
Abstract
The active zone (AZ) is a thickening of the presynaptic membrane where exocytosis takes place. Chemical synapses contain neurotransmitter-loaded synaptic vesicles (SVs) that at rest are tethered away from the synaptic release site, but after the presynaptic inflow of Ca+2 elicited by an action potential translocate to the AZ to release their neurotransmitter load. We report that tissue-type plasminogen activator (tPA) is stored outside the AZ of cerebral cortical neurons, either intermixed with small clear-core vesicles or in direct contact with the presynaptic membrane. We found that cerebral ischemia-induced release of neuronal tPA, or treatment with recombinant tPA, recruits the cytoskeletal protein βII-spectrin to the AZ and promotes the binding of SVs to βII-spectrin, enlarging the population of SVs in proximity to the synaptic release site. This effect does not require the generation of plasmin and is followed by the recruitment of voltage gated calcium channels (VGCC) to the presynaptic terminal that leads to Ca+2-dependent synapsin I phosphorylation, freeing SVs to translocate to the AZ to deliver their neurotransmitter load. Our studies indicate that tPA activates the SV cycle and induces the structural and functional changes in the synapse that are required for successful neurotransmission.
INTRODUCTION
The presynaptic terminal contains neurotransmitter-loaded synaptic vesicles (SVs) distributed in three pools known as readily releasable pool (RRP), recycling pool, and reserve pool. 1 The RRP is assembled by less than 1% of the total population of SVs. They are docked to the active zone (AZ) and therefore are available for immediate release upon stimulation. In contrast, SVs of the recycling and reserve pools represent 10% to 15% and 80% to 90% of the total population of SVs, respectively. However, because they are not docked to the AZ, to release their load of neurotransmitters they need first to translocate to the RRP. Because just a few milliseconds of depolarization deplete the RRP, the mobilization of SVs from the recycling and reserve pools to replenish the RRP is crucial to maintain neurotransmitter release during sustained synaptic activity. 2
Synapsin I is a phosphoprotein that at rest crosslinks SVs in the presynaptic terminal, preventing their mobilization from the reserve and recycling pools to the RRP. However, Ca+2-dependent phosphorylation of synapsin I during an action potential leads to its dissociation from SVs, freeing them to move to the AZ. 3 βII-spectrin is a cytoskeletal protein found in the presynaptic terminal 4 where its exact function is yet unclear. Nevertheless, the identification of a synapsin I-binding site in /βII-spectrin, 5 and the finding that either antibodies against this region 6 or βII-spectrin mutations block synaptic transmission, 7 have led to propose that it has a central role in synaptic function.
Tissue-type plasminogen activator (tPA) is a serine proteinase that in the brain is found in endothelial cells, glia, and neurons, where it has different roles. Indeed, whereas tPA released from endothelial cells into the intravascular space has a fibrinolytic effect mediated by its ability to catalyze the conversion of plasminogen into plasmin,
8
and tPA released from glia activates proinflammatory pathways,
9
induces neuroglial coupling,
10
and regulates the permeability of the blood-brain barrier,
11
the secretion of neuronal tPA has a robust effect on synaptic function. Accordingly, tPA mediates the development of neuronal plasticity in
Early studies showed that under physiologic conditions tPA activity is circumscribed to well-defined areas of the brain, namely the amygdala, the hippocampus, and the hypothalamus, in contrast to a more limited expression in the cerebral cortex. 17 However, subsequent experimental work showed that membrane-depolarizing stimuli such as cerebral ischemia induce the expression of tPA in cerebral cortical neurons, 11 and that its release into the synaptic space promotes neuronal adaptation and survival to metabolic stress.10,18–21 Remarkably, despite the importance of these events, the synaptic location and function of tPA in cerebral cortical neurons are still unclear.
The studies presented here indicate that tPA is found in the presynaptic terminal of cerebral cortical neurons, and that either its release at extrasynaptic sites induced by cerebral ischemia, or treatment with recombinant tPA, recruits the cytoskeletal protein βII-spectrin to the AZ and promotes the binding of SVs to βII-spectrin, bringing them in close proximity to the synaptic release site. We found that tPA also increases the expression of presynaptic Ca+2 channels, leading to Ca+2-dependent phosphorylation of synapsin I, which frees SVs to translocate to the AZ to release their neurotransmitter load. In summary, the studies presented here indicate that the release of tPA from cerebral cortical neurons induces the structural and functional changes in the synapse required to pair membrane depolarization with the presynaptic release of neurotransmitters.
MATERIALS AND METHODS
Animals and Reagents
Strains were 8- to 12-week-old male wild-type (Wt) and T4 mice (with a 10-fold increase in tPA expression in neurons,
22
kindly provided by Professor JD Vassalli and Dr R Mandani; University of Geneva, Switzerland) on a C57BL/6 J background. Experiments were approved by the Institutional Animal Care and Use Committee of Emory University, Atlanta GA, following guidelines established by ARRIVE (Animal Research: Reporting
Neuronal Cultures
Cerebral cortical neurons were cultured from E16 to E18 Wt mice as described elsewhere. 21 Briefly, the cerebral cortex was dissected, transferred into Hanks' balanced salt solution containing 100 units/mL penicillin, 100 μg/mL streptomycin, and 10 mmol/L HEPES, and incubated in trypsin containing 0.02% DNase at 37° for 15 minutes. Then tissue was triturated, and the supernatant was resuspended in B27-supplemented neurobasal medium containing 2 mmol/L L-glutamine and plated onto 0.1 mg/mL poly-L-lysine-coated wells.
Bilateral Common Carotid Artery Occlusion
Wild-type (
Proteomics Analyses
Proteomics analyses were performed as described elsewhere
21
in extracts from synaptoneurosomes prepared from Wt cerebral cortical neurons treated 60 seconds with tPA or vehicle (control). A subgroup of neurons was immunoprecipitated with an antibody against βII-spectrin or an IgG control. Based on our previous data18,23 and to assure that the concentrations of tPA in our experiments resemble as much as possible those found in an
Isolation of Synaptic Vesicles and Synapse-Containing
Fractions Synapse-enriched fractions containing the presynaptic terminal and the apposing postsynaptic membrane (synaptoneurosomes) were prepared according to a modification of published protocols24–27 from either Wt cerebral cortical neurons (days
Live Confocal Microscopy Studies and Quantification of AM1-44 Uptake
Days
Western Blot Analysis
Extracts prepared from Wt cerebral cortical neurons incubated 0 to 5 minutes with 5 nmol/L of either proteolytically active or inactive tPA, or from synaptoneurosomes from neurons treated 60 seconds with 5 nmol/L of tPA or a comparable volume of vehicle (control) and either left intact or subjected to sucrose density fractionation, or from cerebral cortical neurons treated 0 to 60 seconds with 5 nmol/L of tPA, alone or in the presence of 30 μmol/L of BAPTA-AM were homogenized and protein concentration was quantified using the BCA assay. To isolate membranes, neurons and synaptoneurosomes were washed with a buffer containing 0.25 mol/L sucrose/1 nmol/L EGTA and 10 mmol/L Tris-HCl at pH 8.0. Lysates were homogenized in a 2-mL tissue grinder, homogenates were centrifuged at 4° during 5 minutes (2,000 g), and supernatants were transferred to a new tube and centrifuged again at 32,000
Immunoprecipitation Studies
Lysates from synaptoneurosomes prepared from Wt cerebral cortical neurons treated with 5 nmol/L of tPA or vehicle (control) were harvested and lysed in RIPA buffer containing proteinase inhibitor, and incubated first with 0.1 μg of anti-β-II spectrin antibodies at 4° overnight, and then with 500 μg of Dynabeads Protein G (Life Technologies, Grand Island, NY, USA). Beads were washed five times with 300 μL of RIPA buffer, immunoprecipitated proteins were eluted with 30 μL of 2 × Laemmli Sample Buffer (Bio-Rad, Hercules, CA, USA), boiled for 10 minutes, and immunoblotted with antibodies against βII-Spectrin, SYP, and total synapsin I.
Immunocytochemistry and Quantification of p-Synapsin I Expression and βII-Spectrin/Synaptophysin Colocalization
Wild-type cerebral cortical neurons were incubated 0 to 60 seconds with 5 nmol/L of tPA or a comparable volume of vehicle (control), fixed, permeabilized with 50 μg/mL of digitonin, and blocked in 0.25% casein and 10% donkey serum in PBS and colabeled with antibodies against tPA (1:1,200) and either MAP-2 (1:2,500), or PSD-95 (1:2,000), or Tau (1:1,000). A second group of neurons was costained with antibodies against MAP-2 and βII-Spectrin (1:5,000), or SYP (1:2,500) and βII-Spectrin, or synapsin I phosphorylated at Serine-9 (1:2,000). Conjugated secondary antibodies were goat Alexa 488 or 594. Microphotographs were obtained with a Photometric Quantix digital camera connected to an Olympus-BX51 epifluorescence microscope (Center Valley, PA, USA). Each experiment was repeated with neurons from three different cultures. To quantify tPA/MAP-2, tPA/Tau, tPA/basson and p-synapsin I/βII-Spectrin/SYP and p-synapsin-positive puncta, pictures taken from the distal axons of tPA-treated and control neurons were straightened with ImageJ (NIH) and electronically magnified 300%. Images were inverted and merged in Photoshop by placing a copy of the RGB image in the blue channel, and then regions where green and red channel colocalize were copied to the blue channel rendering them white. With this technique, the degree of colocalization varies within a spectrum from light gray to white. In each case, the number of puncta in the distal 50 μm of each axon was quantified with the cell counter of ImageJ.
Electrophysiology Studies
The brain of male Sprague-Dawley rats (16 to 21 days old) was harvested after cardiac perfusion and cut onto 350 μm slices in cold oxygenated cutting solution containing 200 mmol/L sucrose, 2.5 mmol/L KCl, 1.2 mmol/L NaH2PO4, 25 mmol/L NaHCO3, 20 mmol/L dextrose, 0.5 mmol/L CaCl2, 2.4 mmol/L sodium pyruvate, 1.3 mmol/L L-ascorbic acid, and 7 mmol/L MgCl2. Slices were then incubated at 34° in oxygenated artificial cerebrospinal fluid containing 125 mmol/L NaCl, 2.8 mmol/L KCl, 1 mmol/L NaH2PO4, 26 mmol/L NaHCO3, 10 mmol/L D-glucose, 2 mmol/L CaCl2, and 1.5 mmol/L MgSO4. The external solution was continuously bubbled with 95% O2/5% CO2 at pH 7.2, and transferred to the recording chamber attached to an Olympus Optical BX51 microscope (Olympus, Tokyo, Japan). Recordings were made at 32°. Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich. For isolation of glutamatergic miniature excitatory postsynaptic currents (mEPSCs) the bath solution contained 10 μmol/L of bicuculline methiodide to block GABAA receptors, and 1 μmol/L of tetrodotoxin to block Na+ action potentials. Slices were perfused with either tPA at a final concentration of 5 nmol/L or vehicle (control). Using glass electrodes (4 to 8 MΩ), patch-clamp recordings (>1 GΩ) from CA1 pyramidal cells in the whole-cell configuration were acquired (voltage-clamp mode), low-pass filtered (6 kHz), and digitized (20 kHz) using a Multiclamp 700B amplifier and Clampex 10.2 (both from Molecular Devices, Sunnyvale, CA, USA). Resting membrane potential varied from − 57 mV to − 68 mV. Electrodes were filled with K-gluconate intracellular solution (in mmol/L): 94 K-gluconate, 10 NaCl, 36 KCl, 1.1 EGTA, 10 HEPES, 1 MgCl2,1 CaCl2,1 Mg ATP, 0.5 Na-GTP, pH 7.2. Electrophysiologic data were measured and preanalyzed using Clampfit v10.2 (Molecular Devices).
Immunogold Electron Microscopy Studies
Mouse brains were fixed by transcardial perfusion with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.2) for 20 minutes followed by immersion-fixation overnight at 4°. Brains were harvested, sectioned onto 50 μm cuts, and permeabilized in 0.05% Triton X-100 for 10 minutes. After permeabilization, brain sections were incubated in PBS containing 5% rabbit serum, 5% BSA, and 0.1% gelatin to block potential nonspecific interaction between immunoreagents and samples, and incubated overnight with 5 μg/mL of sheep anti-tPA primary antibodies, washed, and incubated with ultrasmall gold particles conjugated to rabbit and sheep antibodies (Aurion, Wageningen, The Netherlands) at 1:100 dilution in PBS/PBS-c. After several washes brain sections were fixed with 2.5% glutaraldehyde in 0.1 mol/L PB. Silver enhancement using the Aurion R-gen SE-EM kit was then conducted following the manufacturer instructions. Brain sections were then fixed with 0.5% osmium tetroxide for 15 minutes, dehydrated and embedded in Eponate 12 resin. Then, areas of the frontal cortex were dissected out from flat embedded vibrating microtome sections and re-embeded, cut onto 70 nm sections, stained with uranyl acetate and lead citrate, and examined with a JEOL JEM-1400 transmission electron microscope (Tokyo, Japan) equipped with a Gatan US100 CCD camera (Pleasenton, CA, USA).
Statistical Analysis
Statistical analysis was performed with two-tailed
RESULTS
Synaptic Expression of Tissue-Type Plasminogen Activator in Cerebral Cortical Neurons
To study the synaptic expression of tPA, Wt cerebral cortical neurons were immunostained with antibodies against tPA, MAP-2 (delineates dendrites), and Tau (identifies axons). Although we detected tPA antigen in MAP-2-positive extensions, we found that most of tPA is expressed in axons wrapped around dendrites (Figures 1A–1D). To determine whether tPA is expressed only in the axonal shaft or also in the synaptic terminal, we performed additional studies with antibodies against PSD-95 (delineates the postsynaptic density) and bassoon (detects the presynaptic terminal). Our data indicate that in the synapse tPA is found mostly in the presynaptic axonal bouton, as denoted by its direct apposition with the PSD (Figure 1E) and colocalization with bassoon (Figures 1F and 1G). Accordingly, we found that approximately one-third (34.30±5.2%) of the total number of tPA-positive puncta located in the distal 50 μm of axons from cerebral cortical neurons colocalize with bassoon (Figure 1H).
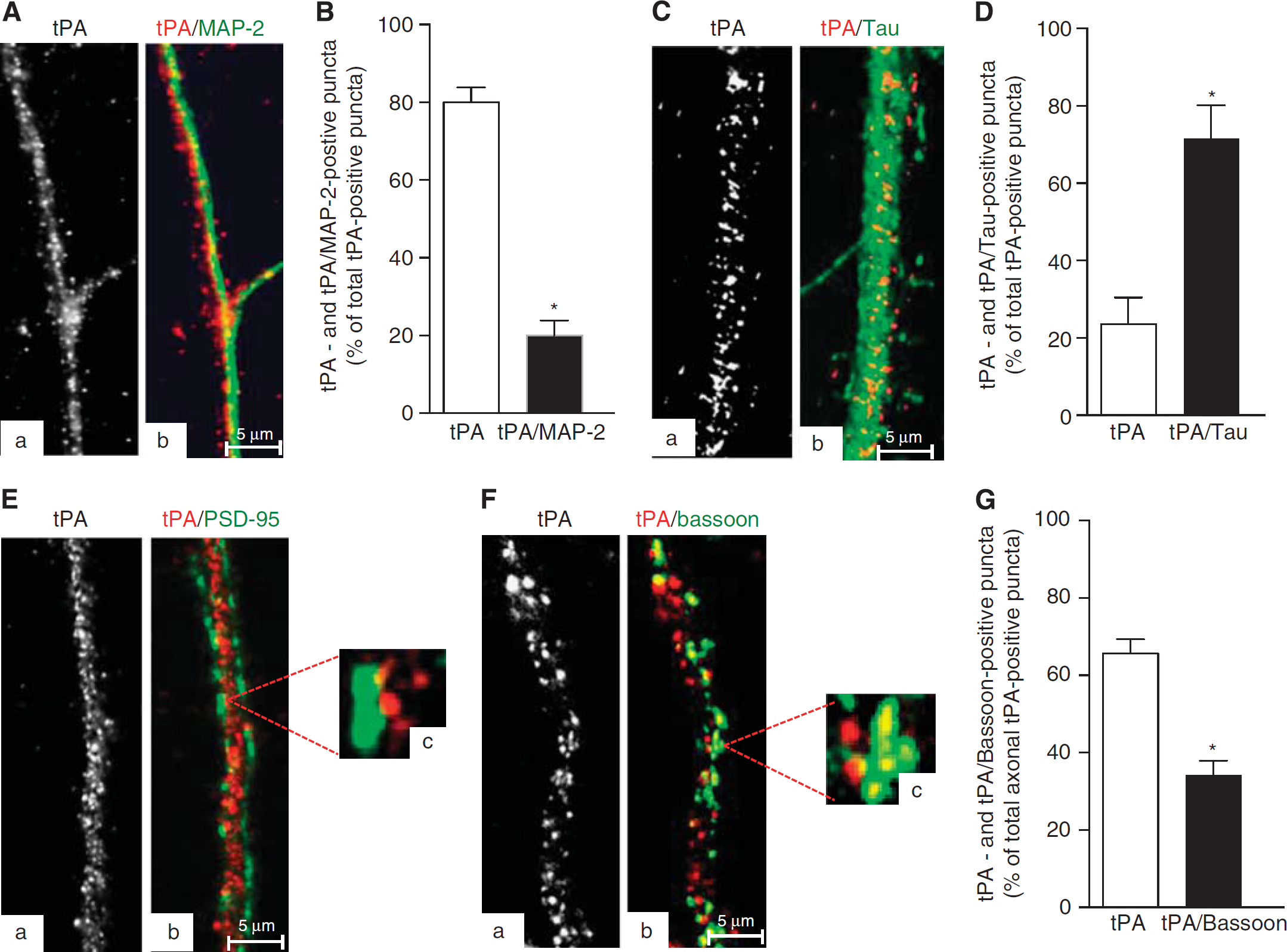
Axonal and dendritic expression of tissue-type plasminogen activator (tPA) in cerebral cortical neurons. (
These data suggest that tPA is localized not only diffusely throughout the axon but also in the presynaptic bouton. To further characterize these observations, synaptoneurosomes prepared from Wt cerebral cortical neurons were subjected to sucrose density fractionation to isolate the synapse, assembled by the presynaptic membrane, SVs docked to the AZ, and the attached PSD of the postsynaptic membrane. Then, each gradient fraction was immunoblotted with antibodies against tPA, SYP (an integral transmembrane protein found in SVs), syntaxin I (a transmembrane protein found in the presynaptic plasma membrane), and PSD-95 (detects the PSD). As previously described, synaptic fractions containing the AZ are identified by their immunoreativity to SYP and syntaxin I.25,26 We found that in the presynaptic terminal tPA is stored outside the AZ (Figure 2A), and in line with these findings our electron microscopy studies detected tPA-containing vesicles in extrasynaptic sites of most of the presynaptic boutons, either intermixed with small clear-core vesicles (arrowheads in Figures 2B–2D), or in direct contact with the presynaptic membrane (asterisk in Figure 2D and dashed square in Figure 2E, magnified in Figure 2F). We also detected tPA-containing vesicles in fewer postsynaptic terminals, either attached to the post-synaptic membrane (continuous square in Figure 2E, magnified in Figure 2G), or in the dendritic spine (arrow in Figure 2E).
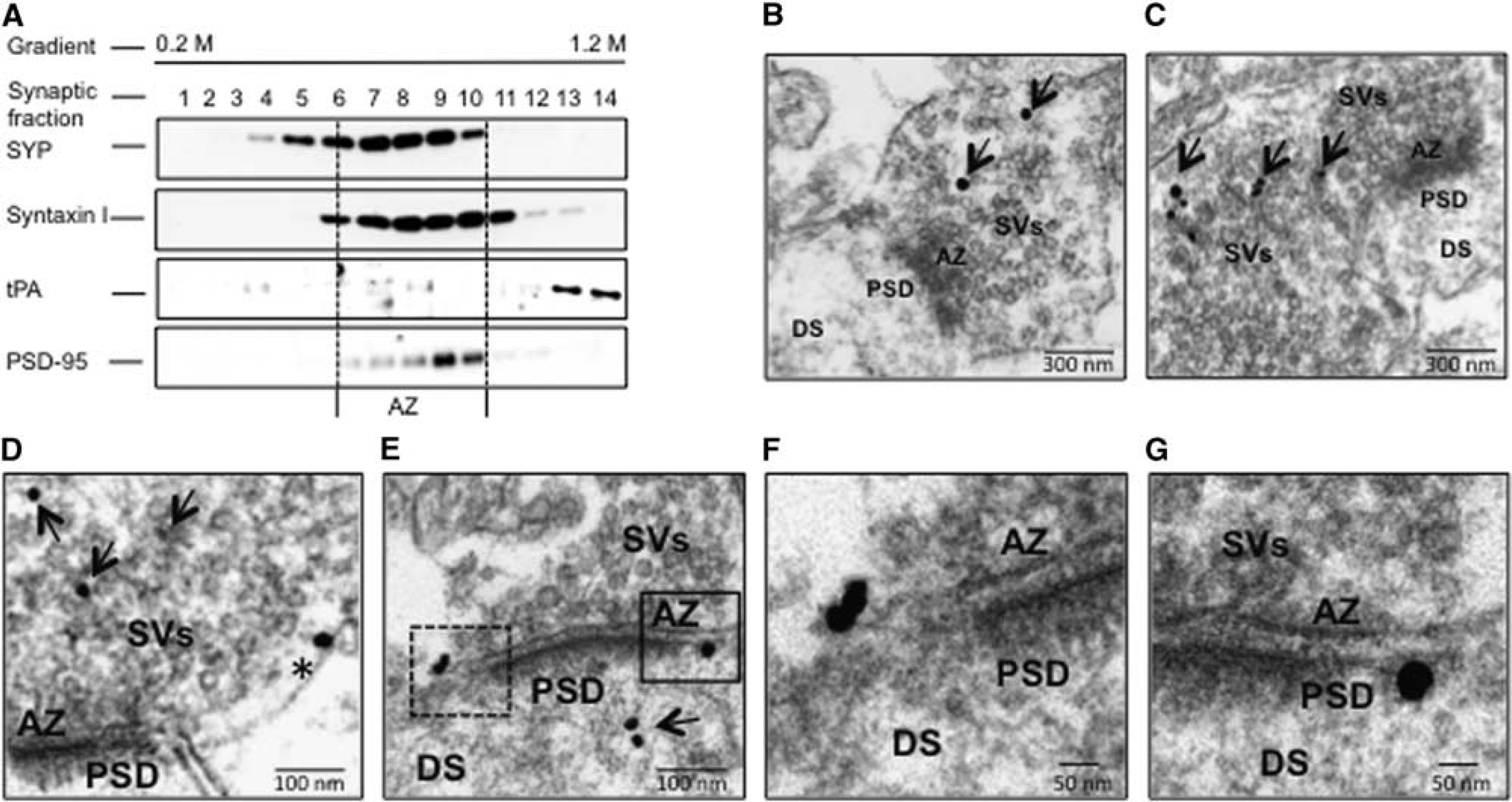
Tissue-type plasminogen activator (tPA) expression in the presynaptic bouton. (
Effect of Tissue-Type Plasminogen Activator on the Presynaptic Terminal
To investigate whether the release of neuronal tPA has an effect on the presynaptic terminal, we used liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) and subsequent analysis with the DAVID Bioinformatics Database to study changes in protein abundance in synaptoneurosomes prepared from Wt cerebral cortical neurons incubated during 60 seconds with 5 nmol/L of tPA or vehicle (control). We found that tPA has a robust effect on the abundance of the cytoskeletal protein βII-spectrin (6.77-fold increase compared with control-treated neurons;
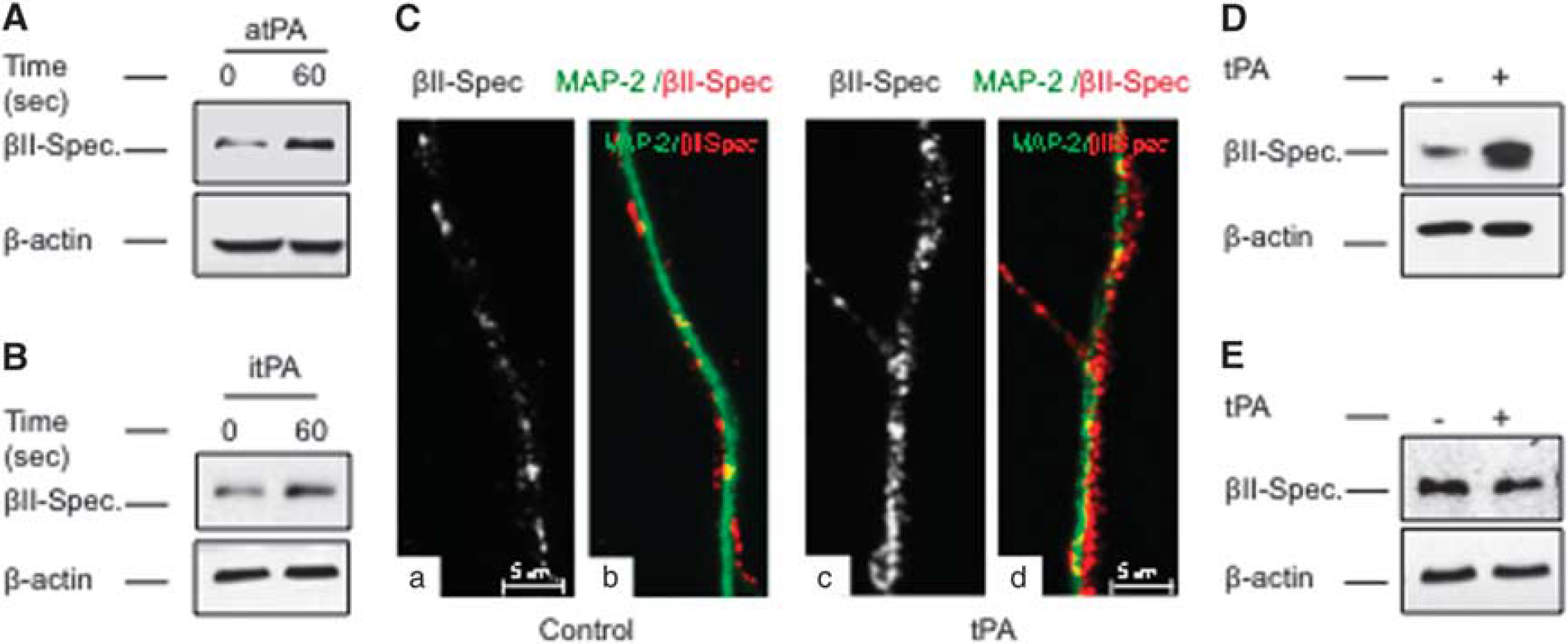
Effect of tissue-type plasminogen activator (tPA) on neuronal βII-spectrin. (
To further study the effect of tPA on βII-spectrin expression in the presynaptic terminal, synapse fractions isolated from synaptoneurosomes prepared from Wt cerebral cortical neurons treated 60 seconds with 5 nmol/L of either proteolytically active or inactive tPA (atPA or itPA, respectively), or with vehicle (control) were immunoblotted with antibodies against SYP, βII-spectrin, syntaxin I, and PSD-95. Our data indicate that tPA induces the recruitment of βII-spectrin to the AZ (Figures 4A and 4B) by a mechanism independent of its ability to catalyze the conversion of plasminogen into plasmin (Figures 4D and 4E). Remarkably, this effect was accompanied by an increase in SYP immmunoreactivity in the AZ (Figures 4A & 4C and 4D & 4F). Cerebral ischemia induces the rapid release of tPA from cerebral cortical neurons. 18 Thus, to study whether endogenous tPA also recruits βII-spectrin to the AZ we performed similar observations in synaptic fractions isolated from the forebrain of Wt mice subjected to either 1 minute of BCCAO, which induces the release of neuronal tPA without causing cell death, 18 or sham-operation (controls). We found that compared with Wt controls BCCAO increases the expression of βII-spectrin in the AZ (Figures 4G and 4H). To further characterize the role of endogenous tPA on this effect, we performed similar observations with synaptic extracts from Wt and T4 mice (with a 10-fold increase in neuronal tPA expression) 22 60 seconds after BCCAO. Our data indicate that compared with Wt animals ischemia causes a larger increase in βII-spectrin expression in the AZ of T4 mice (Figures 4J and 4K).
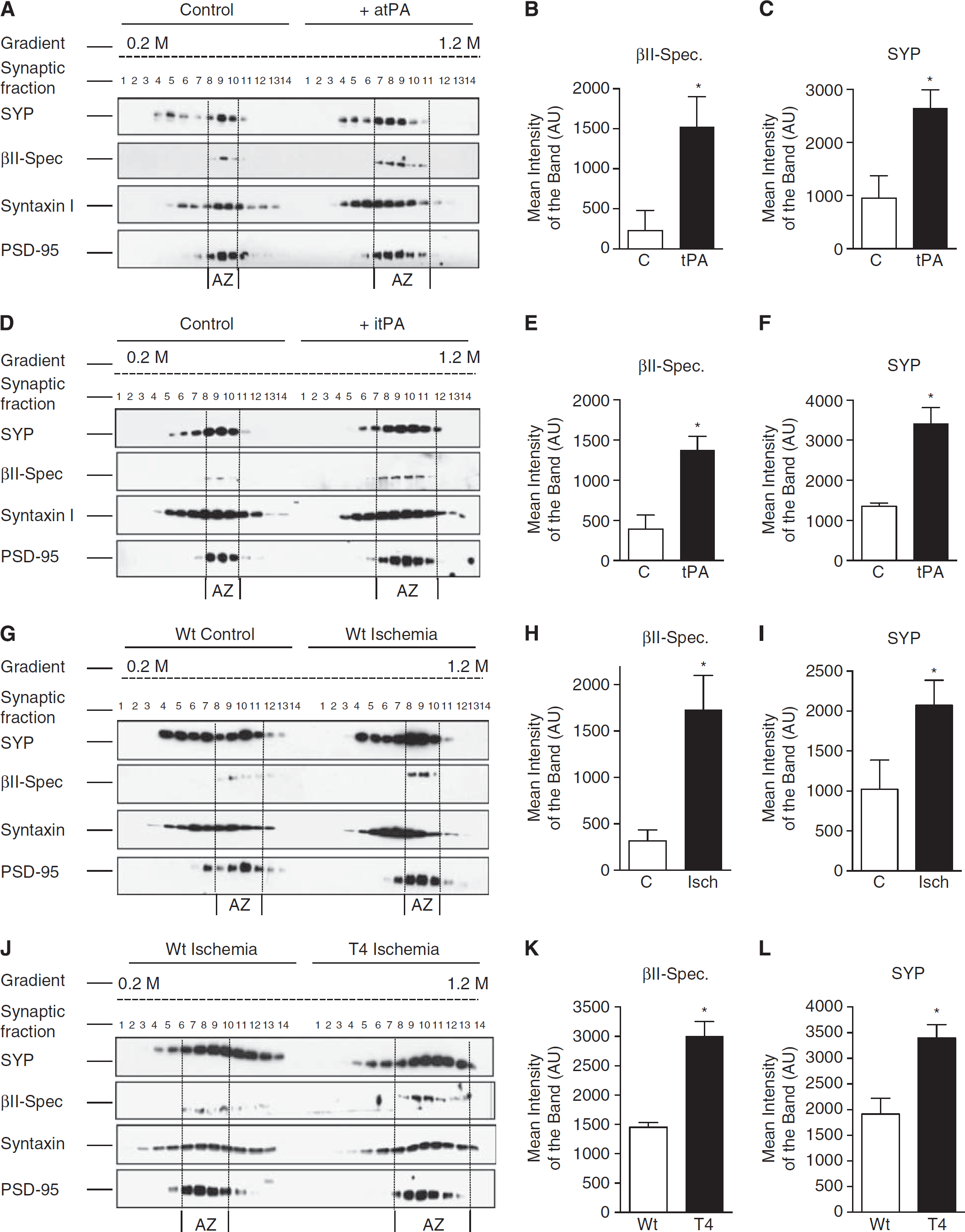
Tissue-type plasminogen activator (tPA) recruits βII-spectrin to the active zone (AZ). Representative western blot analysis (
Tissue-Type Plasminogen Activator Promotes the Binding of Synaptic Vesicles to βII-Spectrin
Synapsin I is a phosphoprotein that clusters SVs in the presynaptic terminal.
28
Because βII-spectrin has a synapsin I-binding site,
5
and based on our finding that tPA increases the expression of both βII-spectrin and SYP in the AZ (Figure 4), we postulated that by inducing the recruitment of βII-spectrin, tPA also enlarges the population of βII-spectrin-bound SVs in close proximity to the synaptic release site. To test this hypothesis, we performed immunofluorescence studies to quantify the colocalization of SYP and βII-spectrin in the distal 50 μm of Wt axons of neurons treated 60 seconds with either 5 nmol/L of tPA or vehicle (control). Our data show that tPA increases the number of SYP/βII-spectrin-positive puncta from 17.46 ±3.56 /50 μm to 33.91 ±4.83/50 μm; Figures 5A and 5B;
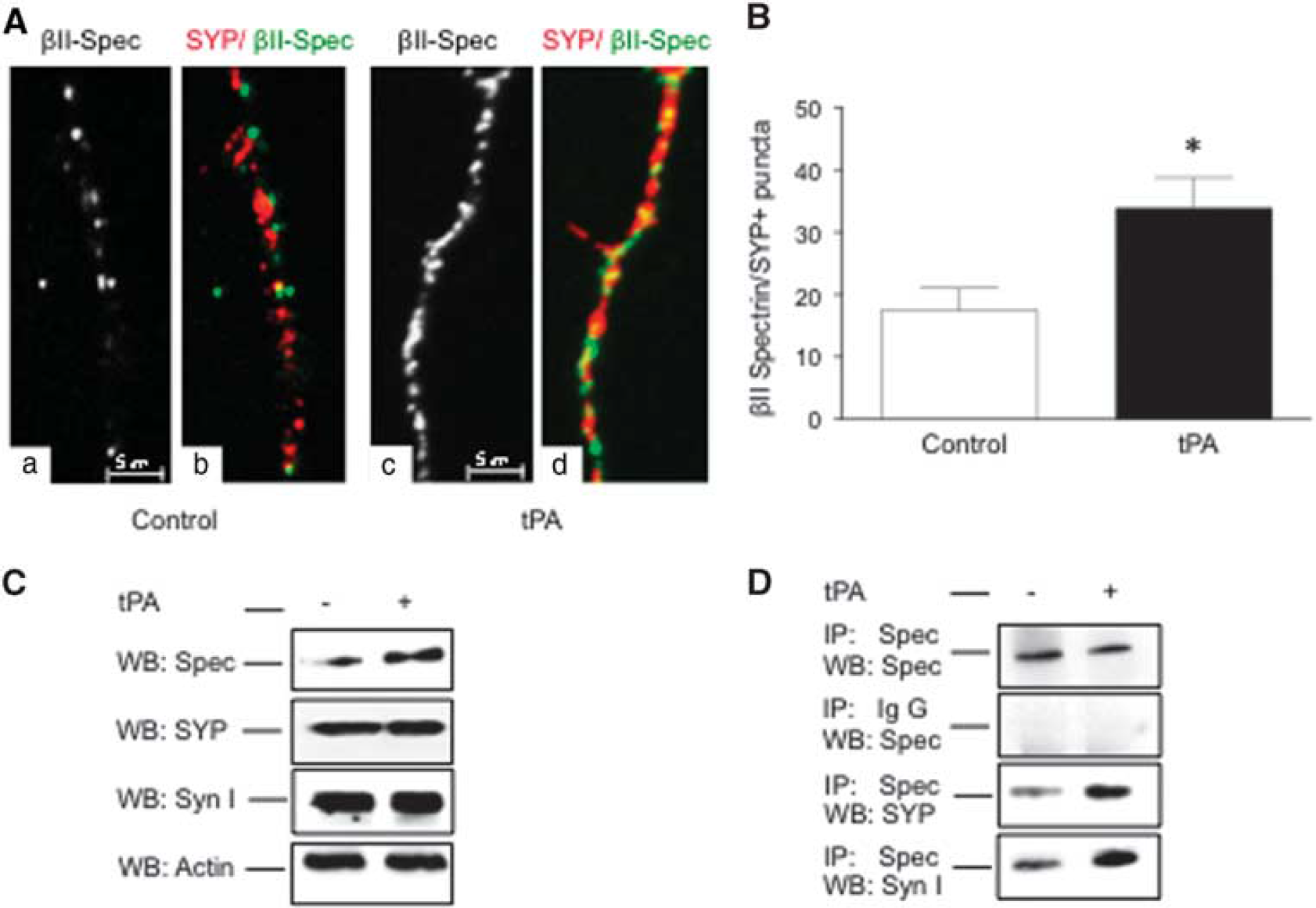
Tissue-type plasminogen activator (tPA) induces the binding of synaptic vesicles to βII-spectrin. (
To further characterize these observations, extracts from synaptoneurosomes prepared from Wt cerebral cortical neurons treated 60 seconds with 5 nmol/L of tPA or vehicle (control) were divided into two groups. The first was immunoblotted with antibodies against βII-spectrin, SYP, or synapsin I. The second was immunoprecipitated with antibodies against βII-spectrin and immunoblotted with antibodies against either SYP or synapsin I. We found that while tPA does not have an effect on the abundance of SVs in the presynaptic terminal, as denoted by unchanged SYP and synapsin I expression in immunoblots from control- and tPA-treated samples (Figure 5C), it enlarges the abundance of βII-spectrin-bound SVs, as indicated by an increase in SYP and synapsin I immunoreactivity in samples of tPA-treated neurons immunoprecipitated with anti-βII-spectrin antibodies (Figure 5D).
Tissue-Type Plasminogen Activator Induces SVs Mobilization to the AZ
Under resting conditions synapsin I clusters SVs and tethers them to the cytoskeleton. However, during synaptic activity synapsin I phosphorylation at Ser 9 leads to its dissociation from SVs freeing them to move to the AZ. 29 Our data indicate that tPA increases the population of SVs bound to βII-spectrin attached to the AZ. To investigate whether tPA also facilitates their mobilization to the synaptic release site, we performed western blot analysis and immunofluorescence studies of synapsin I phosphorylated at Ser 9 (p-Syn I) in Wt cerebral cortical neurons treated 0 to 60 seconds with 5 nmol/L of tPA. We found that tPA induces synapsin I phosphorylation (Figures 6A–6C) and that this effect does not require tPA's-catalyzed conversion of plasminogen into plasmin (Figure 6D).
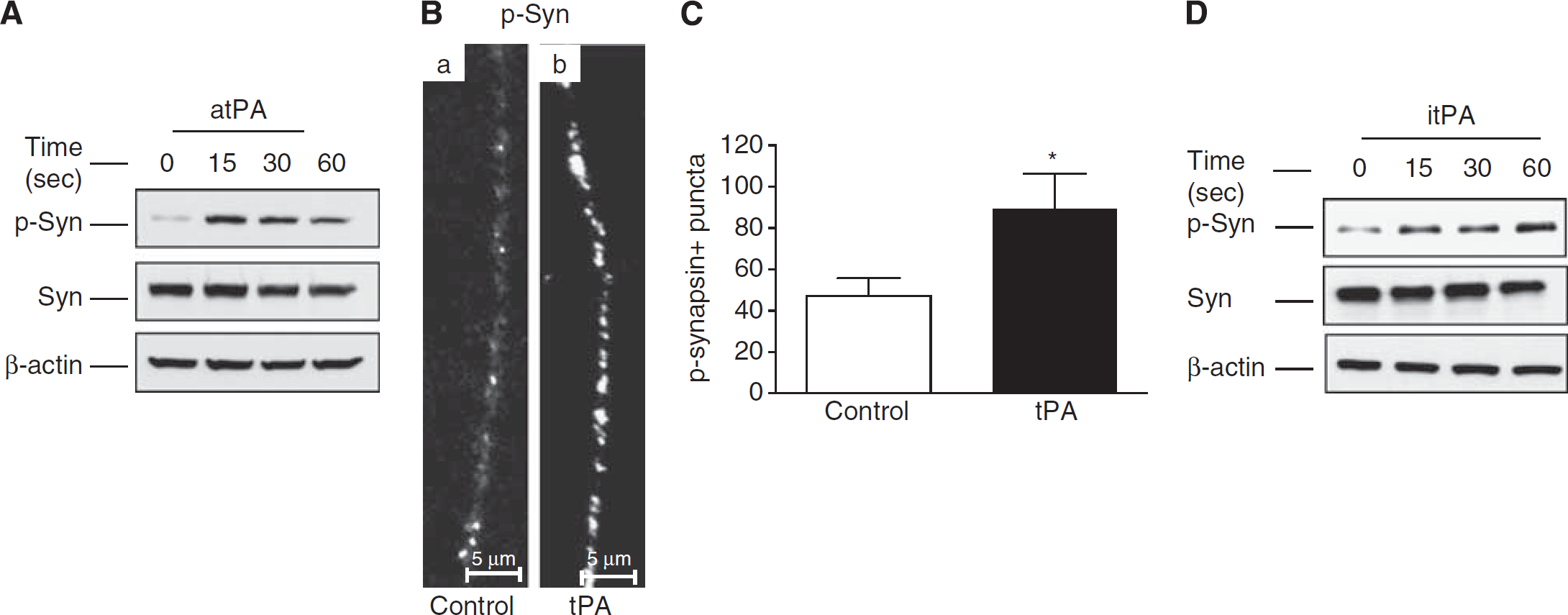
Tissue-type plasminogen activator (tPA) induces synapsin I phosphorylation. (
Because synapsin I phosphorylation requires the inflow of Ca+2 into the presynaptic terminal, 30 we decided to investigate the effect of the fast cell permeable Ca+2 chelator BAPTA on tPA-induced p-Syn I (Ser 9) expression. Our observation that tPA-induced synapsin I phosphorylation is abrogated by BAPTA (Figure 7A) suggests an effect of tPA on presynaptic Ca+2 channels. To test this hypothesis, we studied the expression of the α1 pore-forming subunit of presynaptic VGCC in either synaptoneurosomes or whole-cell extracts prepared from Wt cerebral cortical neurons treated during 60 seconds with 5 nmol/L of tPA or a comparable volume of vehicle (control). Our data indicate that tPA increases the abundance of the α1 subunit of VGCC in the presynaptic terminal (Figure 7B) but not in whole-cell extracts (Figure 7C).
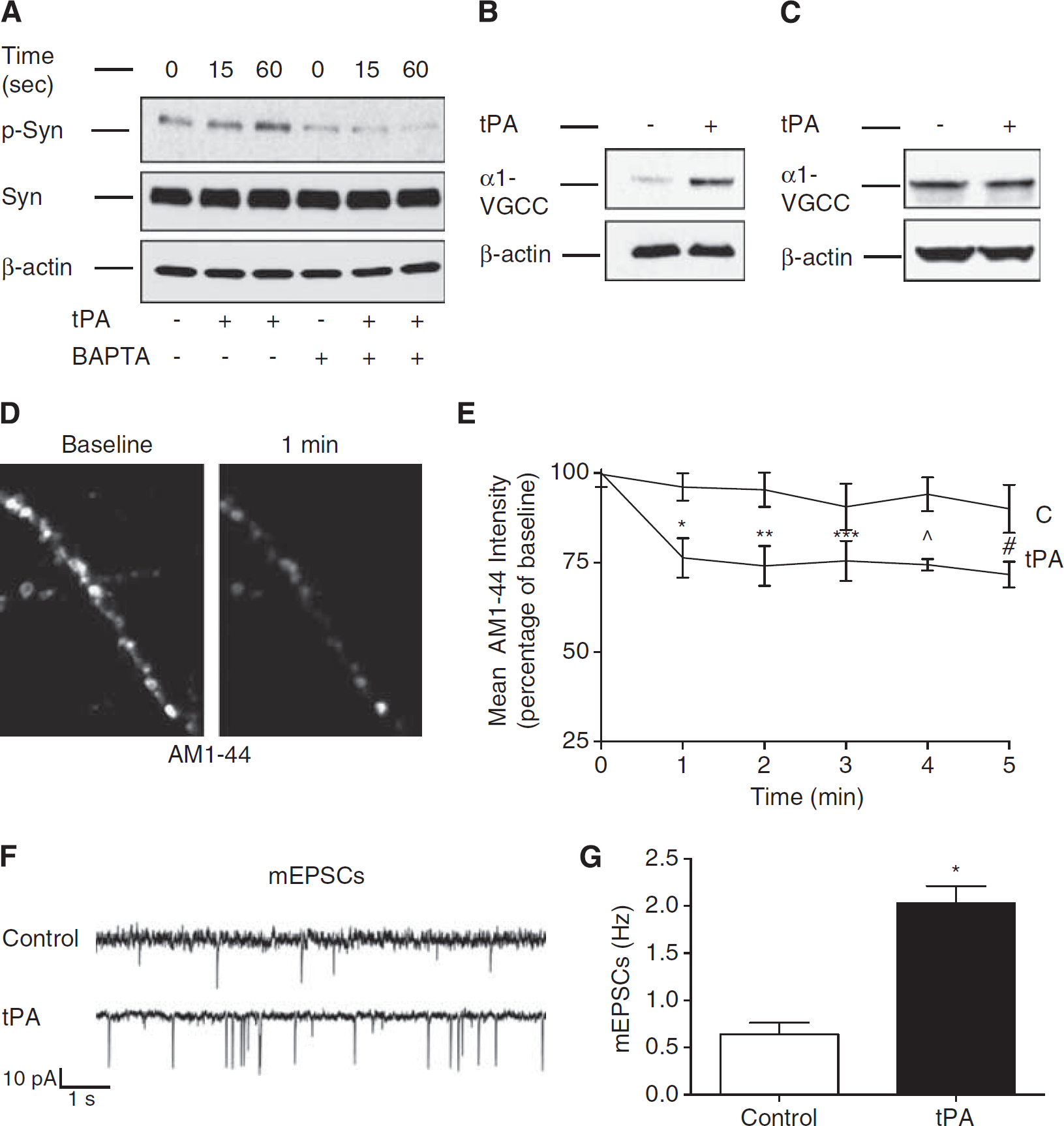
Tissue-type plasminogen activator (tPA) promotes the presynaptic release of neurotransmitters. (
To study whether besides coupling SVs with Ca+2 channels in the AZ, tPA also promotes the release of their load of neurotransmitters, Wt cerebral cortical neurons with their SVs previously loaded with the styryl dye AM1-44 were treated with 5 nmol/L of tPA or a comparable volume of vehicle (control). The release of AM1-44 from SVs was then continuously monitored during 60 minutes with live confocal microscopy as described in Materials and Methods. Our data indicate that tPA induces the rapid release of AM1-44 from SVs. This effect (~19.77% decrease in AM1-44 intensity compared with presynaptic terminals from vehicle-treated neurons) was observed within the first 60 seconds of treatment and remained unchanged thereafter (Figures 7D and 7E;
DISCUSSION
The capacity to undergo experience-dependent changes in synaptic structure and function is one of the most important features of the central nervous system. This property, known as synaptic plasticity, is fundamental not only for the performance of highly complex functions such as perception, learning, and memory, but also to promote neuronal adaptation to an ischemic injury.
31
A substantial body of experimental evidence indicates that neuronal tPA mediates the development of synaptic plasticity either by plasmin-induced degradation of extracellular matrix components,
32
or by plasmin-mediated activation of neurotrophins that have a direct effect on dendritic structure.
33
Likewise, it has been postulated that the interaction of tPA with
Early studies with PC-12 cells found that tPA is stored in catecholamine storage vesicles, 34 and in agreement with these observations, later studies showed that in hippocampal neurons tPA is stored in postsynaptic large-dense core vesicles. 35 Surprisingly, the synaptic location of tPA in cerebral cortical neurons has not been studied yet. Our electron microscopy analyses indicate that although tPA is detected in dendrites, most of it is found in the axonal shaft and in presynaptic vesicles either intermixed with clear-core SVs of the recycling and reserve pools, or in direct contact with the presynaptic membrane. In contrast, we detected tPA in significantly fewer postsynaptic terminals, and in some of them it was attached to the post-synaptic membrane, supporting the possibility postulated by others in hippocampal cells, that dendritic spines also release tPA. 35 In line with these observations, our studies with synaptic fractions containing only the presynaptic membrane, SVs, and attached PSD (these preparations do not contain the post-synaptic terminal24,25) indicate that in cerebral cortical neurons tPA is found in the presynaptic terminal outside the AZ. This finding is important because it indicates that tPA is released at nonsynaptic sites suggesting that, as it has been described for presynaptic neuropetides stored in large-dense core vesicles outside the synaptic release site, tPA may also be able to regulate synaptic function in a large number of neurons, even those located at long distance within the brain. Also, because the release of large-dense core vesicles content is proportional to the magnitude of the stimulus, it is possible that by inducing structural and functional changes in the presynaptic terminal, tPA ensures that the intensity of the depolarizing stimulus is matched by a proportional release of excitatory neurotransmitters.
Spectrin is a cytoskeletal protein with an (αβ)2 tetrameric subunit composition critical for membrane structural integrity. 36 Early studies described the expression of βII-spectrin in the presynaptic terminal, 37 and our data indicate that tPA increases its expression in the AZ by a mechanism that does not require the conversion of plasminogen into plasmin. Importantly, we found that tPA does not have an effect on βII-spectrin mRNA, and that tPA-induced βII-spectrin protein expression in the AZ is not abrogated by inhibition of protein synthesis. These observations and our data indicating that tPA increases the expression of βII-spectrin in membranes, synaptoneurosomes, and presynaptic fractions, but not in whole-cell extracts, suggest that tPA induces the recruitment of βII-spectrin to the AZ.
Only a fraction of the total population of SVs in the presynaptic terminal are docked to the AZ, and therefore are available for immediate release. 1 Thus, to ensure successful neurotramission during sustained neuronal activity this pool needs to be constantly replenished with SVs from another cluster of vesicles tethered away from the AZ, namely the recycling and reserve pools. We found that by increasing the expression of βII-spectrin in the AZ, tPA also enlarges the population of βII-spectrin-bound SVs in close proximity to the synaptic release site. More importantly, our biochemical, live microscopy, and electrophysiological studies show that by inducing synapsin I phosphorylation tPA frees these SVs to translocate to the synaptic release site where they release their neurotransmitter load.
Synapsins are the most abundant phosphoproteins in brain synapses. 38 Three synapsins have been identified: synapsin I and II localized almost exclusively to presynaptic terminals, and synapsin III found in growth cones and cell bodies. 28 The release of neurotransmitters from SVs is a Ca+2-dependent process that requires the coupling of presynaptic Ca2+ channels with Ca2+ sensors in SVs. 39 Our data suggest that tPA recruits Ca2+ channels to the presynaptic terminal. These observations, coupled to our finding that BAPTA abrogates tPA-induced synapsin I phosphorylation, indicate that by increasing the abundance of SVs bound to βII-spectrin in the AZ, tPA also promotes their coupling with presynaptic Ca+2 channels in the synaptic release site. More importantly, our live microscopy and electrophysiology data indicate that tPA not only brings SVs to the AZ but also promotes their release of neurotransmitters into the synaptic cleft.
In summary, based on our data we propose a model where the extrasynaptic release of neuronal tPA during the early phases of an ischemic injury induces the structural and functional changes in the presynaptic terminal required for the translocation of SVs from the reserve and recycling pools to the RRP and the subsequent release of their neurotransmitter load. Our results suggest that tPA is a neuromodulator that matches the intensity of the depolarizing stimulus with the corresponding activation of the SV cycle.
Footnotes
FW, ET, DC-G, LC, HY, and EKB performed experiments; PSG assisted with experimental design and performed statistical analysis; MY designed experiments, performed statistical analysis, and wrote manuscript.
The authors declare no conflict of interest.