
Abstract
Traumatic brain injury (TBI) activates the NALP1/NLRP1 inflammasome, which is an important component of the early innate inflammatory response to injury. We investigated the influence of therapeutic hypothermia on inflammasome activation after TBI. Adult male Sprague–Dawley rats were subjected to moderate fluid percussion brain injury. Temperature manipulation (33°C or 37°C) was initiated 30 minutes after TBI and maintained for 4 hours. At 4 or 24 hours after TBI, traumatized cortex and hippocampus were prepared for immunoblot or immunohistochemical analysis. In the normothermic groups, caspase-1, caspase-11 and expression of the purinergic receptor P2X7 increased at 24 hours after TBI. Posttraumatic hypothermia lead to decreased expression of these proteins at 24 hours compared with normothermic levels. Immunocytochemical studies showed that posttraumatic hypothermia also decreased caspase-1 staining in cerebral cortical neurons compared with normothermic TBI. Cultured cortical neurons subjected to stretch injury demonstrated significant secretion of caspase-1 into the culture medium and caspase-3 activation, both results reduced by hypothermic treatment. Posttraumatic hypothermia decreases inflammasome signaling in neurons and reduces the innate immune response to TBI at 24 hours after injury. Therapeutic hypothermia may protect the injured central nervous system by targeting the detrimental consequences of the innate immune response to injury.
Introduction
Traumatic brain injury (TBI) is a complex and devastating clinical condition mediated at least in-part by proinflammatory cytokines that produce neuronal loss, axonal destruction, and demyelination during the secondary injury cascade (Bramlett and Dietrich, 2007; Helmy et al, 2011; Ziebell and Morganti-Kossmann, 2010). Increased production of cytokines of the interleukin-1 (IL-1) family, such as IL-1β, is well documented, providing clear evidence for a pivotal role of this cytokine in triggering TBI-induced inflammatory process (Ciallella et al, 2002; Hutchinson et al, 2007; Kinoshita et al, 2002; Knoblach and Faden, 2000; Shohami et al, 1994). These cellular and molecular changes represent therapeutic targets for reducing the devastating consequences of TBI. Indeed, improved behavioral and structural protection has been noted using various pharmacological approaches aimed at reducing the inflammatory response to trauma (de Rivero Vaccari et al, 2009; Lloyd et al, 2008; Ziebell and Morganti-Kossmann, 2010).
Moderate hypothermia initiated after a traumatic insult has been reported by several laboratories to improve behavioral and histopathological outcomes (Clifton et al, 1992; Dietrich et al, 1994; Dietrich and Bramlett, 2010; Dixon et al, 1998; Koizumi and Povlishock, 1998). The mechanisms underlying the beneficial effects of posttraumatic hypothermia are multifactorial and include excitotoxicity, calcium-dependent intercellular signaling, apoptosis, and edema formation (Dietrich and Bramlett, 2010; Yenari and Han, 2012). In addition, hypothermia has been reported to attenuate various inflammatory events including proinflammatory gene expression and protein levels when compared with normothermic TBI in some (Chatzipanteli et al, 2000; Goss et al, 1995; Kinoshita et al, 2002; Lotocki et al, 2006; Marion et al, 1997; Vitarbo et al, 2004; Whalen et al, 1997) but not all studies (Buttram et al, 2007; Truettner et al, 2005). Thus, a current area of research is directed to the continued investigation of the cellular and molecular events associated with the beneficial effects of hypothermia on posttraumatic inflammatory processes.
The inflammasome is a multiprotein complex of the innate immune response involved in the activation of caspase-1 after brain injury (de Rivero Vaccari et al, 2009). Upon inflammasome activation, caspase-1 cleaves pro-IL-1β into its active form. Interleukin-1β mediates inflammation by activating or amplifying host responses to tissue injury and infections (Dinarello, 2005; Finsen and Owens, 2011). Our laboratory has previously provided biochemical and immunocytochemical data showing abnormal inflammasome activation in brain cells after TBI, spinal cord injury, and focal cerebral ischemia (Abulafia et al, 2009; de Rivero Vaccari et al, 2008; de Rivero Vaccari et al, 2009). Treatment with selective antibodies that target inflammasome activation and caspase-1 expression reduce some of the detrimental consequences of these acute insults (de Rivero Vaccari et al, 2009). Thus, innate inflammation may be an important therapeutic target for future investigations.
The effects of therapeutic hypothermia on patterns of inflammasome activation have not been reported. The purpose of this study was to test the hypothesis that early cooling after parasagittal fluid percussion brain injury would reduce activation of several components of the inflammasome in brain regions vulnerable to this traumatic insult. We also used an in-vitro cell stretch injury approach to determine if evidence for inflammasome activation could be demonstrated in neurons and whether hypothermia also reduced that response to cellular injury. We report for the first time that inflammasome activation in neurons is modulated by moderate hypothermia in the cerebral cortex after TBI.
Materials and methods
Traumatic Brain Injury
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Miami carried out according to the NIH Guide for the Care and Use of Laboratory Animals. Male Sprague–Dawley rats (250 to 350 g) were subjected to parasagittal fluid percussion injury as previously described in de Rivero Vaccari et al (2009). In this study, rats underwent moderate head injury ranging from 1.7 to 2.2 atmospheres. Animals groups were as follows: Sham (n=6), TBI-Normothermia-4 hours (n=6), TBI-Hypothermia-4 hours (n=6), TBI-Normothermia-24 hours (n=6), and TBI-Hypothermia-24 hours (n=6). Catheters were placed within the tail artery for physiological monitoring of blood pressure and arterial blood gases. The brain temperature was indirectly monitored with a thermistor probe inserted into the temporalis muscle and rectal temperature was also measured with a rectal probe. Readings were taken 30 minutes before trauma and were maintained at a constant level of ∼37°C.
In the hypothermic groups, temperature manipulation (33°C) was initiated 30 minutes after TBI, achieved within 15 minutes and maintained using cooled air blown directly onto the skull and heating lamps for a period of 4 hours followed by a slow rewarming period. The normothermic groups were maintained at a constant level of ∼37°C throughout the procedure. Following these procedures, the animals were returned to their home cages with food and water available ad libitum. At the end of the temperature manipulation or at 24 hours after TBI, rats were killed. Sham animals were subjected to all procedures except for the actual insult.
Immunoblotting
Tissue samples were snap-frozen in liquid nitrogen and stored at −80°C until the time of assay. A 2-mm section of cortex was homogenized in extracton buffer (20 mmol/L Tris-HCl, pH: 7.5, 150 mmol/L NaCl, 1% Triton X-100; 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethyleneglycoltetraacetic acid, 2.5 mmol/L pyrophosphate, 1 mmol/L β-glycerophosphate) containing protease and phosphatase inhibitor cocktails (Sigma, St Louis, MO, USA). Proteins were resolved in 4% to 20% Tris-TGX Criterion precasted gels (Bio-Rad, Hercules, CA, USA) transferred to polyvinylidene difluoride membranes (Bio-Rad) placed in blocking buffer [phosphate-buffered saline, 0.1% Tween 20, and 0.4% I-Block (Applied Biosystems, Austin, TX, USA)]. Membranes were incubated for 1 hour with primary antibodies as following: monoclonal antibody to caspase-1 (1:1,000; Imgenex, San Diego, CA, USA), caspase-11 antibody (Novus Biologicals, Littleton, CO, USA), rabbit anti-Rattus novegicus, affinity-purified antibody ASC (Santa Cruz, Santa Cruz, CA, USA), antibody to P2X7 Receptor (extracellular) (Alomone Labs, Jerusalem, Israel), cleaved IL-1β antibody (1:5,000; Cell Signaling Technology, Beverly, MA, USA), and then appropriate secondary horseradish peroxidase (HRP)-linked antibodies (Cell Signaling Technology). Visualization of signal was enhanced by chemiluminescence using a phototope-HRP detection kit (Cell Signaling Technology). To control for protein loading, immunoblots were stripped with Restore, Western blot stripping buffer (Pierce, Rockford, IL, USA), and blotted for β-actin using monoclonal anti-β-actin antibody (1:5,000; Sigma-Aldrich, St Louis, MO, USA). Quantification of band density was performed using UN-SCAN-IT gel 5.3 software (Silk Scientific, Orem, UT, USA), and data were normalized to β-actin.
Perfusion Fixation and Immunohistochemistry
Animals were anesthetized with isoflurane (3%) in 30% oxygen to 70% nitrous oxide gas mixture and perfused with 500 mL of 4% paraformaldehyde. The brains were removed and placed in 4% paraformaldehyde at 4°C for 20 hours. Then they were transferred to 20% sucrose in 0.1 M phosphate-buffered saline and stored at 4°C until cryostat sectioning (Leica SM 2000R Sliding Microtome, Buffalo Grove, IL, USA). Sections (50 μm) were blocked by treatment with normal goat serum (Vector Laboratories, Burlingame, CA, USA). To determine the precise cellular distribution of caspase-1, sections were double stained with the neuronal marker NeuN (Cell Signaling) and anti-caspase-1 (1:500, Millipore, Billerica, MA, USA) as described (de Rivero Vaccari et al, 2009). Controls using an irrelevant antibody of the same isotype were run in parallel to evaluate antibody specificity. Immunostained brain sections of rats at 4 hours were examined with a Zeiss laser scanning confocal microscope (Zeiss, Inc., Thornwood, NY, USA).
In-Vitro Stretch Injury
Primary cortical neurons were grown on poly-
Statistical Analysis
Data are expressed as mean±standard error of the mean (±s.e.m.). Physiological parameters were analyzed with two-way repeated measures analysis of variance. Other outcome measures were analyzed by one-way followed by appropriate posthoc analysis using Tukey's test or one-tailed t-test. P values of significance used were ∗P<0.05.
Results
Physiology
The physiological parameters were all within the normal ranges during the surgical procedures. No significant differences were found among the study groups except for the temperature values between the hypothermia animals and normothermic and sham groups (P<0.05; Table 1).
Physiological variables
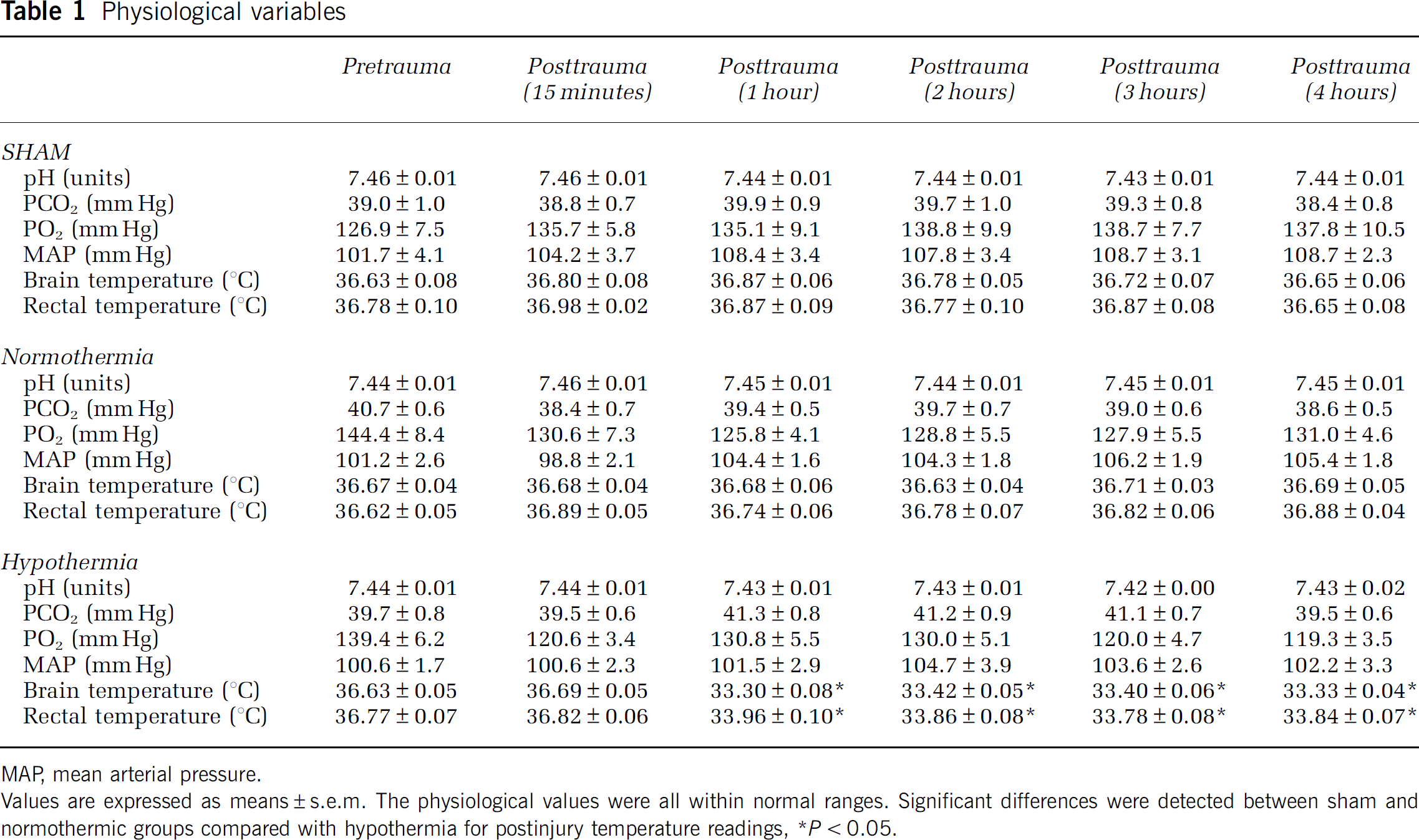
MAP, mean arterial pressure.
Values are expressed as means±s.e.m. The physiological values were all within normal ranges. Significant differences were detected between sham and normothermic groups compared with hypothermia for postinjury temperature readings, ∗P<0.05.
Hypothermia Decreases Inflammasome Activation in the Cortex but not the Hippocampus After Traumatic Brain Injury
In order to investigate the effects of hypothermia on IL-1β processing after TBI, we performed immunoblot analysis of cortical (Figure 1A) and hippocampal (Figure 1B) lysates from sham-operated animals, normothermic animals and hypothermic-treated animals at 4 and 24 hours after TBI. Sham animals were pooled since no difference was found between the two temperature groups. As shown in Figure 1, TBI significantly increases IL-1β processing in the cortex at 24 hours after TBI, whereas hypothermia following TBI significantly reduces IL-1β processing. These results are consistent with previous findings showing that hypothermia lowers the levels of IL-1β after TBI (Kinoshita et al, 2002; Truettner et al, 2005). In contrast, no statistical significant changes in IL-1β protein expression were detected in the hippocampus at any of the time points tested.
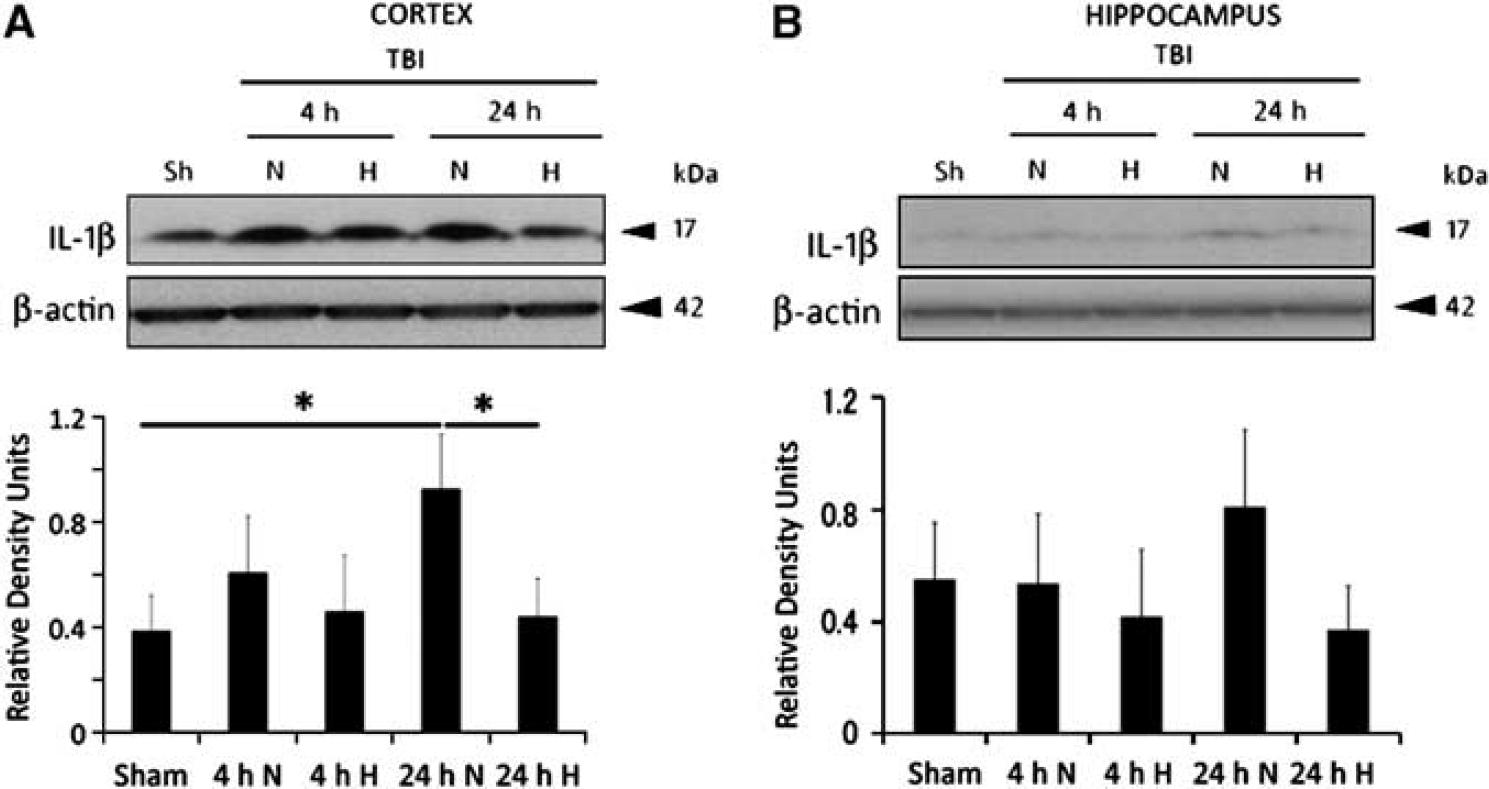
Hypothermia reduces interleukin (IL)-1β processing in the injured cortex at 24 hours after traumatic brain injury (TBI). Immunoblot analysis of IL-1β in lysates of cortex (
Furthermore, to investigate the effects of hypothermia on inflammasome activation, we performed immunoblot analysis of traumatized cortex (Figure 2) and hippocampus (Figure 3) from sham-operated animals, normothermic animals and hypothermic animals at 4 and 24 hours after TBI. In traumatized cortices, caspase-1 (Figure 2A) and caspase-11 (Figure 2B) levels from normothermic animals were higher at 24 hours after TBI, whereas the hypothermic group demonstrated a decrease at 24 hours after trauma when compared with the 24-hour normothermia group. The protein levels of caspase-1 and caspase-11 were not found to change significantly at 4 hours on both, the normothermic and hypothermic groups.
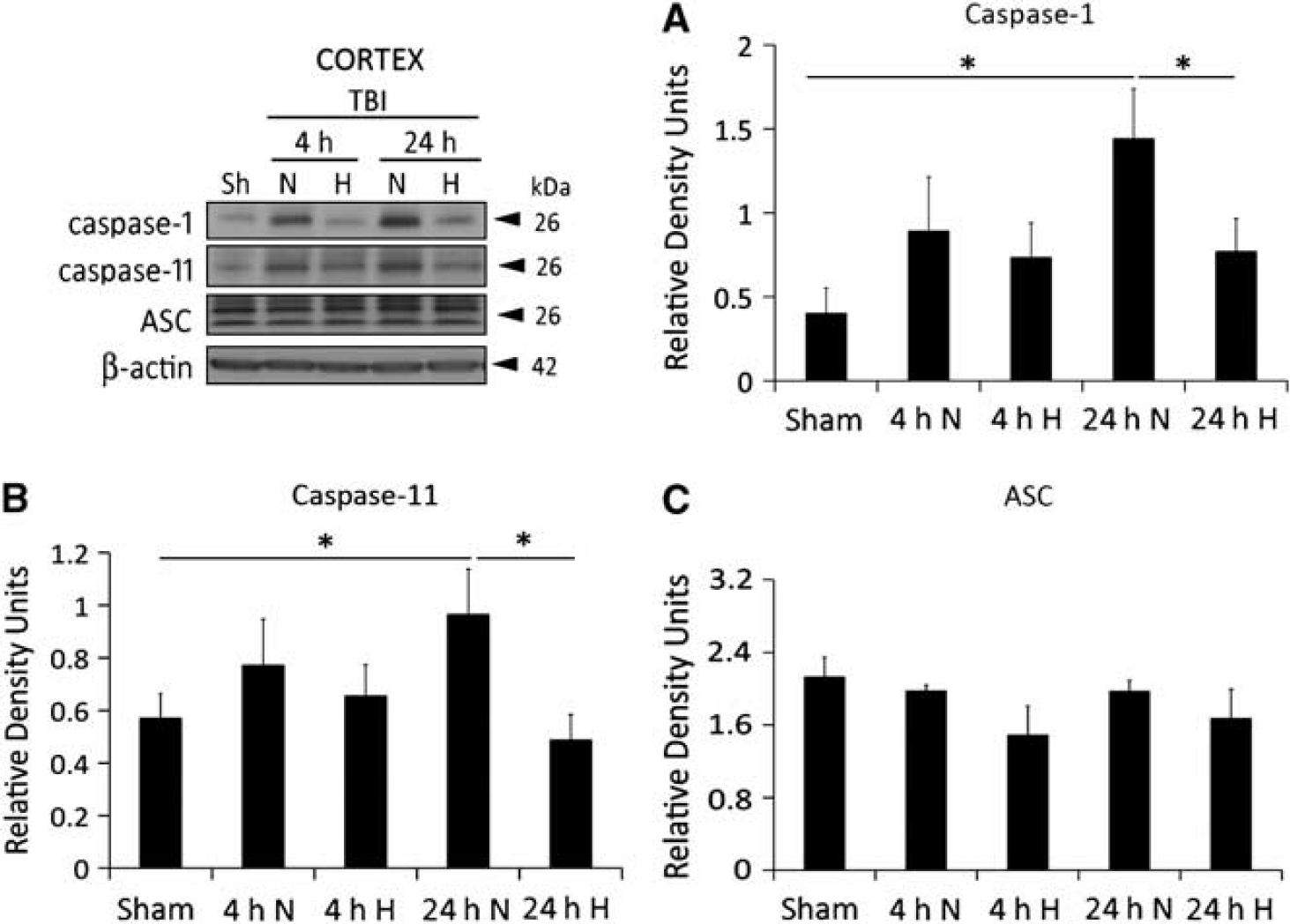
Hypothermia reduces caspase-1 and caspase-11 processing independently of ASC in the injured cortex at 24 hours after traumatic brain injury (TBI). Representative immunoblots of cortical lysates of sham-operated animals (sh), normothermic group (N), and hypothermic group (H) at 4 and 24 hours after injury. Representative immunoblots of caspase-1 (
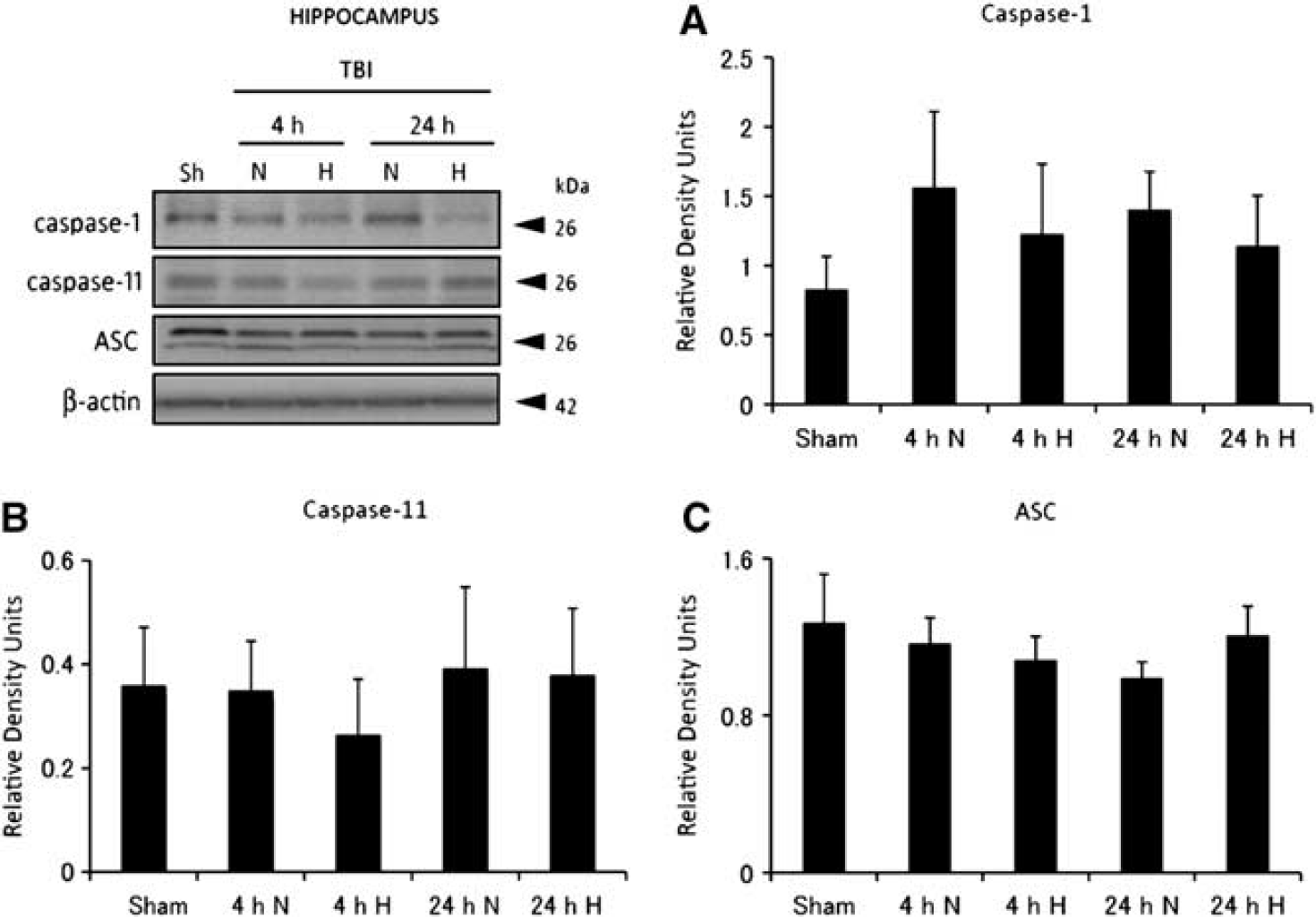
Hypothermia does not affect inflammasome protein expression in the injured hippocampus. Representative immunoblots of lysates of hippocampus of sham-operated animals (sh), normothermic group (N), and hypothermic group (H) at 4 and 24 hours after injury. Representative immunoblots of caspase-1 (
To test if hypothermia effects protein levels of caspase-1 in uninjured brain, we analyzed hypothermic sham and normothermic sham animals. Accordingly, there was no significant difference between the caspase-1 expression of hypothermic sham animals and normothermic sham animals and therefore sham data were pooled.
In addition, no significant difference was detected in the inflammasome adaptor protein ASC (Figure 2C) in cortical samples from different groups of animals, and in the injured hippocampus we found no statistical differences in the levels of either caspase-1, caspase-11, and ASC among the groups tested (Figure 3).
Hypothermia Decreases Capase-1 Immunoreactivity in Cortical Neurons After Traumatic Brain Injury
To determine the cellular effects of hypothermia on caspase-1 expression and cell type distribution after injury, we performed immunohistochemical analysis followed by confocal microscopy in the brain sections of sham and injured (normothermia and hypothermia) animals. Since the effects of hypothermia on the inflammasome are only detected in the cortex and not the hippocampus, we focused our studies on a region of the ipsilateral cerebral cortex corresponding to the pericontusional site of injury. Figure 4 shows confocal images of the cell type expression and regional distribution of caspase-1 in neurons in the cortex of sham, normothermic and hypothermic animals at 24 hours after injury. Sections were stained for caspase-1 (red) and the neuronal marker NeuN (green). In sham animals, caspase-1 immunoreactivity was seen in the cytoplasm of neurons. In normothermic animals, caspase-1 staining was increased at 24 hours after injury. In contrast, less caspase-1 staining was seen in neurons in the hypothermia group. These findings are consistent with our data of decreased inflammasome activation in hypothermia-treated animals in the cortex (Figure 2).
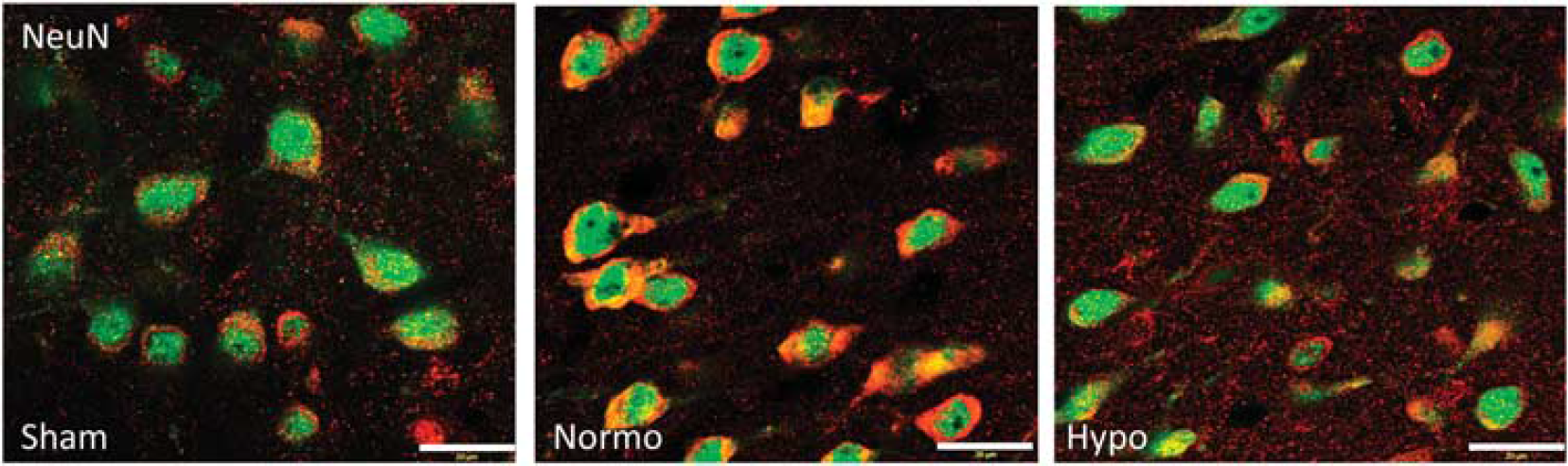
Hypothermia reduces caspase-1 expression in neurons in the injured cortex at 24 hours after traumatic brain injury (TBI). Confocal images show cortex neurons of sham, normothermic group, and hypothermic group. Sections were stained for neurons (green) and caspase-1 (red).
Hypothermia Prevents Traumatic Brain Injury-Induced Increases in Cortical P2X7R
P2X7R has been shown to interact with the inflammasome and regulate inflammasome regulation in cells of the CNS (central nervous system) (Silverman et al, 2009; Mawhinney et al, 2011). To test the effects of hypothermia on P2X7R expression, cortical lysates of sham, normothermic and hypothermic animals after TBI were resolved by immunoblotting. In traumatized cortices, P2X7R levels from normothermic animals significantly increased at 24 hours after TBI compared with sham animals, whereas hypothermia following TBI significantly reduces the level of P2X7R expression (Figure 5).
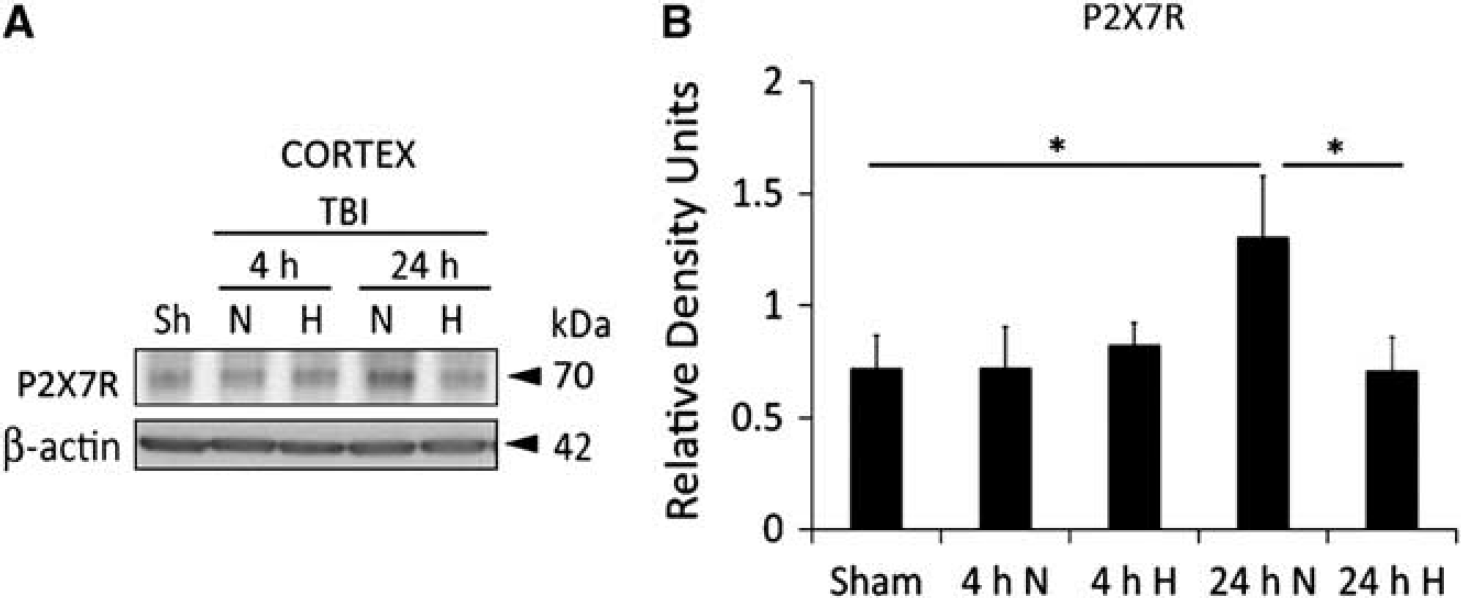
Hypothermia reduces P2X7R expression in the cortex after TBI. (
Hypothermia Prevents Caspase-1 and Caspase-3 Expression in Injured Cortical Neurons
In order to investigate the effects of hypothermia on neurons in vitro, we performed stretch injury and a 2-hour temperature manipulation on cultured cortical neurons followed by immunoblotting. Interestingly, the protein levels of the active fragment of caspase-1 (p26) were lower in the culture media of the hypothermia-treated cultures when compared with the normothermic group (Figure 6A). These findings are consistent with our in-vivo results showing decreased caspase-1 activation after TBI with hypothermia treatment. In contrast, no significant effect of hypothermia treatment was seen in cleaved caspase-1 levels in cell lysates (data not shown).
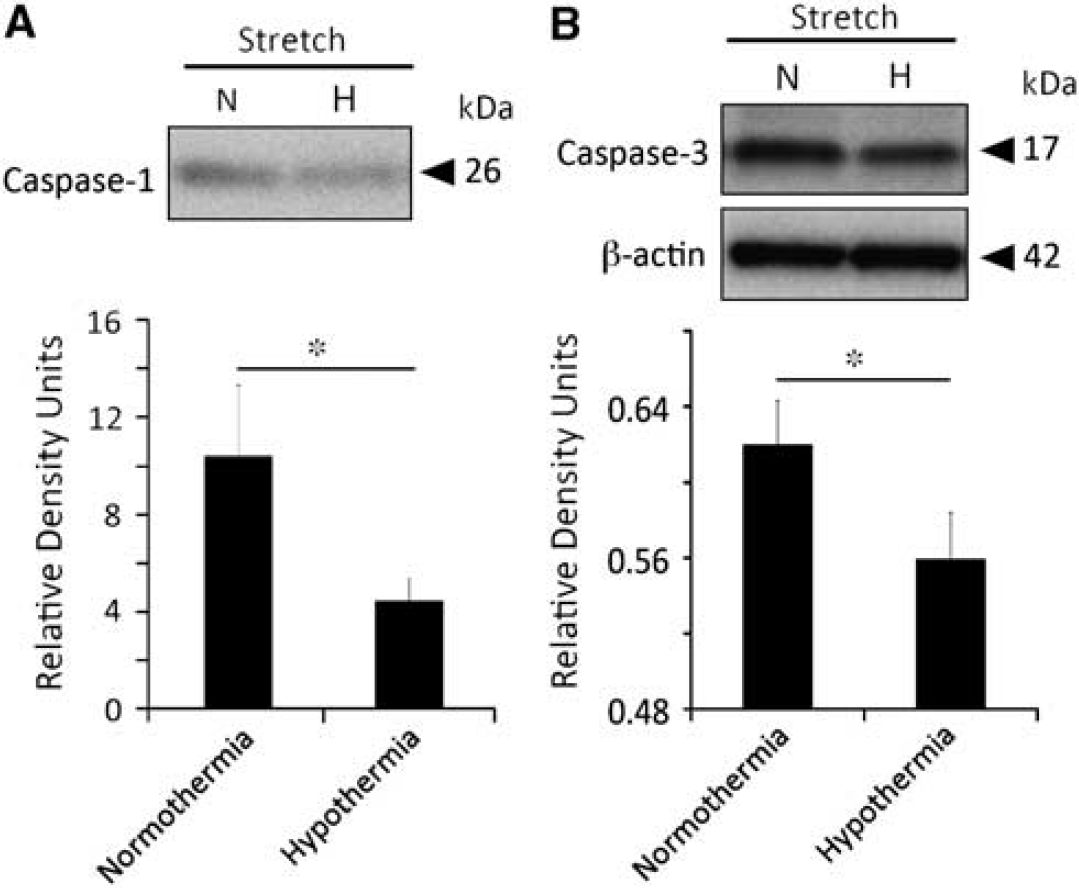
Hypothermia reduces active caspase-1 expression in the media of stretched injured neurons and caspase-3 activation is decreased by hypothermia in stretched injured neurons. Immunoblot analysis of caspase-1 in the media of cortical neurons grown in culture (
In addition, we analyzed the protein levels of active caspase-3 to determine if hypothermia in culture protects cells from caspase-3-mediated cell death. Accordingly, the protein levels of active caspase-3 (p17) in the cell lysates were lower in the hypothermia group when compared with normothermia (Figure 6B). These data are consistent with the previous report that demonstrates hypothermia attenuates caspase-3 in the traumatized cortex in vivo (Lotocki et al, 2006).
Discussion
Increased production of cytokines of the IL-1 family, such as IL-1β, is well documented in human TBI and in animal models of brain injury, providing clear evidence for a role of this cytokine in triggering TBI-induced inflammatory processes (Helmy et al, 2011). Interleukin-1β is a potent mediator of inflammation and initiates and/or amplifies a wide variety of effects associated with innate immunity, host responses to tissue injury, and microbial invasion (Dinarello, 2005). We have previously shown that the NLRP1 inflammasome, a multiprotein complex involved in the activation of caspase-1, which results in the processing of IL-1β, is activated after TBI and that pharmacological inhibition of the inflammasome results in a histopathological improvement (de Rivero Vaccari et al, 2009). Thus, inflammasome activation appears to be a novel therapeutic target for protecting against traumatic injury as well as other human conditions (Strowig et al, 2012).
In the present study, immunoblot analysis of traumatic brain injured tissue obtained from normothermic animals confirmed that TBI induced the increase in IL-1β processing and that therapeutic hypothermia attenuated this increase (Kinoshita et al, 2002; Marion et al, 1997). Most importantly, we show that hypothermia treatment prevented TBI-induced increase in cortical caspase-1, caspase-11 and the purinergic receptor P2X7 at 24 hours after trauma. These data are consistent with the described decreased immunoreactivity of caspase-1 in neurons in hypothermia-treated animals following TBI. Upon inflammasome activation, IL-1β is secreted as the result of cleavage of their proforms by active caspase-1 (cleaved caspase-1). Our findings indicate that therapeutic hypothermia prevents caspase-1 protein expression as well as IL-1β processing independently of ASC expression in the injured cerebral cortex. These effects of early cooling on inflammasome activation after TBI could therefore underlie some of the published findings regarding the effects of therapeutic hypothermia on postinjury inflammatory events (Dietrich and Bramlett, 2010).
The NLRP1 inflammasome interacts with the purinergic receptor P2X7 (Silverman et al, 2009), which is involved in the sensing of ATP and the activation of the inflammasome (Pelegrin and Surprenant, 2006). To the best of our knowledge, our study is the first to demonstrate that TBI increases P2X7 receptor expression and that posttraumatic hypothermia significantly reduces P2X7 receptor protein levels. Interestingly, we have shown that high levels of extracellular potassium activate the inflammasome in neurons and astrocytes (Silverman et al, 2009). In that in-vitro study, high extracellular K+ activated caspase-1 through pannexin 1 channels in primary neurons and astrocytes. Thus, strategies that inhibit inflammasome formation or pannexin 1 channel function may be a potential therapy for ATP-induced cell death (Silverman et al, 2009).
As previously described, therapeutic hypothermia has been reported to protect CNS tissues from irreversible injury by targeting multiple cellular and molecular mechanisms of cell death (Dietrich and Bramlett, 2010; Yenari and Han, 2012). In addition, it is known that moderate hypothermia causes reductions in potassium levels (Clifton et al, 1992; Mirzoyev et al, 2010). Because posttraumatic hypothermia may delay or prevent energy depletion and subsequent neuronal depolarization after brain injury (Prieto et al, 2011; Welsh et al, 1990), the effect of moderate hypothermia on the metabolic status of the posttraumatic brain may be one reason for the described effects on P2X7 receptor protein levels reported in our study. In this regard, hypothermia may potentially stabilize ionic gradients and decrease extracellular potassium and ATP levels, thereby reducing ATP-induced increased P2X7 receptor protein after TBI. Posttraumatic hypothermia may preserve energy homeostasis and thereby result in lower levels of extracellular ATP, hence decreasing activation of the inflammasome and the innate immune response to trauma. Since hypothermia decreases the levels of P2X7 receptor expression then the available injury-induced extracellular ATP may not be able to bind to the P2X7 receptors at high enough concentration to activate the inflammasome. Future studies are required to clarify the effects of temperature modifications on inflammasome biology.
We also investigated the effects of injury and hypothermia on cultured neurons. Using primary cortical neurons and a stretch injury device, we showed that compared with the normothermic setting, levels of the active fragment of caspase-1 (p26) in the culture media and caspase-3 (p10) cell lysates were decreased under hypothermia conditions. After neuronal stretch injury, inflammasome activation may initiate membrane channel or receptor perturbations that allow ATP or ions to alter intracellular signaling pathways and activate danger/alarm signals including damage associated molecular pattern molecules (Seong and Matzinger, 2004). These findings support the established concept that different types of neuronal injury can lead to an acute innate immune response and abnormal inflammasome activation. In this regard, Satchell et al (2005) reported elevations in caspase-1 protein levels in cerebrospinal fluid samples from infants and children after TBI. The fact that under hypothermic conditions caspase-1 levels are reduced in cultured neurons reinforces the concept that after brain or spinal cord injury, posttraumatic hypothermia may protect neurons from dying after TBI by targeting innate immunity (de Rivero Vaccari et al, 2009).
Stretch injury of cultured neurons under hypothermic conditions also reduced active caspase-3 levels compared with normothermic levels observed after stretch injury. Previous studies have reported that vulnerable neurons can die by apoptotic mechanisms after fluid percussion brain injury (Keane et al, 2001). Also, previous studies have reported that apoptotic mechanisms are temperature sensitive (Dietrich and Bramlett, 2010; Yenari and Han, 2012) and that posttraumatic hypothermia reduces caspase-3 expression in the vulnerable cerebral cortex in the present model of TBI (Lotocki et al, 2006). The present in-vitro studies provide additional data regarding the importance of this cell death mechanism after mechanical stretch injury and the ability of hypothermia to target this molecular injury pathway.
Although our current investigation presents novel in vitro and in vivo data regarding the effects of hypothermia on secondary injury mechanisms after TBI, there were several limitations to the study. First, we studied a relatively short posttraumatic time period (24 hours). Thus, there is the possibility that the restricted duration of cooling used in this study may have simply delayed the inflammasome response to injury. Second, there is a possibility that the hypothermia effect on caspase-1 cleavage in cortex demonstrated at 24 hours might not translate into improved neurological outcome in a direct cause-and-effect relationship manner. Additional studies are therefore required to answer this important mechanistic question regarding improved function. Finally, as demonstrated by previous investigations, there are multiple effects of hypothermia on postinjury inflammatory cascades (Dietrich and Bramlett, 2010) and the current data showing a depressed inflammasome activation by early cooling may be one of several targets for this experimental therapy.
In conclusion, the present study indicates that therapeutic hypothermia attenuates TBI-induced changes in inflammasome-associated protein levels including cleaved caspase-1 and caspase-11. These studies are important because they show that early cooling after injury reduces the activation of the innate immune response to TBI at 24 hours after injury. The fact that hypothermia targets this multiprotein inflammasome complex identifies a mechanism by which CNS temperatures can alter the diverse and prolonged inflammatory response to a variety of insults that influence cell death, repair and immunity. Finally, the present results reemphasize this large multimeric danger-sensing complex as a potential therapeutic target for new drug development for TBI patients.
Footnotes
Acknowledgements
The authors thank Mr Jeremy Lytle for his editorial assistance.
Disclosure/conflict of interest
The authors declare no conflict of interest.