the NLRP1 inflammasome that consists of NLRP1, ASC, caspase-1, caspase-11, XIAP, and pannexin-1, leading to activation of caspase-1 and cleavage of XIAP. This finding is the first report that the NLRP1 inflammasome is present in traumatized cortex and the first demonstration that pannexin-1 is part of the NLRP1 inflammasome complex.
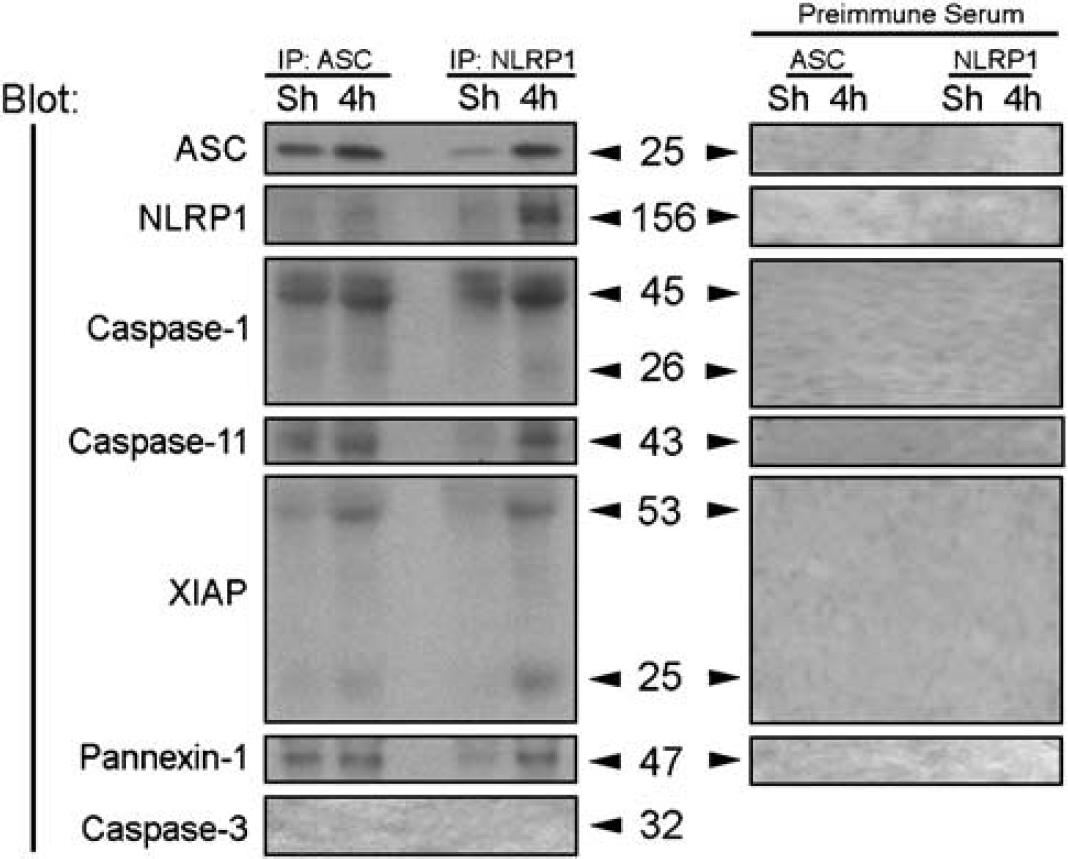
TBI induces association of NLRP1 inflammasome proteins, processing of caspase-1, and cleavage of X-linked inhibitor of apoptosis protein (XIAP). Coimmunoprecipitation with anti-ASC, anti-NLRP1, and preimmune serum of cortical lysates obtained from sham animals (Sh) and traumatized animals at 4 h after TBI. Immunoprecipitates were blotted for ASC, NLRP1, caspase-1, caspase-11, XIAP, pannexin 1, and caspase-3 (control). Anti-NLRP1 and anti-ASC immunoprecipitated NLRP1, processed caspase-1, caspase-11, pannexin 1, and the pro- and cleaved forms of XIAP, thus indicating association of these proteins in a multiprotein complex. Preimmune serum did not immunoprecipitate inflammasome proteins and was used as control.
NLRP1 Inflammasome Proteins are Present in Cortical Neurons, and Traumatic Brain Injury Induces Alterations in Protein Expression Pattern
Figure 4 shows confocal images of the cell-type expression and regional distribution of NLRP1 inflammasome proteins in cortical neurons of sham animals and proteins near the cortical injury epicenter at 4 h after injury. Sections were stained for caspase-1, caspase-11, ASC, NLRP1 (red), and the neuronal marker MAP2. Caspase-1 immunoreactivity was seen in MAP2 positive cells, indicating that caspase-1 is expressed in neurons in the cerebral cortex of sham animals. Intense caspase-1 immunoreactivity was seen in the nucleus (arrow), and patchy staining was present in the cell cytoplasm (arrow) and processes. In contrast, caspase-11 immunoreactivity showed diffuse punctate staining confined to the neuronal soma and processes. Intense ASC and NLRP1 staining was detected in the soma of cortical neurons and exhibited a patchy distribution pattern (arrows), whereas weak NLRP1 immunoreactivity was detected in the nucleus.
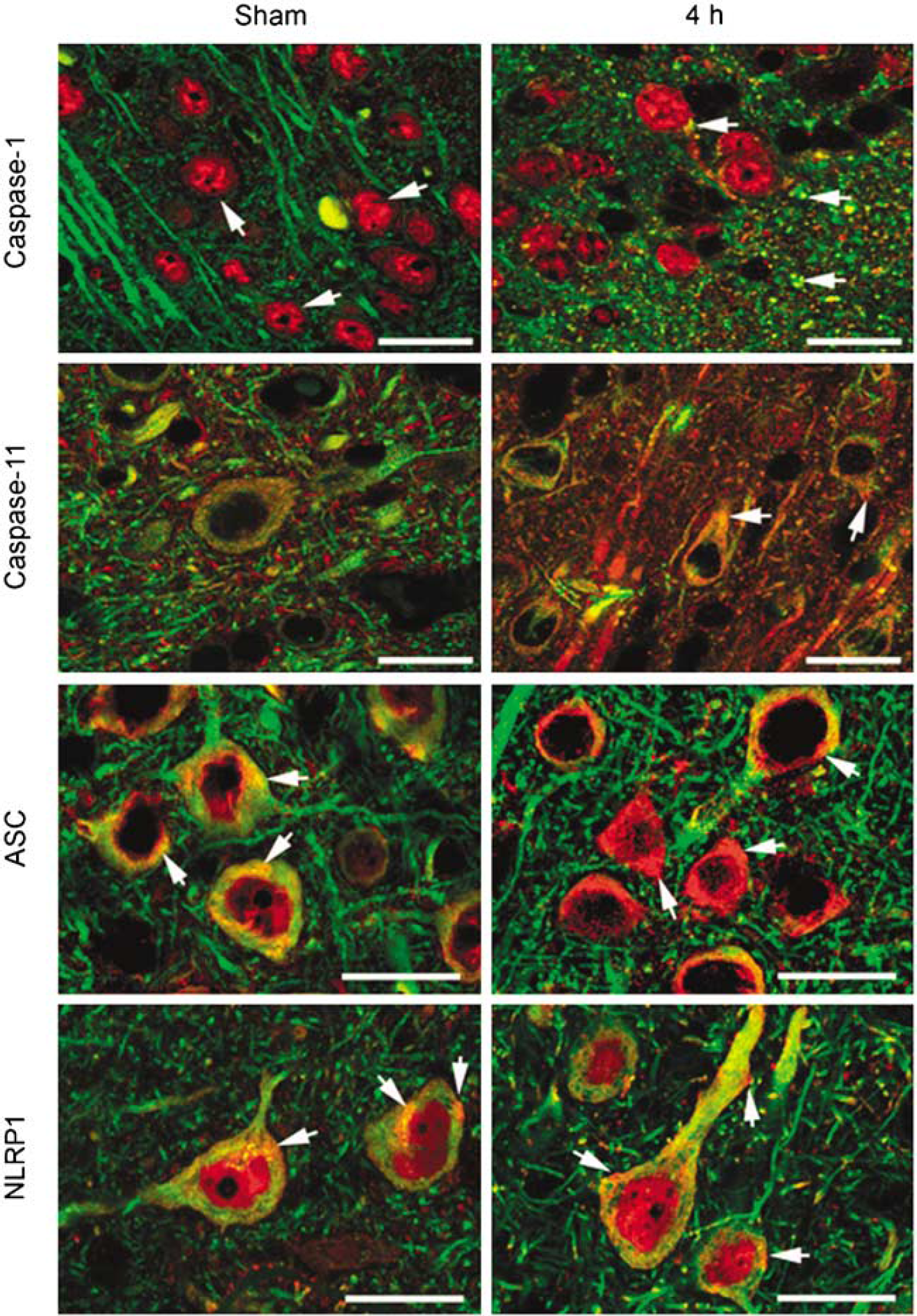
NLRP1 inflammasome proteins are present in cortical neurons and TBI induces alterations in protein expression patterns. Confocal images of cortical neurons in sham and injured brains at 4 h posttrauma. Sections were stained for caspase-1, caspase-11, ASC, NLRP1 (red), and the neuronal marker MAP2. In sham animals, caspase-1 immunoreactivity was seen in the nucleus (arrow). By 4 h after injury, increased caspase-1 staining was present in neuronal nuclei and patchy staining was present in the cell cytoplasm and processes near the plasma membrane. Caspase-11 immunoreactivity showed diffuse punctate staining confined to the neuronal soma and processes (arrow). Increased caspase-11 staining was present by 4 h posttrauma in the neuronal soma in a patchy distribution (arrow). Intense ASC and NLRP1 staining was detected in the soma of cortical neurons and exhibited a patchy pattern in the cytoplasm (arrow). Both inflammasome proteins showed increased expression as evidenced by intense patchy staining located near or associated with the plasma membrane (arrows) by 4 h posttrauma. Bar = 20 μm.
Moderate FPI resulted in altered staining patterns of inflammasome proteins in cortical neurons (Figure 4). At 4 h after injury, increased caspase-1 immunoreactivity was present in neuronal nuclei, whereas intense caspase-1 staining was seen in the cell cytoplasm as large patches (arrow) near the plasma membrane. Increased caspase-11 staining was present in the neuronal soma that was localized in a patchy distribution (arrow). A more striking alteration was observed in the immunostaining of ASC and NLRP1 after TBI. By 4 h, immunoreactivity of both inflammasome proteins was markedly enhanced, and intense patchy staining was seen in the neuronal soma near or associated with the plasma membrane (arrows). As shown earlier, XIAP was present in the perinuclear region and cell processes of cortical neurons and TBI induced alterations in the expression pattern (Lotocki et al, 2003). The cellular distribution and location of NLRP1 inflammasome proteins near the plasma membrane of neurons after trauma is consistent with their role in the processing and secretion of IL-1β. Anti-ASC and anti-NLRP1 antibody specificity was evaluated by preabsorption of antiserum with immunogen peptides to remove specific antibody binding (de Rivero Vaccari et al, 2008). Antigen-depleted antiserum did not stain sections of sham and traumatized brains and served as a negative control (de Rivero Vaccari et al, 2008). Of importance is the fact that the intensity and pattern of inflammasome protein expression in neurons was altered by TBI and is consistent with the idea that neurons process and secrete IL-1β through activation of the inflammasome complex.
ASC Neutralization Reduces Traumatic Brain Injury-induced Activation and Processing of Caspase-1 and X-Linked Inhibitor of Apoptosis Protein Cleavage
To dissect the contribution of the NLRP1 inflammasome to TBI-induced inflammation, we blocked the activity of the NLRP1 inflammasome with antibodies against the inflammasome adaptor protein ASC. Antibody treatment was started immediately after trauma. One group of animals received 15 μg of anti-ASC delivered intracerebroventricularly (Figure 5, ASC). Control groups received a similar treatment regimen, but using IgG of the same isotype corresponding to anti-ASC. Cortices were removed at 24 h after treatment and lysates were prepared and immunoblotted for caspase-1 and XIAP (Figure 5). As shown earlier, antiinflammasome antibodies cross the blood-brain barrier and are taken up by neurons in the traumatized CNS (Abulafia et al, 2009; de Rivero Vaccari et al, 2008). Moreover, neutralization of ASC after TBI interfered with inflammasome signaling significantly reduced processing of caspase-1, IL-1β and decreased XIAP cleavage (Figure 5).
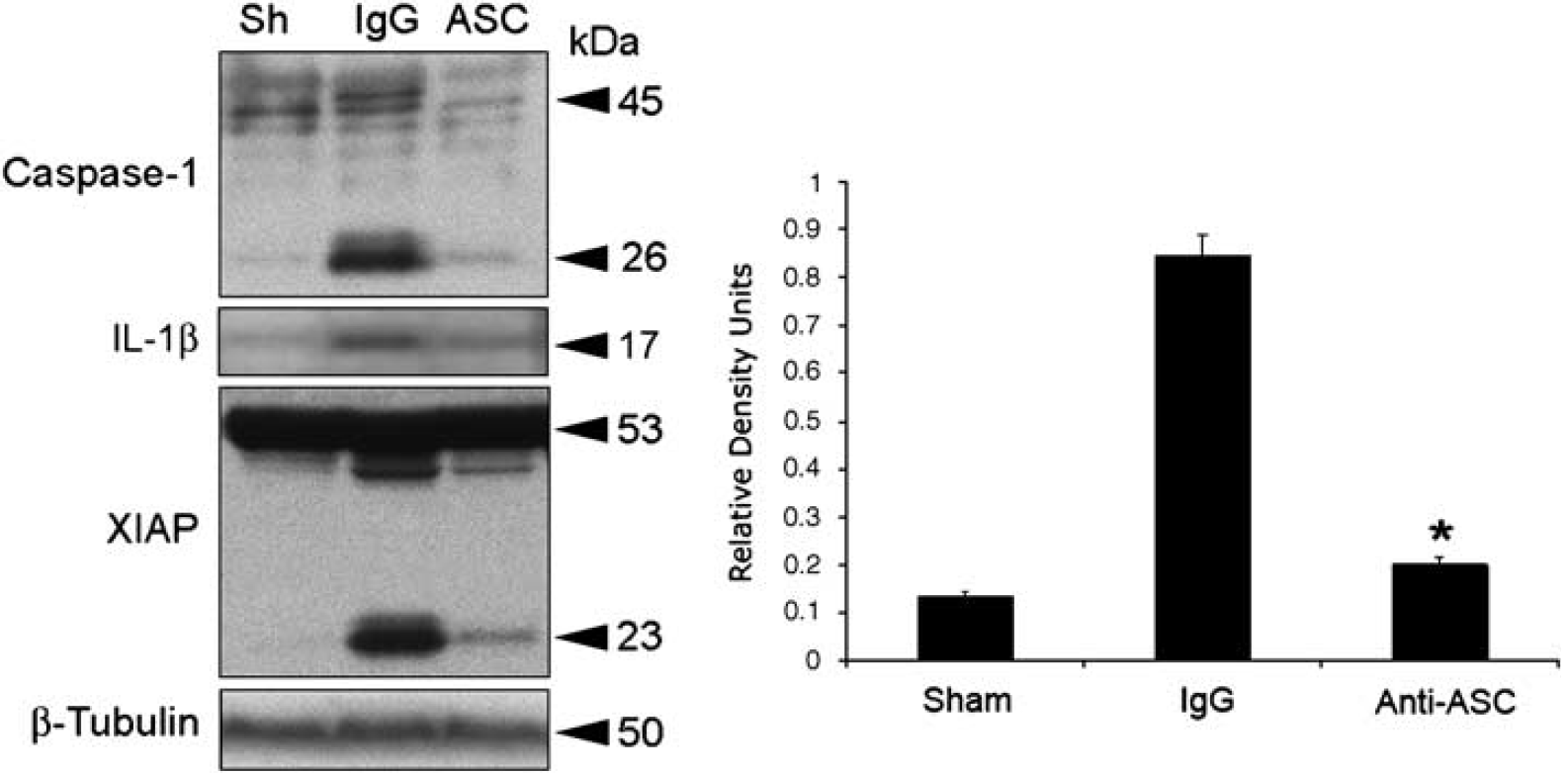
ASC neutralization decreases TBI-induced activation and processing of caspase-1, interleukin-1β (IL-1β) and X-linked inhibitor of apoptosis protein (XIAP) cleavage. Representative immunoblots of injured cortices from animals subjected to TBI and treated intracerebroventricularly with antibodies to ASC (ASC), IgG controls (IgG), or left untreated (Sh) after injury. Animals were killed 24 h after treatment. Treatment resulted in inhibition of inflammasome activation as detected by a decrease in the processing of procaspase-1, IL-1β, and cleavage of XIAR Anti-ASC treatment also resulted in a significant reduction in caspase-1 processing (right panel). *P < 0.05 versus sham, **P < 0.05 versus IgG.
ASC Neutralization Decreases Brain Contusion Volume
To determine whether inflammasome signaling was causally linked to tissue damage during TBI in vivo, we blocked the activity of ASC with neutralizing antibodies and measured the lesion volumes at 3 days after injury. Rats were subjected to moderate FPI, treated immediately with 15 μg of anti-ASC intracerebroventricularly, and then injected with 50 μg of the anti-ASC antibody intraperitoneally at 24 and 48 h after injury. Another group of rats was treated with IgG and served as control. Figure 6 shows representative coronal brain sections of the lesion at 3 days after trauma. Brains from animals treated with anti-ASC after the FPI showed significantly reduced contusion volume compared with vehicle-treated rats.
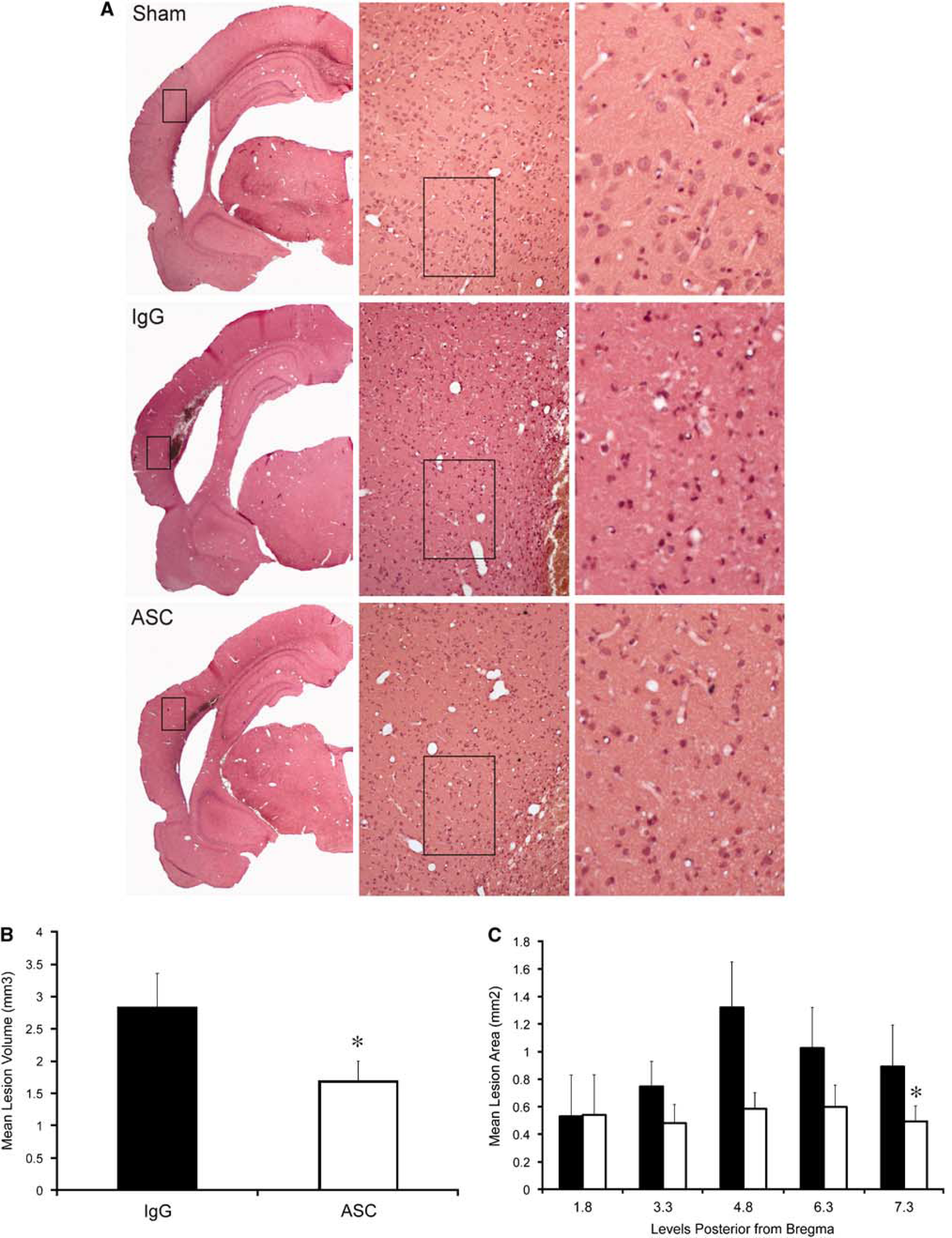
ASC neutralization decreases brain contusion volume. (
Discussion
In this study, we have shown for the first time a role for the NLRP1 inflammasome system in the inflammatory response induced by TBI. Our data show that moderate fluid-percussion brain injury initiates activation of the NLRP1 inflammasome, resulting in processing of caspase-1 and upregulation of caspase-11 and the adaptor protein ASC, leading to maturation of IL-1β. The NLRP1 inflammasome was detected in cerebral cortical neurons in the normal and traumatized brain. The neuronal NLRP1 inflammasome is a multiprotein complex that consists of inflammatory caspase-1, caspase-11, NLRP1, ASC, the inhibitor of apoptosis protein XIAP, and pannexin 1. Neutralization of ASC interferes with NLRP1 inflammasome signaling and leads to a significant reduction in processing of caspase-1, resulting in a significant reduction in contusion volume. These results suggest a protective response by this novel antiinflammatory treatment. Thus, the NLRP1 inflammasome plays an important role in innate CNS immunity after TBI.
Increased production of cytokines of the IL-1 family, such as IL-1β, is well documented in human TBI and in animal models of brain injury, providing clear evidence for a pivotal role of this cytokine in triggering TBI-induced inflammatory processes (Bhat et al, 1996; Dinarello, 2004, 2005a, b, 2006). IL-1β and IL-18 are potent mediators of inflammation and initiate and/or amplify a wide variety of effects associated with innate immunity, host responses to tissue injury, and microbial invasion (Bhat et al, 1996; Dinarello, 2004, 2005a, b, 2006). On cleavage of their proforms by caspase-1, these cytokines become active and are secreted. Thus, caspase-1 activity is critical for the inflammatory response. The generation of proinflammatory cytokines induces the subsequent recruitment of circulating immune cells that may amplify the immune response at the site of injury.
Assembly of the inflammasome depends on NLR family members, such as NLRPs. Our data show that the inflammasome in cortical neurons is a protein complex containing NLRP1 as a scaffolding protein that activates caspase-1 to promote IL-1β maturation. Although the total levels of NLPR1 in lysates did not change significantly after TBI (Figure 2), the proportion of NLRP1 that forms the inflammasome increases (Figure 3). The NLRP1 inflammasome in cortical neurons is similar in composition to the NLRP1 inflammasome in spinal cord motor neurons (de Rivero Vaccari et al, 2008). In addition, we report that the NLRP inflammasome contains pannexin 1. Recent evidence indicates that pannexin 1 plays a crucial role in inflammation, explicitly through its association with the P2X7 receptor (Kanneganti et al, 2007; Pelegrin and Surprenant, 2006). The pannexin 1 channel as well as the inflammasome can be activated by high extracellular K+ (unpublished data). After TBI, high concentrations of K+ ions required for channel activation may occur in the vicinity of dying cells and the subsequent release of cytoplasmic contents into a confined space. Alternatively, efflux of K+ through pannexin 1 channels could create a locally high K+ concentration sufficient to activate the channel. Thus, TBI-induced high extracellular K+ may serve as a stimulus to activate pannexin 1 in the neuronal NLRP1 inflammasome and may serve as a novel upstream target for therapeutic interventions targeting TBI-induced inflammatory responses. Additional studies with pannexin-1 deficient animals are needed to determine the precise role of pannexin 1 in TBI-induced inflammasome signaling to test this hypothesis.
Although several studies have investigated the role of NLR family members using biochemical or genetic approaches, data on the cell types and tissues that express these proteins are still lacking. Our recent work shows that the NLRP1 inflammasome is present in spinal cord neurons in rats (de Rivero Vaccari et al, 2008) and cortical neurons in mice (Abulafia et al, 2009) and plays an important role in the innate CNS inflammatory response after experimental spinal cord injury (SCI) and thromboembolic stroke. In this study, we show that the NLRP1 inflammasome is also present in cortical neurons of the rat. A recent report using monoclonal antibodies against NLRP1 and NLRP3 revealed distinct tissue distribution patterns of these NLR family members, where NLRP1 is expressed in human neurons (Kummer et al, 2007). These findings indicate that microglial cells, which share properties with tissue macrophages, are not the main source of IL-1β in the brain. Indeed, IL-1β has been implicated in the pathogenesis of several neurologic diseases including TBI, Alzheimer's disease, epilepsy, Parkinson's disease, and stroke (Allan et al, 2005). Given the large number of NLR family members and their distinct, but separate, expression profiles in tissue, we hypothesize that CNS cells contain a number of yet undiscovered inflammasomes that contribute to a site-specific role in the inflammatory response.
Specific IL-1 antagonizing drugs (e.g., IL-1Ra or anakinra) have been successfully introduced in clinical treatments of autoinflammatory diseases, such as rheumatoid arthritis or gout (Braddock and Quinn, 2004; Dinarello, 2005a; Fisher et al, 1994; Liao et al, 1984; So et al, 2007), and recent results of a phase II study of IL-1Ra treatment in stroke patients are encouraging (Emsley et al, 2005). Moreover, inhibitors of caspase-1 have been shown to attenuate mechanical neuronal injury (Fink et al, 1999) and improve survival of transplanted neurons in rats (Marchionini et al, 2004). However, despite the prominent role of IL-1β in acute brain injury, there is a paucity of specific drugs that block inflammation and improve functional outcomes after TBI. Our recent work on SCI (de Rivero Vaccari et al, 2008) and ischemic stroke (Abulafia et al, 2009) shows that neurons are a source of IL-1β in the CNS and provides in vivo evidence for a direct link between activation of the innate immune response and secondary injury. We also show that blocking IL-1β activation after injury results in tissue sparing and functional improvement. In this study, we extended this approach and tested a novel upstream antiinflammatory therapeutic strategy that involves inhibition of caspase-1 activation by blocking inflammasome signaling. We show that anti-ASC treatment blocks the innate CNS inflammatory response, which is critical for brain homeostasis and injury-induced inflammatory processes. Thus, neutralizing antibodies against the inflammasome are successful in inhibiting inflammation in multiple CNS injury models (Abulafia et al, 2009; de Rivero Vaccari et al, 2008).
Treatment with the neutralizing antibody was also tested on its effect on contusion volume in the present model. In rats receiving anti-ASC, a significant difference was shown between the treated and nontreated groups in terms of lesion volume. The fact that anti-ASC treatment in the present model reduced caspase-1 activation and improved tissue sparing emphasizes the importance of this targeted treatment strategy on limiting inflammatory processes that contribute to pathogenesis of TBI. Our findings suggest a novel upstream target for therapeutic interventions for the inhibition of TBI-induced inflammatory responses. We show that the NLRP1 inflammasome in neurons of the cerebral cortex after TBI is critical for the activation of the innate CNS inflammatory response. As the innate immune response impacts on outcomes of various neurodegenerative diseases, the continued discovery of novel treatment strategies that interfere with the activation of the inflammatory response after CNS injury remains an important research goal.
Footnotes
Acknowledgements
This work was supported by a grant from NIH/NINDS, NS30291 and, NN42133.