
Abstract
Few data are available on the involvement of brain microvascular endothelial cells (BMECs) in excitotoxic neonatal brain lesions. Therefore, we developed an original approach for investigating mouse-derived BMECs in vitro. We hypothesized that newborn and adult BMEC cultures would show age-related differences in phenotype and sensitivity to glutamate. Expression of the monocarboxylate transporter, MCT1, was higher in neonatal than in adult BMECs, whereas expression of the glucose transporter, GLUT1, was higher in adult than in neonatal BMECs that overexpressed the N-methyl-D-aspartate receptor NR1 subunit (NMDAR1) compared with adult BMECs. The ability of neonatal and adult BMECs to be activated by glutamate was confirmed through intracellular calcium ([Ca2+]i) recording. The glutamate-induced [Ca2+]i increase was blocked by the selective NMDAR antagonist, MK-801. Significant glutamate-evoked concentration-dependent release of tissue-type plasminogen activator (t-PA) and matrix metalloproteinases (MMPs) activities was found in supernatants of neonatal, but not in adult BMECs. The glutamate-mediated release of t-PA, MMP-2, and MMP-9 proteolytic activities in neonatal BMECs was blocked by MK-801. Conceivably, this protease release from neonatal BMECs may participate in neonatal brain lesions.
Keywords
Introduction
Despite improvements in obstetrics and newborn intensive care, neurologic disabilities related to perinatal events have not decreased significantly in Western countries. The global incidence of cerebral palsy (i.e., motor disability because of perinatal events) is ∼1 to 2.5/1,000 live-born children (Himmelmann et al, 2005). Half the cases of cerebral palsy occur in full-term newborns. Preterm infants show a predisposition for white matter injuries (periventricular leukomalacia, intraventricular hemorrhage, and posthemorrhagic hydrocephalus); whereas full-term infants are prone to stroke and to diffuse cortical and brainstem-nucleus damage (Ferriero, 2004; Volpe, 1989). Germinal matrix hemorrhage in premature neonates is often attributed to vascular immaturity (Ment et al, 1995). However, the pathophysiology of neonatal brain damage is complex and multifactorial, in contradiction to the earlier view that hypoxia-ischemia was the predominant factor (Vannucci and Hagberg, 2004). Many factors can play a role before conception, during gestation, or in the perinatal period (e.g., genetic factors, growth-factor deficiencies, preeclampsia, and maternal infection with cytokine overproduction). Excitotoxicity is now recognized as a pathway shared by most of the processes responsible for brain damage (Lipton and Rosenberg, 1994). Excess glutamate causes neuronal death through excessive activation of ionotropic N-methyl-d-aspartate receptors (NMDARs) and Ca2+ signaling. Several groups have developed and characterized models of neonatal brain lesions in various species (Gressens et al, 1997; Gunn et al, 1992; Thoresen et al, 2001). Although ischemic events play a major role in the pathophysiology of neonatal brain damage, the interplay between brain microvascular endothelial cells (BMECs) and other brain-cell populations remains poorly understood, probably because relevant experimental tools are scarce. As part of the neurovascular unit, BMECs are sensitive to glutamate (Andras et al, 2007; Sharp et al, 2003, 2005). Primary cultured or immortalized BMECs express functional NMDAR (Krizbai et al, 1998; Scott et al, 2007). In vitro, BMECs respond to glutamate without losing viability, by generating nitric oxide, superoxide, and peroxynitrites, as well as by affecting endothelial barrier integrity through NMDAR activation (Kuhlmann et al, 2008; Scott et al, 2007; Sharp et al, 2005). Glutamate-induced oxidative stress may lead to the activation of secondary messengers, disrupting endothelial tight junctions by an effect on occludin (Andras et al, 2007). Brain microvascular endothelial cells may also interact with neural cells by the release of various endocrine/paracrine factors, which influence the course of excitotoxic brain lesions. Some of these factors, including nitric oxide and tissue-type plasminogen activator (t-PA), may have dual effects. The serine-protease, t-PA, which is secreted by BMECs, converts plasminogen to active plasmin and exerts pleiotropic effects within the central nervous system (Yepes and Lawrence, 2004). Thus, t-PA is thought to regulate many physiologic processes during brain development (e.g., neural migration and plasticity, learning, and memory). However, detrimental effects of t-PA have also been reported in ischemic, inflammatory, and neurodegenerative central nervous system diseases involving neuronal (Wang et al, 1998), microglial (Tahraoui et al, 2001), or endothelial cells (Liu et al, 2004). We showed recently that t-PA contributed to white matter cavitation in an excitotoxic lesion model in neonatal mice (Hennebert et al, 2004; Leroux et al, 2007). Plasminogen proteolysis mediated by t-PA also results in an activation of matrix metalloproteinases (MMPs). In addition, t-PA induces MMP-9 expression by a receptor-mediated effect in response to ischemia (Tsuji et al, 2005; Wang et al, 2003). Both MMP-9 and MMP-2 degrade the basement membrane in blood vessels and protein components of endothelial tight junctions crucial to the integrity of the blood-brain barrier (BBB) (Fukuda et al, 2004). In the immature brain, MMP-9 participates in BBB leakage and inflammation (Svedin et al, 2007). As proteases are also produced by the immature brain microvascular endothelium, efforts are needed to elucidate the role of BMECs in modulating neonatal brain injury. Most of the studies investigating the influence of BMECs on other neural cell populations in vitro used adult preparations, which might differ from neonatal cells. Indeed, the metabolism and vasculature of the immature brain differ from those of the adult brain, in particular regarding the predominant role of monocarboxylates, which is crucial in the pathogenesis of neonatal lesions (Adle-Biassette et al, 2007). Neonatal excitotoxicity is also dependent on the type of ionotropic glutamate receptors, most notably NMDARs, which are activated at each developmental stage. Monocarboxylates and glucose transporters, as well as NMDAR, are expressed in brain microvessels.
Therefore, we designed this study for comparing neonatal and adult BMEC phenotypes and function in vitro with the goal of developing a new model relevant to the neonatal phenotype, which may help to better understand the role of the brain microvascular endothelium in neonatal brain injuries.
Materials and methods
Animals
Swiss Naval Medical Research Institute (NMRI) mice (JANVIER; Le Genest-St-Isle, France) aged 10 days (neonatal) or 5 weeks (adult) were used. The animals were given food and water ad libitum and maintained on a 12:12 h light-dark cycle. Protocols were approved by the Ethical Committee for Animal Experimentation of Normandy, France (agreement no. N/02-09-06/15).
Primary Cultures of Mouse Brain Microvascular Endothelial Cells
Neonatal BMEC culture procedures were modified according to protocols developed earlier for adult BMECs (Coisne et al, 2005; Song and Pachter, 2003). Meninges-free cortices were homogenized in a Dounce tissue homogenizer in 10mL molecular cellular and developmental biology (MCDB) medium 131 supplemented with 2% of FCS (fetal calf serum), 100 U/mL penicillin, and 100 μ/mL streptomycin (all from GIBCO, Invitrogen, Cergy Pontoise, France). The homogenate was centrifuged at 200 g for 10 mins. The pellet was suspended in 18% (w/v) dextran solution (Sigma-Aldrich, Saint Quentin Fallavier, France) and centrifuged at 2,000 g for 45 mins at 4°C. Thereafter, it was suspended and filtered using a 70- μm cell strainer (Falcon, BD Biosciences, Pont de Claix, France). The resulting microvessel filtrate was digested at 37°C in MCDB131 with 0.5mg/mL of collagenase-dispase and 100 μg/mL DNase I (all from Roche Diagnostics, Meylan, France). The digested fragments were purified using antimurine platelet endothelial cell adhesion molecule-1 (PECAM-1) (CD31, clone MEC13.3, BD Biosciences)-precoated Dynabeads (Dynal, Invitrogen) for 30mins. Bead-bound microvessel fragments were collected and washed thrice. The digested microvessels were seeded onto murine collagen type IV (50 μg/mL; BD Biosciences) in MCDB131 supplemented with 10% FCS, 10% horse serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 2mmol/L L-glutamine, 10 μg/mL vascular endothelial growth factor (VEGF165; R&D System, Lille, France), and bFGF (basic fibroblast growth factor) (Invitrogen). The cultures were incubated in this complete medium at 37°C in a humidified atmosphere containing 5% CO2. After 24 h, adherent cells were washed thrice and exposed to a fresh complete medium. This medium was changed every 2 days. When confluence was achieved (DIV (day in vitro) 3 to 5), the cells were used for application.
Isolation of Mouse Brain Microvessels
Microvessels were isolated from neonatal or adult brain cortices using the procedure described for BMEC primary cultures, but omitting the stages after immunomagnetic selection.
Primary Cultures of Cortical Neurons
Cultured neurons were prepared from mouse embryos (E15-E16) from Swiss NMRI. Cell suspensions in DMEM (Dulbecco's Modified Eagle's Medium) (Sigma-Aldrich) were seeded onto poly-D-lysine (0.1 mg/mL; Sigma-Aldrich) and laminin (0.02mg/mL; Invitrogen). Cells were cultured in DMEM containing 5% FCS, 5% horse serum, and 2 mmol/L glutamine (Sigma-Aldrich) and were maintained at 37°C in humidified 5% CO2 atmosphere. Cytosine β-arabinoside furanoside (10 μmol/L; Sigma-Aldrich) was added on DIV 3 to inhibit glial proliferation. Neuronal cultures were used after DIV 11.
Fluorescent-Labeled Acetylated Low-Density Lipoprotein Uptake and Immunocytochemical Characterization
Neonatal and adult BMECs were incubated with 10 μg/mL acetylated low-density lipoprotein (LDL) labeled with Dil-Ac-LDL (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate) (Biomedical Technologies Inc., Stoughton, MA, USA) for 3h at 37°C. The cells were washed thrice and fixed with 4% paraformaldehyde. The nuclei were then labeled with Hoechst 33258 (Sigma-Aldrich). Dil-Ac-LDL-positive cells were observed at 550 nm. For all immunocytochemical experiments, BMECs and brain microvessels were fixed with 4% paraformaldehyde for 10 mins at room temperature. After washing and blocking with 3% bovine serum albumin in a phosphate-buffered saline containing 0.1% Tween, cell monolayers, or brain microvessels on gelatin-coated slides were incubated overnight with primary antibody at 4°C. The following antibodies were used: rat anti-PECAM-1 (1:100, BD Biosciences), rabbit anti-vWF (anti-von Willebrand factor) (1:100; Dako, Trappes, France), chicken antimonocarboxylate type 1 (anti-MCT1) (1:100), rabbit antiglucose type 1 (anti-GLUT1) (1:100), and rabbit anti-NR1Pan (1:100) (all from Millipore, Molsheim, France). Neonatal and adult BMECs and neonatal brain microvessels were washed, incubated with the corresponding secondary antibody (all from Vector, Clinisciences, Montrouge, France) for 90mins, washed, and incubated with ExtrAvidin conjugated to fluorescein isothiocyanate (ExtrAvidin-FITC; Sigma-Aldrich) or to tetramethylrhodamine isothiocyanate (ExtrAvidin-TRITC; Sigma-Aldrich) for 30mins. Neonatal BMECs and microvessels were stained with Extravidin-FITC, whereas adult BMECs were stained with Extravidin-TRITC. Nuclei were stained with Hoechst 33258 (Sigma-Aldrich) for 10 mins at room temperature after the last labeling. To evaluate the proportion of contaminating pericytes, we performed double-labeling using FITC monoclonal α-smooth muscle actin (1:200; Sigma-Aldrich) for pericytes and PECAM-1 immunoreactivity revealed by conjugated Alexa-Fluor 594 antirat secondary antibody (Invitrogen) for BMECs. Pericytes and BMECs were counted manually using a grid placed over ten different pictures taken from three independent experiments for each age group.
Flow Cytometry
Cells were harvested by brief trypsinization to avoid the loss of PECAM-1 expression then washed with fluorescence-activated cell sorting (FACS) buffer containing phosphate-buffered saline solution, 2% FCS, and 0.01% NaN3. Thereafter, the cells were stained with rat anti-PECAM antibody (1:100) for 30mins at 4°C, washed twice with FACS buffer, and finally stained with phycoerythrin-labeled goat antirat Ig (1:200; BD Biosciences) for 30 mins at 4°C. Cells were washed twice with FACS buffer. Cell fluorescence was measured by flow cytometry using a FACS-Calibur instrument (BD Biosciences). Data were analyzed using FLOWJO software (Tree Star, Ashland, OR, USA).
Real-Time Reverse-Transciptase Polymerase Chain Reaction Studies
Total RNAs were extracted from cultured cells or from freshly isolated microvessels using the NucleoSpin RNA II kit (Macherey-Nagel, Hoerdt, France) according to the manufacturer's instructions. Reverse transcription of 1 μg of total RNAs was performed using the Promega RT system (Promega, Charbonnières, France; reverse transcription at 42°C for lh). Two primers were designed for each gene using the Beacon Designer software (Bio-Rad, Marnes-la-Coquette, France). Primer alignment was achieved using the Blast database (NCBI) to ensure primer specificity. The primers were as follows: NR1 subunit (F: CTCTAGC CAGGTCTACGCTATCC; R: GACGGGGATTCTGTAGAAGC CA), NR2A subunit (F: ACATCCACGTTCTTCCAGTTTGG; R: GACATGCCAGTCATAGTCCTGC), NR2B subunit (F: CC AGAGTGAGAGATGGGATTGC; R: TGGGCTCAGGGATGA AACTGT), NR2C subunit (F: GCTTCTGCATCGACATCCTC AA; R: CATACCATTCCACACACCACGAA), NR2D subunit (F: CTGTGTGGGTGATGATGTTCGT; R: GTGAAGGTAGAG CCTCCGGG), NR3A subunit (F: ATCCTCAAGCGCATCG GACA; R: CGACTCTGGCTCATCCCTCTG), NR3B subunit (F: GGCCGTGACCAGCTTCAGTA; R: CAATGGGTGAGGC TGTATCTCG), MCT1 (F: GCAGCCGTCCAGTAATGATCG; R: GCAAGCCCAAGACCTCCAATAAC), GLUT1 (F: ATCCC ATCCACCACACTCACC; R: GCCTGCCAAAGCGATTAAC AAAG), t-PA (F: CTCCGACCCATGCTCAGAA; R: TTGTA CCAGGCCGCTGTTG), and β-microglobulin (F: GCCGAAC ATACTGAACTGCTAC; R: GCTGAAGGACATATCTGACAT CTC). All NMDAR subunits and t-PA primers were validated using cDNA from cortical neuron cultures; whereas MCT1 and GLUT1 were validated using cDNA from immortalized endothelial cells b. End3 (ATCC number CRL-2299).
The PCR mix solutions were prepared with RNase-free water containing primers and IQ SYBR Green Supermix (Bio-Rad). For PCR amplification, 20 μL of mix was added to 5 μL of reverse transcription reaction that was earlier diluted to 1:20 (v/v). Negative controls were performed in each experiment using samples without reverse transcription as a template to control for RNA contamination with genomic DNA and with RNase-free water instead of cDNA to check that qPCR mixes were not contaminated with DNA. Assays were run in triplicate on the iCycler iQ real-time PCR detection system (Bio-Rad). The amplification conditions were Hot Goldstar enzyme activation, 95°C for 3mins; 50 cycles of PCR at 95°C, 15 secs and 60°C, 1 min. Gene expression levels were computed as follows: relative mRNA expression = 2−(Ct of gene of interest), where Ct was the threshold cycle value. The gene expression was also computed relative to the mRNA expression level of the reference gene transcript (β-microglobulin), as follows: relative mRNA expression = 2−(Ct of gene of interest—Ct of gene of reference).
Western Blot Assay
Homogenates from cultured BMECs were prepared on ice in a lysis buffer (50mmol/L Tris-HCl, 150 mmol/L NaCl, 0.5% Triton X-100, Sigma-Aldrich, pH = 7.4) containing protease inhibitors (Complete Mini, Roche Diagnostics). Clear supernatant samples were obtained after centrifugation for 10 mins (10,000 g) at 4°C. Proteins were quantified (BCA protein assay, Pierce, Perbio Sciences, Brébières, France), and immunoblotting assays were then performed using 10 μg protein for MCT1, 15 μg for GLUT1, and 30 μg for NR1. Samples were separated on 4% to 12% gradient BIS-TRIS gel NOVEX (Invitrogen) according to the manufacturer's protocol. Proteins were transferred onto a PVDF membrane (Polyscreen, Perkin Elmer, Courtaboeuf, France), which was blocked for 1 h with a Tris-buffered saline containing 0.5% Tween 20 and 5% nonfat milk. Thereafter, the membrane was incubated overnight at 4°C with the following dilutions of primary antibodies: chicken anti-MCT1 (1:5,000), rabbit anti-GLUT1 (1:2,500), rabbit anti-NR1Pan (1:1,000) (all from Millipore), mouse anti-tPA (1:500; Abcam, Paris, France), and rabbit antiactin (1:1,000; Sigma-Aldrich). After incubation with the corresponding biotinylated secondary antibodies and thereafter with horseradish peroxidase streptavidin (all from Vector, Clinisciences), proteins were visualized using an enhanced chemiluminescence ECL Plus immunoblotting detection system (Perkin Elmer). Densitometry analysis was performed using ImageJ (NIH), and the protein content was normalized for actin content.
[Ca2+]i Measurement/Calcium Imaging
Cells were cultured on glass-bottom dishes coated with the appropriate matrices. Cells were loaded for 45mins at 37°C in the dark in HBSS (Hank's balanced salt solution without magnesium)-buffered saline bathing solution containing 10 μmol/L fura-2-acetoxymethyl ester (FURA-2AM) with 0.1% of pluronic acid F-127 (all from Invitrogen). They were rinsed thrice and superfused continuously with HEPES-buffered saline solution without magnesium to prevent the NMDAR blockade. In BMECs, calcium increase was stimulated by 1 mmol/L of glutamate (Sigma-Aldrich) and 1 mmol/L of adenosine triphosphate (ATP) (Sigma-Aldrich), and glutamate was added together with 50 μmol/L of MK-801 (a selective noncompetitive NMDA antagonist, Tocris, Bristol, UK) to block the NMDAR component. Cortical neurons were subjected to the same procedure with 100 μmol/L of glutamate. Experiments were monitored at room temperature using an inverted LEICA DMI6000 fluorescence microscope equipped with a CCD camera (Retiga Exit, Qimaging, Roper Scientific, Evry, France) and with a variable filter wheel (Sutter Instrument, Roper Scientific). Dual fluorescent images (340 and 380 nm excitation; 510 nm emission) were captured at a magnification of × 40 and collected every 5 secs. The calcium increase was calculated as the F340-F380nm ratio using the Metamorph software (Roper Scientific). The proportion of responsive cells was the proportion of total cells showing increased calcium during stimulation, expressed as the percentage.
Zymography Assay
Brain microvascular endothelial cell-conditioned media were collected after a 6h exposure to increasing doses of glutamate (50 μmol/L to 1 mmol/L). Glutamate was dissolved in serum-free media with 10 μmol/L of glycine (a NMDAR coagonist). Exposure to increasing glutamate doses for 6h was not toxic for neonatal or adult BMECs, as established by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium) cell viability assay (Supplementary Data, Supplementary Figure 1S). Gel zymography was used to quantify t-PA, MMP-9, and MMP-2 in media. Briefly, equal amounts of media (10 μL) were resolved on 12% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) containing casein (1 mg/mL; MP Biomedicals, Illkirch, France) and plasminogen (4.5 μg/mL; Calbiochem, VWR, Fontenay-sous-Bois, France) for t-PA detection or on 10% SDS-PAGE (Invitrogen) containing 0.1% gelatin for MMP-9 and MMP-2 detection. After migration, gels were renatured and developed at 37°C for 90mins (t-PA activity) or 72 h (MMPs activities). Clear bands were visualized after Coomassie staining and quantified using the ImageJ software.
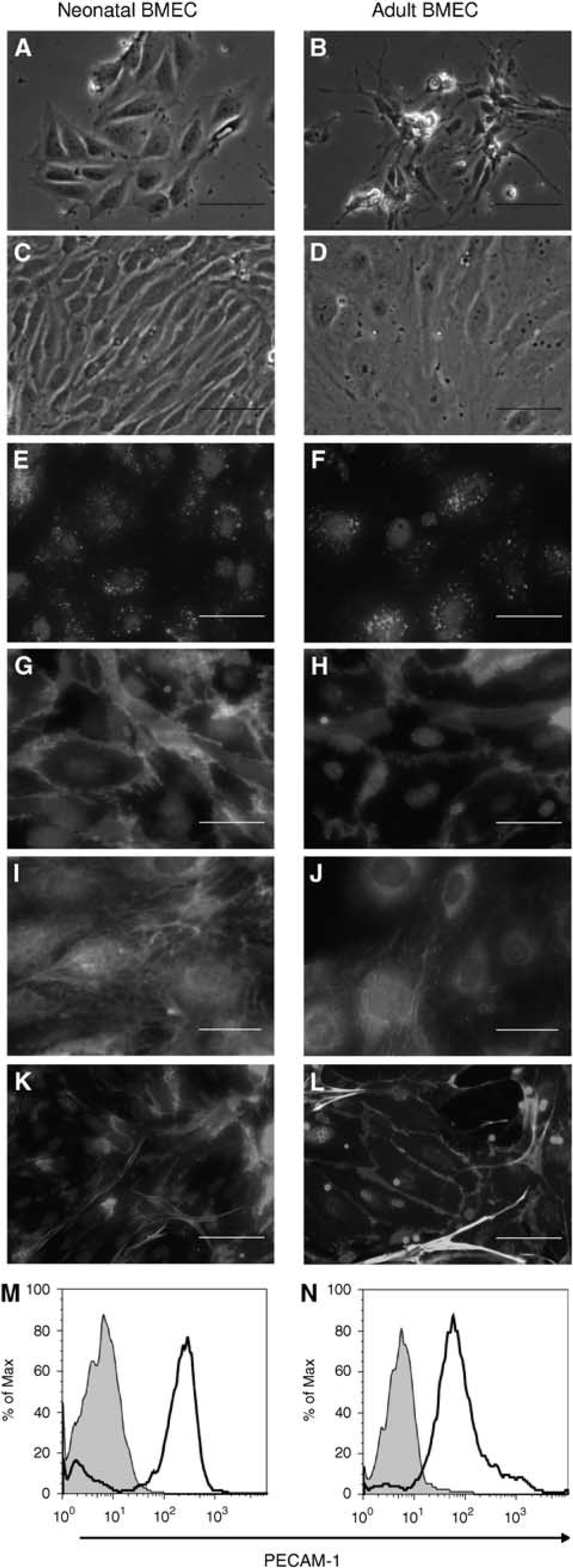
Morphology and phenotype of neonatal (left panel) and adult (right panel) brain microvascular endothelial cells (BMECs) in primary cultures. (
Statistics
Data were described as mean ± s.e.m. for each group. To compare neonatal and adult BMECs, we used the nonparametric Mann-Whitney test. Age-related differences in NMDAR subunit mRNA contents in BMECs were evaluated using two-way ANOVA (analysis of variance). The effects of glutamate on t-PA and MMP activities were compared using the nonparametric Kruskal-Wallis test followed by the Dunn post hoc test. P-values < 0.05 were considered statistically significant.
Results
Brain Microvascular Endothelial Cells From Both Neonatal and Adult Brains Exhibit Specific Endothelial Markers
Cultured neonatal (Figure 1, left panel) and adult (Figure 1, right panel) BMECs showed well-recognized endothelial characteristics. The cells appeared as small clusters 24 h after plating (Figures 1A and 1B) and reached confluence within 2 to 3 days for neonatal BMECs (Figure 1C) and 4 to 5 days for adult BMECs (Figure 1D). Confluent monolayers showed the characteristic ‘cobblestone’ or ‘spindle-shaped’ morphologies depending on seeding densities. Neonatal and adult BMECs took up Dil-Ac-LDL. In the cytoplasm, Dil-Ac-LDL-positive cells showed a strong granular staining (Figures 1E and 1F). Immunocytochemistry analyses confirmed the endothelial phenotype of both types of cultures, with membrane expression of PECAM-1 (Figures 1G and 1H) and cytoplasmic expression of vWF (Figures 1I and 1J). Although endothelial cell monolayers appeared homogeneous by phase-contrast and fluorescence microscopy, few pericytes were detected by α-smooth muscle actin immunostaining (Figures 1K and 1L). The amount of pericytes was similar in neonatal and adult BMEC monolayers (10.01% ± 1.70% versus 9.84% ± 1.70%). Flow cytometry analysis showed that the percentage of PECAM-1 expressing cells did not differ between neonatal and adult BMEC cultures (79.3% ± 2.2% versus 81.3% ± 4.5%) (Figures 1M and 1N). Thus, in our hands, neonatal and adult BMECs both show the phenotype of endothelial cells with a few contaminating pericytes.
BMECs and Brain Microvessels From Neonatal and Adult Mice Show Different Patterns of Expression of Brain Nutrient Transporters MCT1 and GLUT1
The expression of two brain nutrient transporters—MCT and GLUT1—was investigated by immunocytochemistry, real-time PCR, and western blot in neonatal and adult BMECs (Figure 2) and in freshly isolated microvessels (Figure 3). Neonatal and adult BMECs showed immunoreactivity to MCT1 (Figures 2A and 2B) and GLUT1 (Figures 2C and 2D). Both neonatal and adult BMECs expressed MCT1 and GLUTl mRNA (Figures 2E and 2F). However, in cell extracts, MCT1 protein (∼45kDa) was expressed at a significantly higher level in neonatal than in adult BMECs (Figure 2G; 1.50 ± 0.24 versus 0.55 ± 0.22, P < 0.05). In contrast, GLUTl (∼50kDa) expression was significantly higher in adult than in neonatal BMECs (5.54 ± 0.58 versus 1.42 ± 0.09, P < 0.05) (Figure 2H). Control studies of the MCT1 expression were performed in freshly prepared microvessels from neonatal and adult mouse brain samples (Figures 3A and 3B). The MCT1 mRNA levels measured in three independent experiments were 20.6 ± 1.2-fold higher in neonatal than in adult BMECs (Figure 3C). The GLUTl mRNA contents in neonatal and adult microvessels were similar (Figure 3D). A stronger MCT1 protein expression was detected in neonatal microvessels compared with adult microvessels (4.80 ± 2.40 versus 1.77 ± 0.66, P < 0.05) (Figure 3E). Glucose type 1 was significantly more abundant in adult than in neonatal microvessels (7.72 ± 2.59 versus 1.43 ± 0.67, P < 0.05) (Figure 3F).
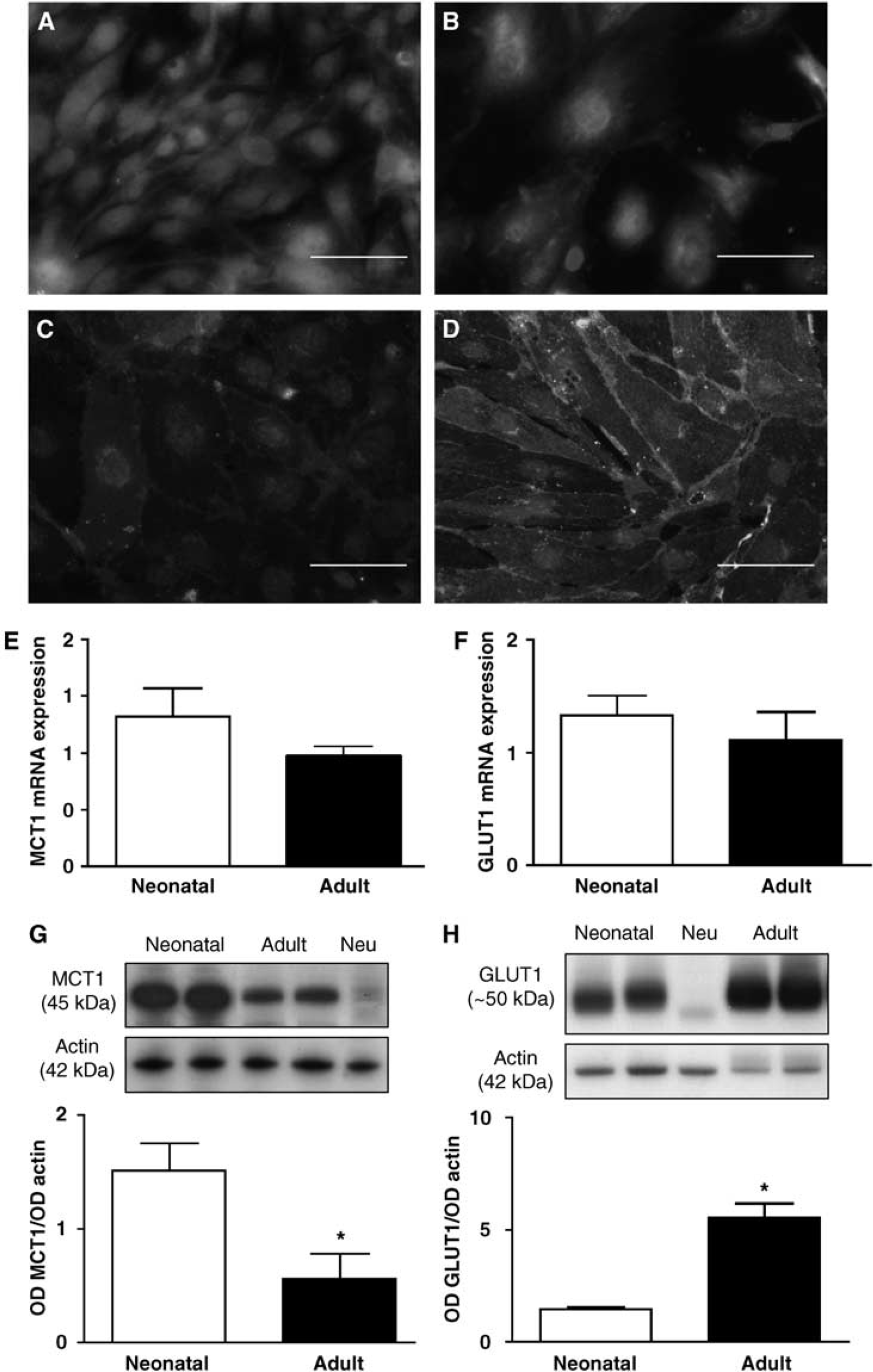
Expression of MCT1 and GLUT1 in primary cultures of neonatal and adult BMECs. (
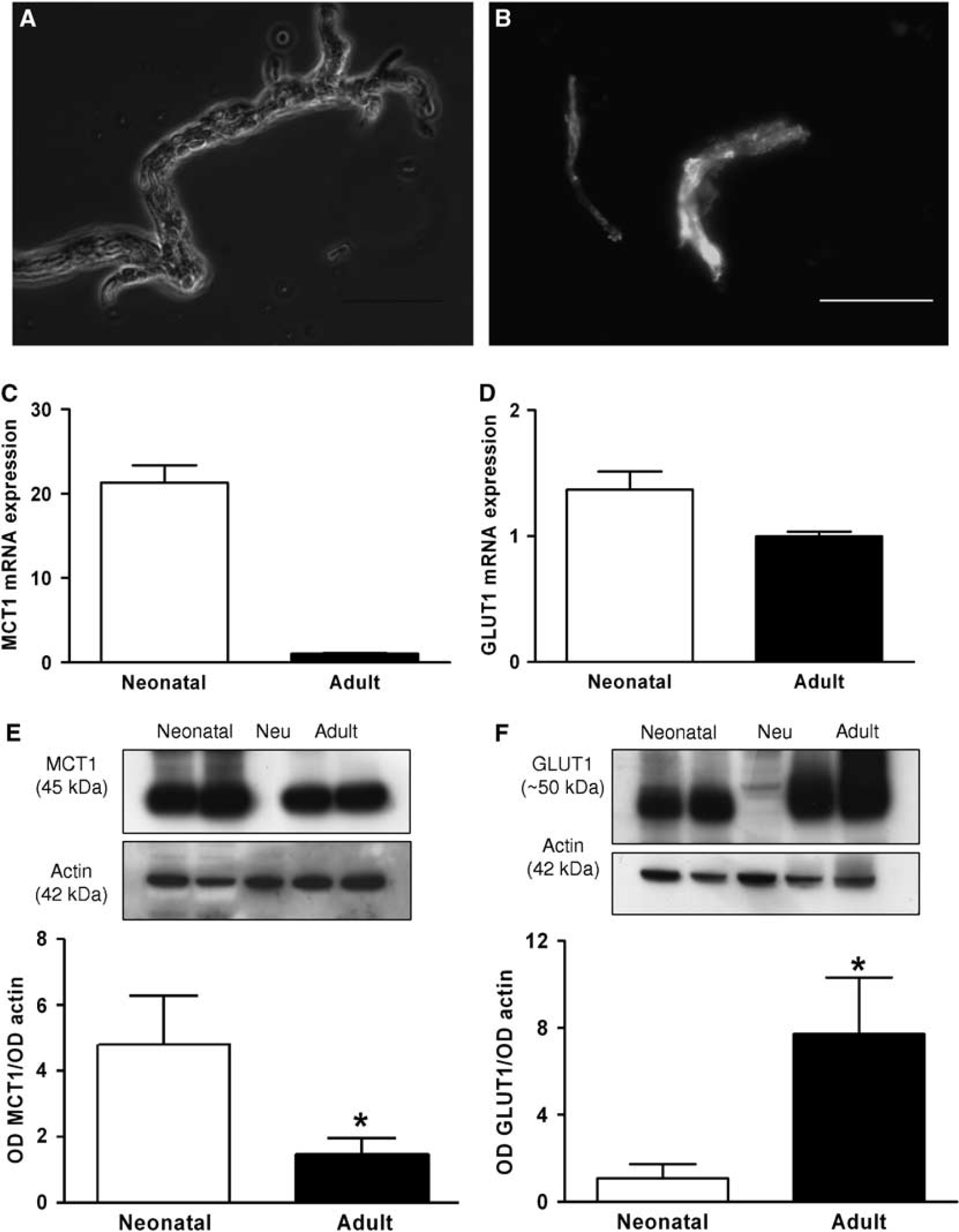
MCT1 and GLUT1 expression in freshly isolated brain microvessels. (
Overexpression of NR1 Subunit in Neonatal BMECs Does not Influence NMDAR Function
The NMDAR expression was investigated with special attention to the NR1 subunit (NMDAR1). Neonatal and adult BMECs showed punctate NMDAR1 and PECAM immunostaining concentrated along the intercellular borders (Figures 4A and 4B). NMDAR1 mRNA was significantly higher in neonatal than in adult BMEC cultures (7.37 ± 3.01 versus 1.15 ± 0.35, P < 0.05) (Figure 4C). A global analysis of mRNAs encoding NMDAR subunits (NR2A-D, NR3A, and NR3B) showed significantly higher expression levels in neonatal than in adult BMECs (Supplementary Data, Supplementary Table 1S; F(1,36) = 4.40, P < 0.05). A 116-kDa NMDAR1 protein was detected in both our models of BMECs and in control extracts (i.e., cortical neurons). The NMDAR1 expression was significantly higher in neonatal than in adult BMECs (0.90 ± 0.16 versus 0.29 ± 0.01, P < 0.05) (Figure 4D). The NMDAR function was then investigated by measuring [Ca2+]i. Application of 1 mmol/L of glutamate to FURA-2-loaded neonatal and adult BMECs caused [Ca2+]i increase in 14.63% ± 3.57% and 15.48% ± 3.68% of neonatal and adult cells, respectively (Table 1). When 50 μmol/L of MK-801 was added together with glutamate, the proportion of responsive cells was < 3% for neonatal BMECs and 5% for adult BMECs (Table 1). The positive controls were 1 mmol/L of ATP added to neonatal BMECs, adult BMECs, and cortical neurons; and glutamate ± MK-801 added to cortical neurons (Table 1).
Proportion of neonatal brain microvessel endothelial cells (BMECs), adult BMECs, and cortical neurons showing [Ca2+]i mobilization on exposure to glutamate alone, glutamate with MK-801, or ATP
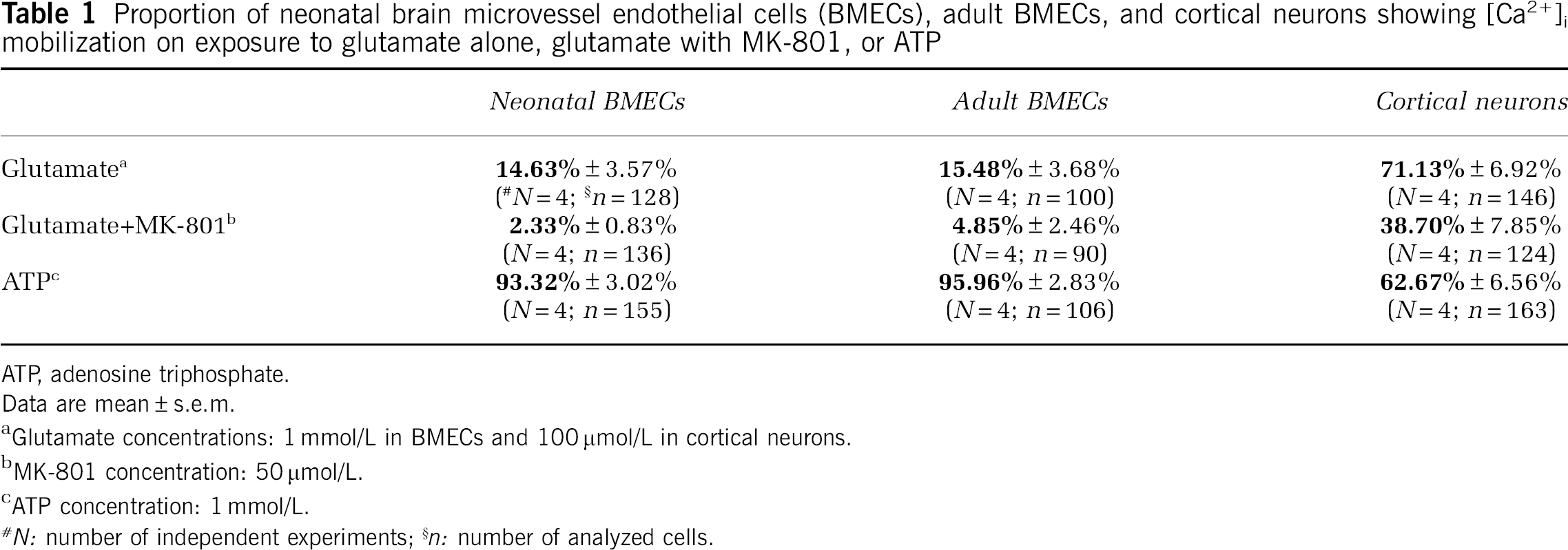
ATR adenosine triphosphate.
Data are mean ± s.e.m.
Glutamate concentrations: 1 mmol/L in BMECs and 100 μmol/L in cortical neurons.
MK-801 concentration: 50 μmol/L.
ATP concentration: 1 mmol/L.
N: number of independent experiments;
n: number of analyzed cells.
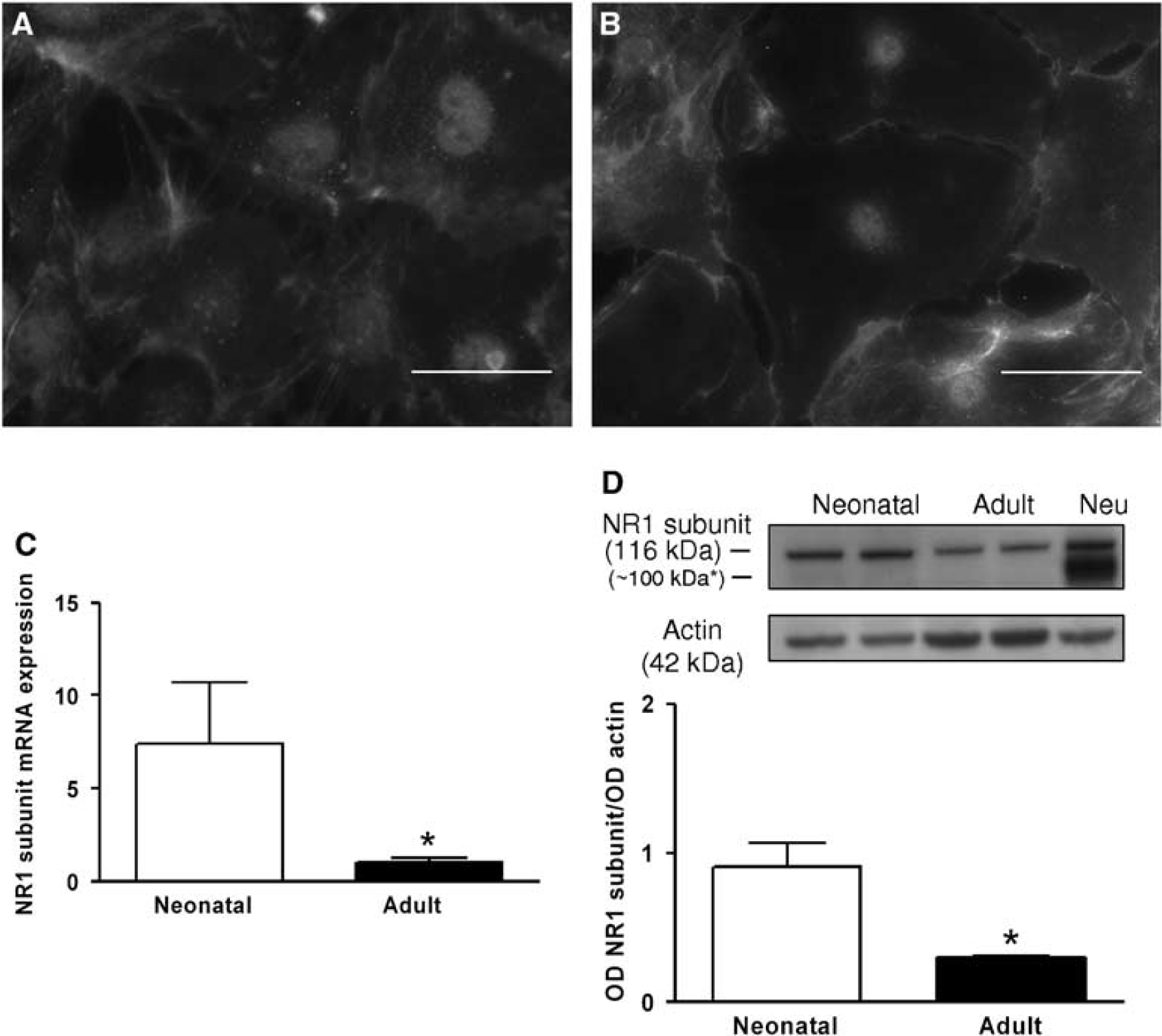
NMDAR1 subunit expression in primary cultures of neonatal and adult BMECs. (
Higher t-PA Protein Expression in Neonatal BMECs, and Glutamate-Induced t-PA Elevation Only in Neonatal BMECs
The mRNA for t-PA was not significantly different between neonatal and adult BMECs (Figure 5A). However, western blot analysis showed a larger amount of t-PA (∼ 69 kDa) in neonatal than in adult BMECs (1.11 ± 0.55 versus 0.38 ± 0.19, P < 0.05) (Figure 5B). Increasing doses of glutamate applied for 6 h induced a dose-dependent increase in t-PA proteolytic activity in supernatants from neonatal, but not adult, BMEC cultures (Figures 5C and 5D). In neonatal BMECs, the t-PA activity increased significantly in response to 1 mmol/L glutamate (100% ± 29.27% versus 227.36% ± 14.16%, P < 0.05) (Figure 5C), and this effect was reversed by 10 μmol/L of MK-801 (227.36% ± 14.16% versus 107.64% ± 16.79%, P < 0.05) (Figure 5C).
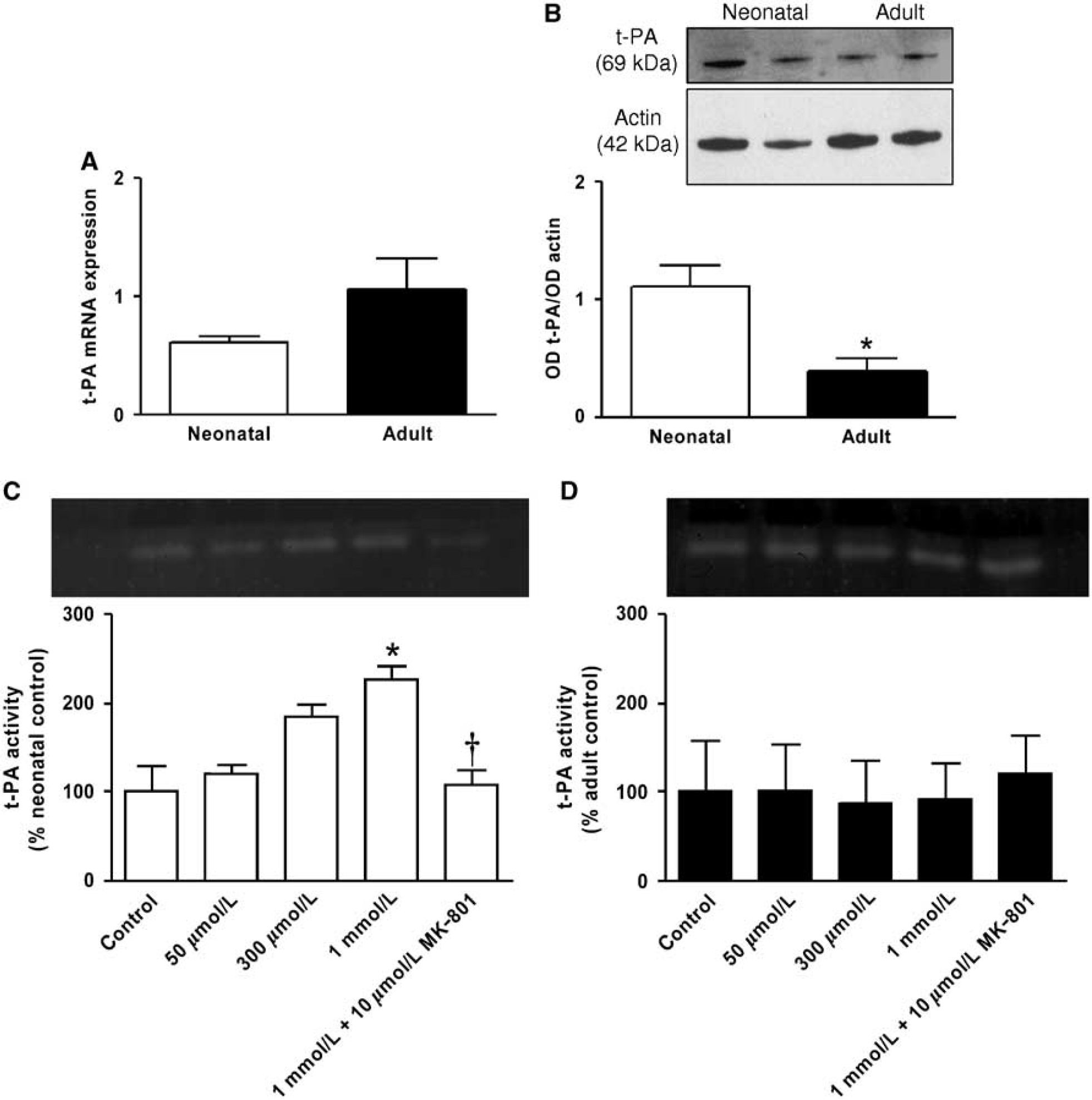
Quantification of t-PA expression and glutamate-mediated proteolytic activity in primary cultures of neonatal and adult BMECs. (
Glutamate Enhances MMP-9 and MMP-2 Proteolytic Activity in Neonatal BMECs
Glutamate tested over a wide concentration range for 6 h significantly increased MMP-9 activity in media (100% ± 3.62% versus 154.06% ± 8.5%, P < 0.001) (Figure 6). In contrast, MMP-2 activity showed a significant increase only with 1 mmol/L of glutamate (100% ± 16.55% versus 179.57% ± 16.47%, P < 0.05) (Figure 6). MK-801 (10 μmol/L) prevented the glutamate-induced increases of MMP-9 and MMP-2 activities in neonatal BMEC supernatants (154.06% ± 8.5% versus 113.87% ± 5% and 179.57% ± 16.47% versus 96.48% ± 13.01%, respectively, P < 0.05) (Figure 6). Under the same conditions, we found no quantifiable MMP proteolytic activities in adult BMECs (not shown).
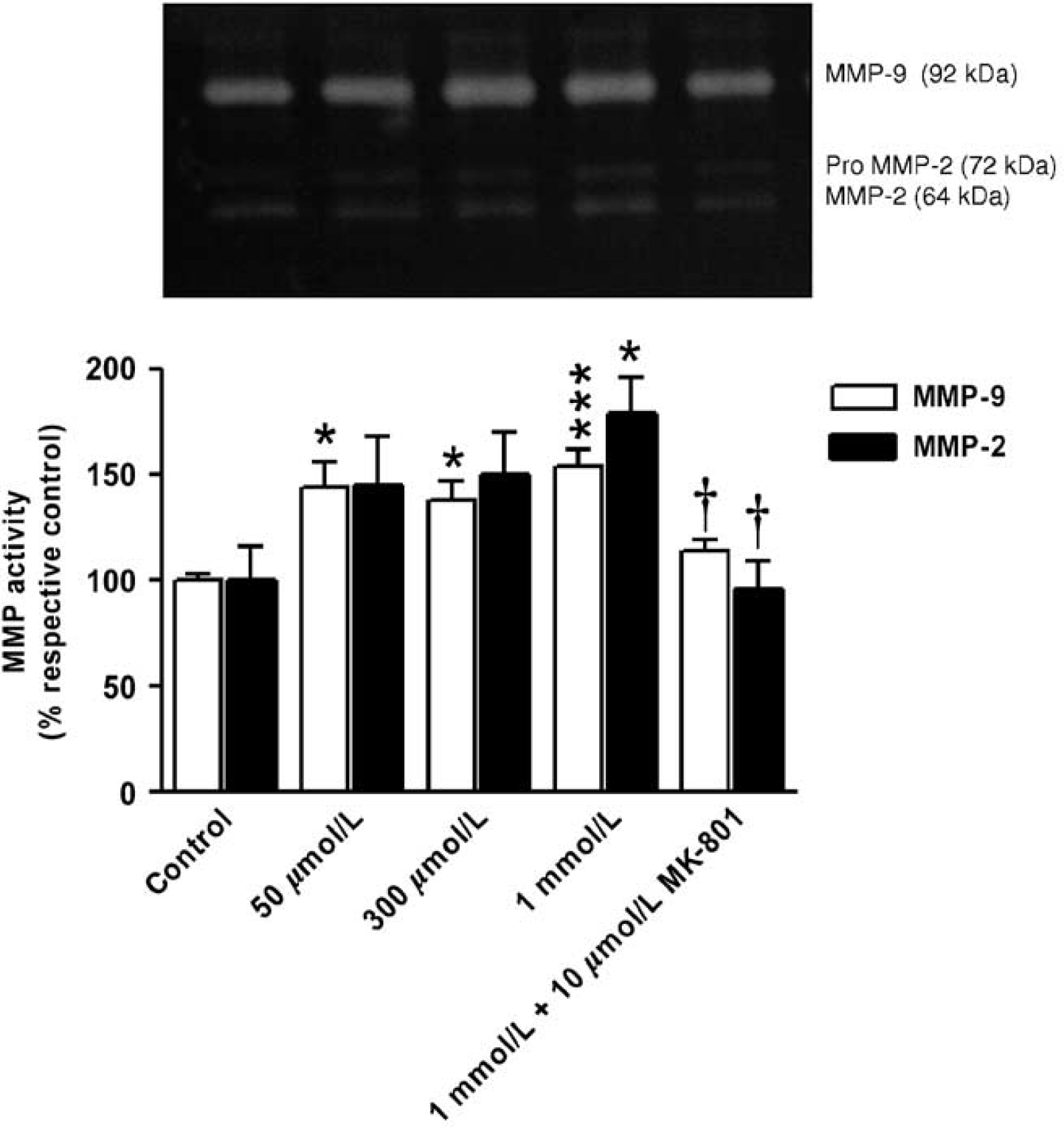
Effect of glutamate challenge on MMP proteolytic activity release from primary cultures of neonatal BMECs. MMP-9 (open bars, n = 6) and MMP-2 (black bars, n = 7) proteolytic activities in supernatants collected from neonatal BMECs exposed to glutamate (50 μmol/L to 1 mmol/L) ± MK-801 (10 μmol/L) for 6 h. Representative gelatin zymogram of MMP activities is shown in the upper panel. Results are reported as percentage activity with the respective control level set at 100% (mean ± s.e.m.). *P < 0.05 versus control; ***P < 0.001 versus control, †P < 0.05 versus 1 mmol/L glutamate.
Discussion
We compared BMECs from 10-day-old and adult mice with the goal of developing an in vitro tool for investigating endothelial cell-neural cell interactions. Currently available endothelial cell models for glutamatergic excitotoxicity studies consist of immortalized BMECs (Scott et al, 2007) and primary cultures of adult BMECs (Andras et al, 2007; Kuhlmann et al, 2008; Sharp et al, 2003, 2005). We found that cultured neonatal and adult BMECs differed regarding both phenotype and function compared with adult BMECs, neonatal BMECs showed evidence of metabolic immaturity, with higher MCT1 and lower GLUTl expression, in agreement with earlier published data (Baud et al, 2003; Vannucci and Simpson, 2003); higher NMDA subunit expression, with no difference, however, in the receptor-evoked [Ca2+]i increase; and greater glutamate-evoked release of t-PA, MMP-9, and MMP-2.
N-methyl-d-aspartate receptors are thought to be major mediators of neuronal cell death in the immature brain. Maturation patterns of NMDAR and alpha-amino-3-hydroxy-5-methylisoxazol-4-propionate (AMPA)/kainate receptors correlate with age-specific regional susceptibility to brain injury. N-methyl-d-aspartate receptors predominate in the immature brain, whereas AMPA/kainate receptors increase throughout brain maturation (Jensen, 2002). There is a general agreement that brain NMDARs are developmentally regulated according to physiologic functions (e.g., synaptogenesis and learning), regional location, cell type, and subunit coassembly. Increased levels of mRNAs encoding NR1 and NR2 subunits were found in rat cerebral cortex between P7 and P20 (Zhong et al, 1995). Moreover, the NR1 subunit protein increased gradually in the neocortex until P20 and was lower at the adult stage (P60) (Babb et al, 2005). Interestingly, alterations in NR2 subunit mRNA expression during brain development led to a differential sensitivity of NMDAR receptors to antagonists and glycine (Zhong et al, 1996). In contrast, the ontogeny of NMDARs within the brain endothelium is still unclear, probably because of a decade of controversy regarding the NMDAR expression in endothelial cells. Now, it is well established that primary or immortalized adult BMECs express functional NMDARs (Kuhlmann et al, 2008; Scott et al, 2007; Sharp et al, 2005) and AMPA receptors (Andras et al, 2007; Sharp et al, 2003). The results of our study showing the expression of functional NMDARs in neonatal BMECs may shed light on the role of NMDARs in the BBB dysfunction that characterizes neonatal brain damage. Earlier studies using BMEC models showed that glutamate-induced BBB disruption involved NMDARs, Ca2+ signaling, and changes in junctional organization (Andras et al, 2007; Kuhlmann et al, 2008; Sharp et al, 2003). In adult BMECs, BBB breakdown was prevented by MK-801 but not by AMPA/kainate inhibitors (Andras et al, 2007). We showed in vitro that NMDAR mRNA and NMDAR1 protein expression levels were higher in neonatal than in adult BMECs (Figure 4 and Table 1). We cannot explain why BMECs expressed only the N-glycosylated form (116 kDa) of NMDAR, instead of the two expected forms (116 and ∼ 100 kDa, N-glycosylated and N-deglycosylated forms, respectively) (Chazot et al, 1992; Kopke et al, 1993; Nicole et al, 2001). Further investigations are needed to determine whether NMDAR1 turnover and/or addressing differ between the two cell types according to their function. We showed that the age-related difference in NMDAR expression was not caused by contaminating cells. Microvessel digestion recruits some contaminating cells in BMEC cultures, such as pericytes (the most common type) and astrocytes. As astrocytes also express NMDAR, we used Glial Fibrillary Acidic Protein (GFAP) immunostaining to check that none were present in our cultures (data not shown). We achieved a compromise between the presence of some pericytes (∼10% in both neonatal and adult BMECs) and the risk of decreased BMEC viability caused by attempts to eliminate contamination. However, to our knowledge, pericytes express metabotropic glutamatergic receptors but not NMDARs (Gillard et al, 2003). Furthermore, PECAM-1 and NMDAR1 colabeling showed cobblestone-like cells that did not have the oblong morphology characteristic of pericytes (see α-smooth muscle actin staining, Figures 1K and 1L). We also showed that a high glutamate dose (1 mmol/L) increased intracellular [Ca2+]i in newborn BMECs, in accordance with a recent study using calcium-green-fluorescence imaging in an adult BMEC model (Kuhlmann et al, 2008). Despite the difference in NMDAR expression, neonatal and adult BMECs responded similarly to glutamate, as assessed by calcium imaging approaches and MTT viability assay. Glutamate exposure under normoxic conditions did not induce adult endothelial cell death in an earlier study (Sharp et al, 2003) or in this study (see Supplementary Data on the MTT assay). In contrast, oxygen and glucose deprivation caused apoptosis of adult BMECs (Liu et al, 2004).
Importantly, we found age-related differences in NMDAR function manifesting as differences in glutamate-evoked t-PA, MMP-9, and MMP-2 release. Expression of t-PA occurs mainly in BMECs, glial cells, and neurons in the central nervous system. In addition to many beneficial effects on physiologic processes, t-PA may also have a variety of deleterious effects within the brain, including cleavage of the NMDAR1 subunit (Nicole et al, 2001) and amplification of extracellular MMPs (Tsuji et al, 2005) (e.g., MMP-9 and MMP-2). These effects may increase excitotoxicity, further damage the BBB, and cause exacerbation of edema and cerebral hemorrhage. Our findings in neonatal BMECs of increased t-PA, MMP-9, and MMP-2 activities are consistent with these possibilities. A still unanswered question is the manner by which the brain endothelium relates to adjacent neurons during an excitotoxic challenge and vice versa. If t-PA, MMP-9, and MMP-2 activities are upregulated after a glutamate overload, as suggested by our study, then investigations into the effects of these factors on neuron death would be of considerable interest (e.g., by coculture and/or conditioned media). Obviously, we cannot exclude the contribution of other endothelial factors, which may be neuroprotective or deleterious. For example, conditioned media from BMECs protects neurons from a wide range of insults (hypoxia, oxygen-glucose deprivation, oxidative stress) that act by brain-derived neurotrophic factor (Guo et al, 2008). However, conditioned media from microvessels from patients with Alzheimer's disease kill neurons via a protein-mediated pathway (Grammas et al, 2000). Finally, the role of endothelial secretions in brain insults remains to be elucidated.
Taken together, our findings strongly suggest that glutamate-induced BMEC signaling may be involved in neonatal excitotoxic brain injury. As neonatal BMECs expressed higher levels of NMDAR and protease activity (i.e., t-PA, MMP-2, and MMP-9) reversed by MK-801, we believe our data support a predominant role of endothelial NMDARs. These data support a link between glutamate, NMDAR, and proteases in the immature brain endothelium that may be relevant for understanding neonatal brain injury. Neonatal BMECs may be potential targets for neuroprotective strategies. Further investigations are needed to better understand the crosstalk between factors and cell types (e.g., neurons, glial cells, and oligodendrocytes) within the neonatal neurovascular unit.