
Abstract
Studies in animal models of cerebral ischemia indicate that besides its thrombolytic effect, treatment with tissue-type plasminogen activator (tPA) also induces an increase in matrix metalloproteinase-9 (MMP-9) activity in the ischemic tissue associated with the development of cerebral edema. Earlier, we had shown that the low-density lipoprotein receptor-related protein 1 (LRP1) is a substrate for tPA in the brain. In this study, we investigated the effect of the interaction between tPA and microglial LRP1 on MMP-9 activity after middle cerebral artery occlusion (MCAO). We found that exposure to oxygen–glucose deprivation (OGD) conditions increases MMP-9 activity in wild-type (Wt) and plasminogen-deficient (Plg−/−) microglia, but not in tPA (tPA−/−) or LRP1-deficient (macLRP−) cells. Treatment with tPA increases MMP-9 expression in tPA−/− but not in macLRP− microglia. Middle cerebral artery occlusion increases MMP-9 expression and activity in Wt but not in tPA−/− or macLRP− mice, and treatment with tPA increases MMP-9 activity in tPA−/− mice but not in macLRP− animals. Finally, MCAO-induced ischemic edema and degradation of the interendothelial right junction protein claudin-5 were significantly attenuated in tPA−/− and macLRP− mice. The results of our study indicate that the interaction between tPA and microglial LRP1 increases MMP-9 expression and activity resulting in the degradation of claudin-5 and development of cerebral edema.
Keywords
Introduction
The neurovascular unit (NVU) is a dynamic structure assembled by endothelial cells, the basal lamina, astrocytic end-feet processes, surrounding neurons, and perivascular microglia (Abbott et al, 2006). Ischemic stroke is the third cause of mortality and is the leading cause of disability in the world (World Health Organization, 2006). Early after the onset of cerebral ischemia, there is an increase in the permeability of the NVU associated with the development of cerebral edema. Matrix metalloproteinase-9 (MMP-9) is a member of the metalloproteinase family classically recognized as matrix-degrading enzymes. After the onset of cerebral ischemia, there is a progressive increase in the activity of MMP-9 in the ischemic tissue associated with an increase in the permeability of the NVU (Asahi et al, 2000, 2001).
Tissue-type plasminogen activator (tPA) is a highly specific serine proteinase and is one of the two main plasminogen activators. In the intravascular space, tPA is primarily a thrombolytic enzyme and its substrate is plasminogen (Plg). However, tPA is also expressed within the central nervous system (CNS), where it is associated with learning, synaptic plasticity (Krystosek and Seeds, 1981), cell death (Tsirka et al, 1995), and regulation of NVU permeability (Yepes et al, 2003; Polavarapu et al, 2007). We (Yepes et al, 2003) along with others (Wang et al, 2003; Lee et al, 2007) have shown that tPA increases the levels and activity of MMP-9 both in cell cultures and in an animal models of cerebral ischemia.
The low-density lipoprotein receptor-related protein-1 (LRP1) is a member of the low-density lipoprotein (LDL) receptor gene family composed of a 515-kDa heavy chain that is noncovalently bound to an 85-kDa light chain containing a transmembrane and cytoplasmic domains (Herz and Strickland, 2001). Low-density lipoprotein receptor-related protein-1 mediates the internalization of apoE-enriched lipoprotein particles, α-2-macroglobulin–protease complexes, and several other ligands, including plasminogen activators, proteinase–inhibitor complexes, clotting factors, the amyloid precursor protein, and MMP-9 (Herz and Strickland, 2001).
Our earlier studies indicate that the onset of ischemic insult is followed by an increase in the expression of LRP1 in the ischemic tissue, and that treatment with either the receptor-associated protein or anti-LRP antibodies after middle cerebral artery occlusion (MCAO) decreases the magnitude of ischemic edema (Polavarapu et al, 2007). Tissue-type plasminogen activator is a ligand for LRP1 (Bu et al, 1992a, 1992b), and evidence of the fact that the effect of tPA on the permeability of the NVU is mediated by its interaction with LRP1 comes from earlier studies, indicating that the intraventricular injection of tPA into wild-type (Wt) mice induces a dose-dependent increase in the permeability of the NVU, and that this effect is attenuated when tPA is coadministered with either receptor-associated protein or anti-LRP antibodies (Yepes et al, 2003). Importantly, the binding of tPA to LRP1 increases the synthesis of MMP-9 (Hu et al, 2006).
Microglia are immune cells of the CNS that become activated in response to changes in the microenvironment induced by multiple pathologic situations, such as cerebral ischemia (Hanisch and Kettenmann, 2007). Indeed, the onset of cerebral ischemia induces the activation of microglial cells that results in the generation of a local inflammatory reaction mediated, among others, by the induction of MMP-9 expression and activity (Stoll et al, 1998). Our earlier work indicates that LRP1 is expressed in microglial cells (Zhang et al, 2009). In the studies presented herein, we show that the effect of tPA on the expression and activity of MMP-9 in the ischemic tissue is largely mediated by its interaction with microglial LRP1, and that this process leads to degradation of the tight junction protein claudin-5 and increase in the permeability of the NVU. This is not only a novel mechanism for the development of ischemic edema but is also a potential target for the development of new therapeutic strategies aimed at preserving the integrity and barrier function of the NVU during cerebral ischemia.
Materials and methods
Animal Model of Cerebral Ischemia and Analysis of Vascular Permeability
Murine strains were Wt C57BL/6J, tPA deficient (tPA−/−), and plasminogen deficient (Plg−/−). LRP-floxed mice on an LDL receptor-deficient background were kindly provided by Dr Joachim Herz (University of Texas Southwestern, Dallas, TX, USA). These mice were crossed with LysMCre mice as described previously (Lillis et al, 2008), and then backcrossed with Wt C57BL/6J mice to generate LRPflox+/− Cre+/− mice on an LDL receptor Wt background. These mice were crossed with each other to generate LRPflox+/+ Cre+ (designated macLRP−) or LRP flox+/+ Cre− (designated Wt) mice, which were used as littermate controls. Transient MCAO (tMCAO) was induced with a 6-0 silk suture advanced from the external carotid artery into the internal carotid artery until the origin of the MCA as described elsewhere (Belayev et al, 1999). Briefly, after making a midline skin incision, the external carotid artery was isolated and ligated proximally with a 6-0 silk suture. A nylon monofilament (6-0, Ethicon, Issy Les Moulineaux, France), coated with a mixture of silicone resin (Xantopren Mucosa; Heraeus Kulzer, Hanau, Grunerweg, Germany) and a hardener (Universal activator; Heraeus Kulzer) was introduced through the incision in the external carotid artery and advanced gently up to the origin of the MCA. The suture was tightly fixed at the final position and withdrawn after 60 mins of cerebral ischemia. Cerebral perfusion in the distribution of the MCA was monitored throughout the surgical procedure and after reperfusion with a laser Doppler (Perimed, North Royalton, OH, USA), and only animals with a >70% decrease in cerebral perfusion after occlusion and complete recovery after suture withdrawal were included in this study. The rectal and masseter muscle temperatures were controlled at 37°C with a homoeothermic blanket. Heart rate, systolic, diastolic, and mean arterial blood pressures were controlled throughout the surgical procedure using an IITC 229 System (IITC-Life Science; Woodland Hills, CA, USA). Immediately after tMCAO, a subgroup of animals was intracortically injected at the bregma (−1 mm), mediolateral (3 mm), and dorsoventral (3 mm) with 2 μL of either phosphate-buffered saline or murine tPA (60 nmol/L; Molecular Innovations, Royal Oak, MI, USA). For permeability studies, Wt and macLRP− mice were injected intravenously with 2% Evans blue dye followed by tMCAO. Twelve hours later, the brains were harvested and the extravasation of Evans blue dye was quantified in the ischemic hemisphere as described elsewhere (Yepes et al, 2003). Each observation was repeated 10 times. Statistical analysis was performed with Student's t-test.
Cell Cultures and Exposure to Oxygen–Glucose Deprivation Conditions
Microglial cells obtained from 1-day-old Wt, tPA−/−, Plg−/−, and macLRP− C57BL/6J mice were cultured as described elsewhere (Fedoroff and Richardson, 2001). Briefly, the cells were dissociated into a single-cell suspension by triturating through a Pasteur pipette and plated onto 6-well plates coated with 0.05 mg/mL poly-
Quantitative Real-Time PCR Analysis
Wild-type, tPA−/−, Plg−/−, and macLRP− microglial cultures were exposed to OGD conditions for 6 h as described above. Wild-type and macLRP− mice underwent tMCAO, and the brains were harvested 6 h later. In both cases, we performed quantitative real-time PCR (RT-PCR) analysis for MMP-9 as described elsewhere (Yepes et al, 2003). Briefly, for the quantitative measurement of mRNA, 2 μg of DNase I-treated total RNA was used for cDNA synthesis. Reverse transcription was performed using High Capacity cDNA Archive Kit (Applied Biosystems; Foster City, CA, USA) with random oligonucleotide primers. TaqMan Gene Express Assays of TaqMan probes and primers were purchased from Applied Biosystems (Mm00442991-m1). Polymerase chain reactions were carried out in an ABI Prism 7000 system (Applied Biosystems) under the following conditions: 50°C for 2 mins, 95°C for 10 mins, 40 cycles at 95°C for 15 secs, and 60°C for 1 mins. Each observation was repeated 6 times. The PCR results were analyzed as described elsewhere (Pfaffl, 2001), and statistical analysis was performed with Student's t-test.
Western Blot Analysis
Monoclonal antibodies to claudin-5 were purchased from Zymed Laboratories (San Francisco, CA, USA). Polyclonal antibodies to β-actin were obtained from Sigma-Aldrich (St Louis, MO, USA). Wild-type, tPA−/−, and macLRP− mice underwent tMCAO, and the brains were extracted 12 h later. Tissue was processed and gels were loaded as described. A total of three observations were made for each time point.
Matrix Metalloproteinase-9 Activity Analysis
Wild-type, tPA−/−, and macLRP− mice underwent tMCAO. A subset of tPA−/− and macLRP− mice was intracortically injected with murine tPA (1 μmol/L) immediately after tMCAO at the coordinates described above. Six hours later, the brains were removed, divided into ipsilateral and contralateral hemispheres, and homogenized in a 2:1 volume to weight ratio of lysis buffer containing 0.5 mol/L NaCl, 1% Triton X-100 (Sigma-Aldrich), 0.05% Tween 20 (Sigma-Aldrich), and 50 mmol/L sodium acetate. The samples were centrifuged at 10,000 g for 10 mins and the supernatant was added to Gelatin Sepharose 4B beads (Amersham Biosciences, Piscataway, NJ, USA). After 3 h of incubation at room temperature, the beads were washed thrice in phosphate-buffered saline, incubated in the sample buffer for 30 mins, and centrifuged. The supernatant was then loaded in a gelatin zymography gel (Invitrogen, Carlsbad, CA, USA). After electrophoresis, the gel was washed twice during 45 mins with 2.5% Triton X-100 (Sigma-Aldrich), followed by a 30-min wash with Tris-buffered saline containing 5 mmol/L CaCl2, then incubated overnight at 37°C in the same buffer after which the gel was stained with 0.5% Coomassie Brilliant Blue R-250 (Amersham Biosciences) for 30 mins and then destained. Purified murine MMP-9 from Chemicon (Temecula, CA, USA) was used as a marker for MMP-9. The density of the band was quantified using the NIH Image Analyzer System and statistical analysis was performed with Student's t-test.
Results
Tissue-Type Plasminogen Activator Induces the Expression of Microglial Matrix Metalloproteinase-9 Through a Plasminogen-Independent Mechanism
To study the role of tPA on microglial MMP-9 expression during hypoxic conditions, we performed a quantitative RT-PCR analysis for MMP-9 mRNA in microglial cultures obtained from Wt, tPA-deficient (tPA−/−), and plasminogen-deficient (Plg−/−) mice exposed to OGD conditions for 6 h. A subset of tPA−/− microglial cultures was incubated with murine tPA. We found that exposure to OGD conditions induces a sharp increase in MMP-9 expression in Wt and Plg−/− microglia (4.6±0.33 and 3.48±0.38, mRNA fold increase, respectively; Figure 1). In contrast, the increase in MMP-9 mRNA expression in tPA−/− microglia was minimal (1.1±0.032, P<0.001). Importantly, incubation of tPA−/− microglia with murine tPA increased the expression of MMP-9 mRNA to 3.48±0.38 (P<0.001 compared with untreated tPA−/− microglia; n=6 for each observation).
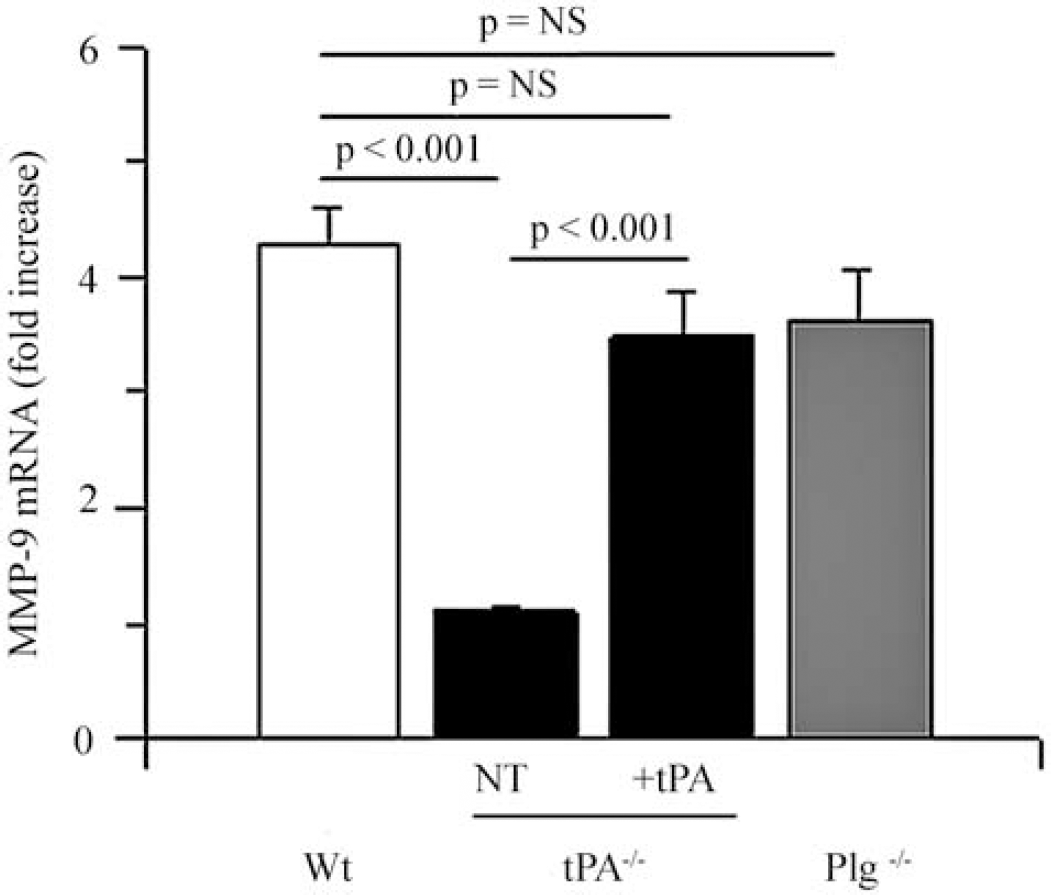
Role of tPA and plasminogen on hypoxia-induced microglial MMP-9 expression. Quantitative real-time (RT)-PCR analysis of MMP-9 expression in microglial cultures from wild-type (Wt, white bar), tPA-deficient (tPA−/−, black bars), and plasminogen-deficient (Plg−/−, gray bar) mice after 6 h of exposure to oxygen–glucose deprivation (OGD) conditions. A subset of tPA−/− microglia was either left untreated (NT: no treatment) or incubated with murine tPA (+tPA). Error bars describe s.e.m. n=6.
Low-Density Lipoprotein Receptor-Related Protein 1 Mediates the Effect of tPA on Microglial MMP-9 Expression
Previous studies have indicated that tPA induces the expression and activity of MMP-9 in the CNS (Wang et al, 2003; Yepes et al, 2003; Lee et al, 2007). As microglial cells are one of the most important sources of MMP-9, we decided to study whether microglial LRP1 mediates the effect of tPA on MMP-9 expression. To study this, we performed a quantitative RT-PCR analysis in Wt and macLRP− microglial cultures exposed to OGD conditions for 6 h. A subset of macLRP− microglial cultures was incubated with murine tPA. The results of our study indicate that exposure to OGD conditions induces a sharp increase in MMP-9 mRNA expression in Wt microglia (3.3±0.38), and that this effect is significantly attenuated in macLRP− microglia (1.3±0.44, P=0.001 compared with Wt microglia; Figure 2). Importantly, incubation with tPA failed to induce an increase in microglial MMP-9 mRNA expression in macLRP− microglial cultures (1.77±0.23; nonsignificant compared with untreated macLRP− cultures; n=6 for each observation).
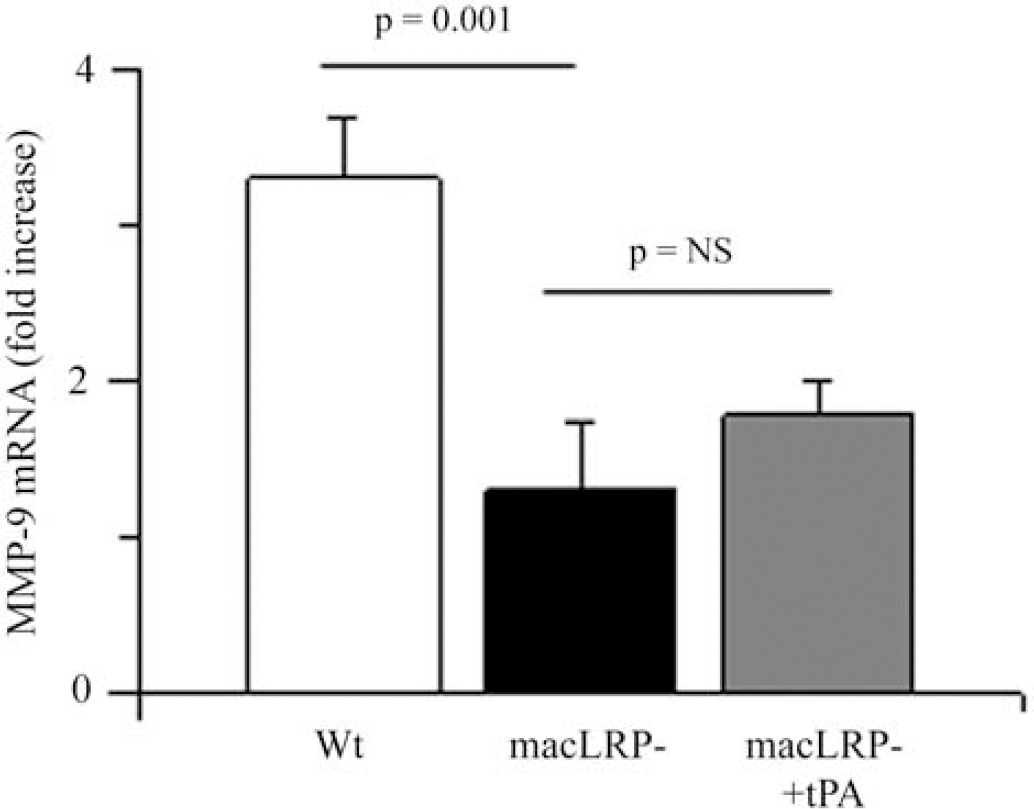
Effect of LRP1 deficiency on hypoxia-induced microglial MMP-9 expression. Quantitative real-time (RT)-PCR analysis of MMP-9 expression in microglial cultures from wild-type (Wt, white bar) and microglial LRP-deficient (macLRP−, black and gray bars) mice after 6 h of exposure to oxygen–glucose deprivation (OGD) conditions. A subset of macLRP− microglia was incubated with murine tPA (gray bar, macLRP− plus tPA). Error bars describe s.e.m. n=6.
Effect of Microglial LRP1 Deficiency on Cerebral Ischemia-Induced MMP-9 Expression and Activity
To study the effect of microglial LRP1 deficiency on cerebral ischemia-induced MMP-9 expression and activity, we performed quantitative RT-PCR analysis and gelatin zymography assays in Wt and macLRP− mice 6 h after tMCAO. Our results indicate that 6 h after tMCAO, there is a sharp increase in MMP-9 mRNA in Wt mice (5.16±0.31 mRNA fold increase; Figure 3A, white bar) that is significantly attenuated in macLRP− mice (1.56±0.39 mRNA fold increase; P<0.001 compared with Wt mice). Each observation was repeated 6 times. Similarly, the gelatin zymography assay showed a significant decrease in MMP-9 activity in macLRP1− mice (Figure 3B).

Effect of microglial LRP1 deficiency on cerebral ischemia-induced MMP-9 expression and activity. (
Microglial LRP1 Mediates the Effect of tPA on MMP-9 Activity During Cerebral Ischemia
To study the effect of the interaction between tPA and microglial LRP1 on MMP-9 activity in the ischemic brain, we performed a gelatin zymography assay in brain extracts obtained from Wt and tPA−/− mice 6 h after tMCAO. Our results indicate that cerebral ischemia induces an increase in MMP-9 activity in Wt mice and that this effect is significantly attenuated in tPA−/− mice (Figure 4A). To determine whether the effect of tPA on MMP-9 activity is mediated by microglial LRP1, we performed a gelatin zymography assay in brain extracts obtained from Wt, tPA−/−, and macLRP− mice 6 h after tMCAO and the intracerebral injection of recombinant tPA. Our data indicate that treatment with tPA increases the levels of MMP-9 activity in tPA−/− but not in macLRP− mice (Figure 4B), showing that the effect of tPA on MMP-9 activity in the ischemic brain is mediated by microglial LRP1.
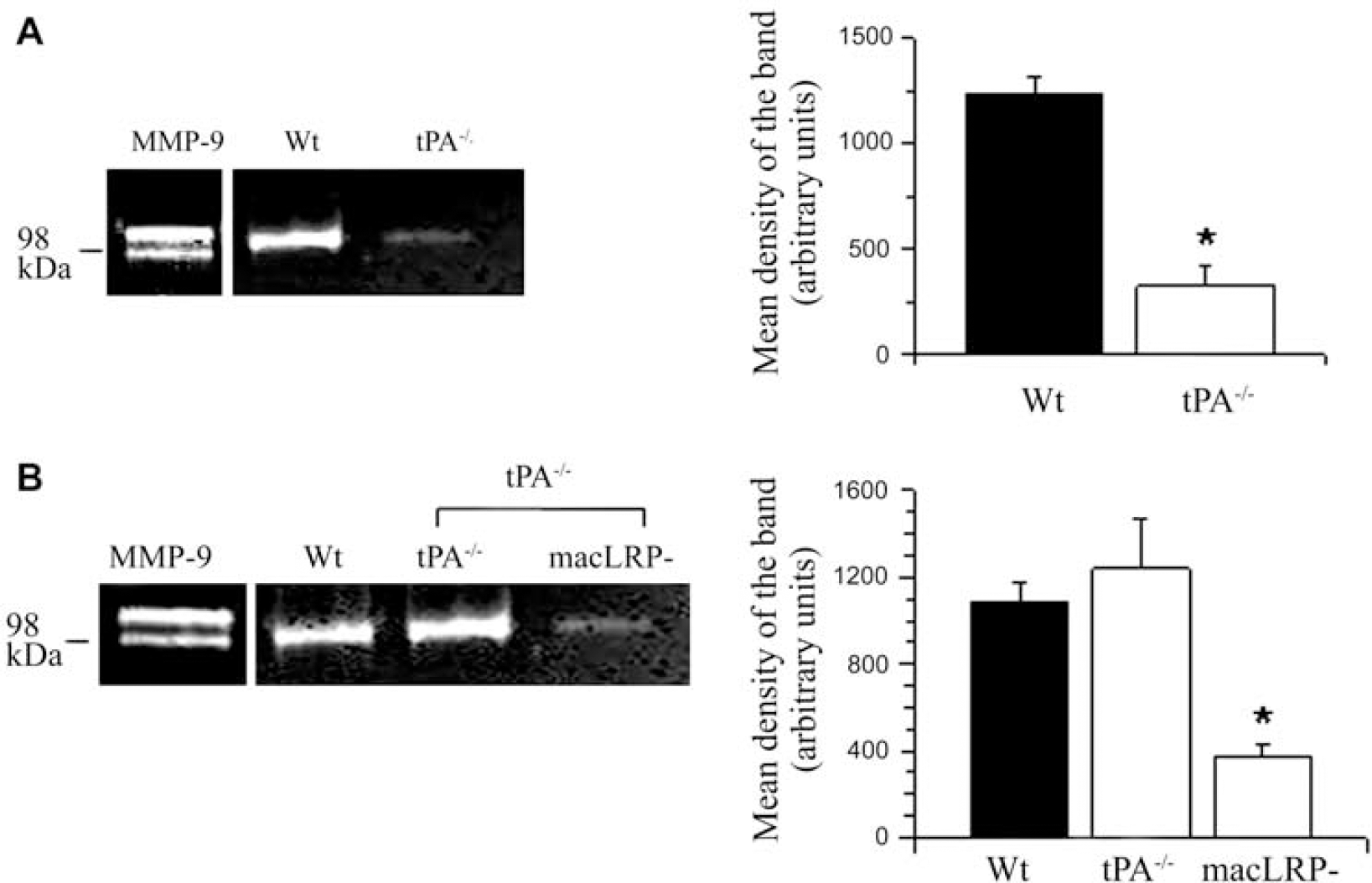
Microglial LRP1 mediates tPA-induced MMP-9 activity after MCAO. (
Effect of the Interaction Between tPA and Microglial LRP1 on the Permeability and Composition of the Neurovascular Unit
Our earlier studies indicate that tPA increases the permeability of the NVU (Yepes et al, 2003; Polavarapu et al, 2007; An et al, 2008). To investigate the role of microglial LRP1 in this process, we quantified the extravasation of Evans blue dye in Wt and macLRP− mice 12 h after tMCAO and the intracerebral injection of either murine tPA or vehicle (control). The results of our study show that genetic deficiency of microglial LRP1 is associated with a preservation of the barrier function of the NVU (75±3% reduction in Evans blue dye extravasation), and that this effect is only partially reversed by the intracerebral injection of tPA (Figure 5A). Each observation was repeated 12 times. As it has been described that claudin-5 is a substrate for MMP-9, we performed a western blot analysis for claudin-5 in brain extracts obtained from Wt, tPA−/−, and macLRP− mice 12 h after tMCAO. Our data indicate that cerebral ischemia induces a significant degradation of claudin-5 in the ischemic tissue, and that this effect is significantly attenuated in tPA−/− and macLRP− mice (Figure 5B).
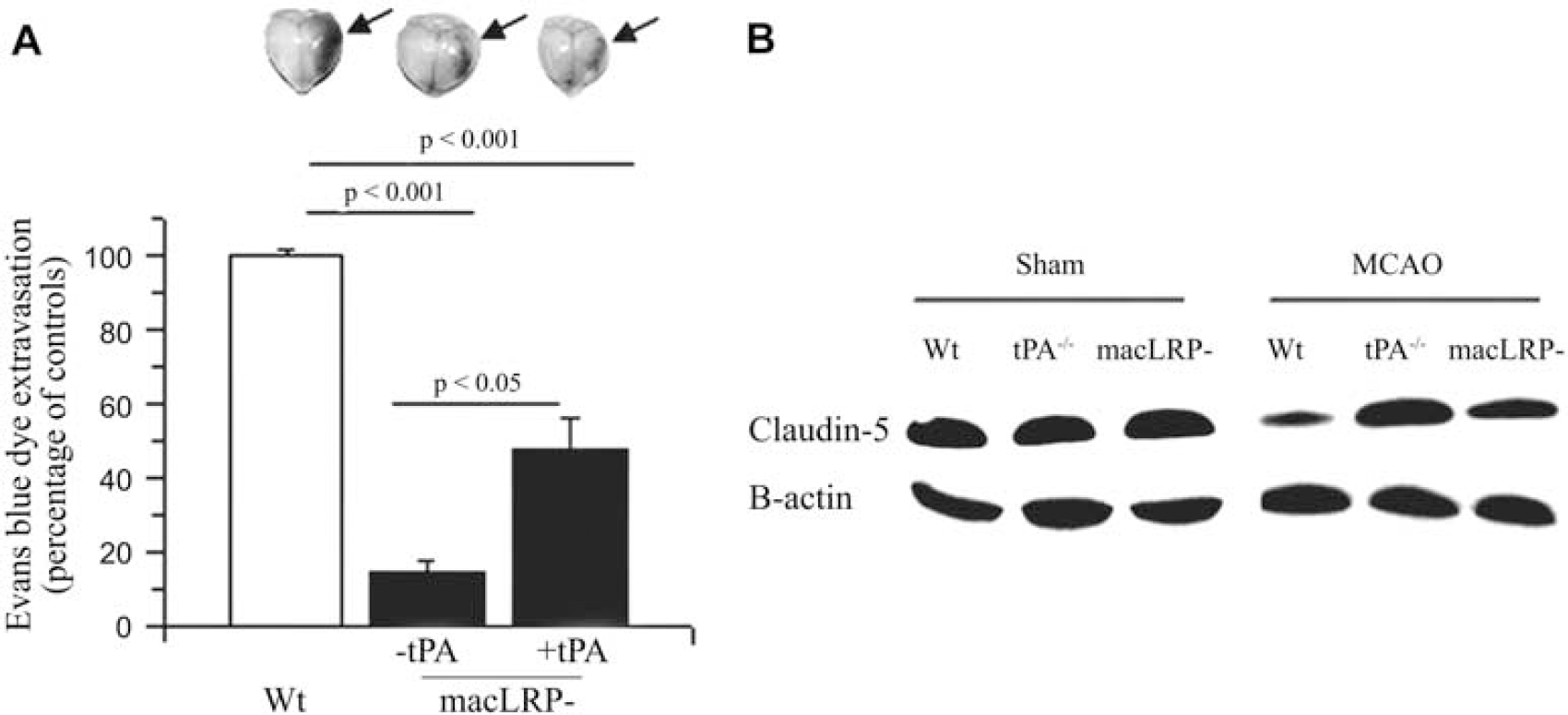
Effect of microglial LRP1 deficiency on the permeability of the neurovascular unit. (
Discussion
Low-density lipoprotein receptor-related protein 1 is a member of the LDL receptor gene family implicated in the internalization of multiple ligands, including tPA and MMP-9 (Herz and Strickland, 2001). Low-density lipoprotein receptor-related protein 1 is also involved in cellular signal transduction pathways and neurotransmission. It is expressed in smooth muscle cells, and specific deletion of the LRP1 gene from vascular smooth muscle cells on a background of LDL receptor deficiency causes smooth muscle cell proliferation, increased susceptibility to cholesterol-induced atherosclerosis, and aneurysm formation (Boucher et al, 2003). In the CNS, LRP1 is found in neurons, perivascular astrocytic end-feet processes (Wolf et al, 1992), and in microglial cells (Zhang et al, 2009). In neurons, LRP1 has been found to mediate events such as long-term potentiation and calcium influx through NMDA (N-methyl-
Tissue-type plasminogen activator is expressed in endothelial cells, neurons, and glia (Yepes et al, 2009). Owing to the fact that in the intravascular space tPA is primarily a thrombolytic enzyme, recombinant tPA is used for the treatment of patients with acute ischemic stroke (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). However, multiple studies with animal models of cerebral ischemia indicate that the onset of ischemic insult also induces an increase in tPA activity in the ischemic tissue associated with cell death (Wang et al, 1998) and the development of cerebral edema (Yepes et al, 2003). Importantly, some of these deleterious effects of tPA have also been observed in acute stroke patients (Kidwell et al, 2008). Therefore, the understanding of the mechanism(s) whereby tPA injures the ischemic brain is of pivotal importance for improving the safety of the treatment of patients with acute ischemic stroke. Tissue-type plasminogen activator is a ligand for LRP1 (Bu et al, 1992a, 1992b), and a growing body of evidence indicates that LRP1 is the natural substrate for tPA within the CNS (Yepes et al, 2003, 2009; Zhang et al, 2007). In this study, we describe a novel mechanism whereby the interaction between tPA and microglial LRP1 during cerebral ischemia leads to the development of cerebral edema.
The activation of microglial cells involves a number of features, including morphologic changes and the release of free radicals and proinflammatory molecules. Injury to the brain tissue or to parenchymal blood vessels triggers the activation of microglial cells. Importantly, the passage of blood-derived factors into the brain parenchyma also leads to microglial activation (Nimmerjahn et al, 2005). In earlier work, we showed that tPA induces microglial activation through a plasminogen-independent pathway (Zhang et al, 2009). As it has been previously shown by others that during cerebral ischemia, tPA crosses over from the intravascular space into the ischemic tissue (Benchenane et al, 2005), it is possible that recombinant tPA administered for the treatment of acute ischemic stroke not only contributes to the lysis of the clot in the intravascular space (beneficial role) but also to the activation of microglia in the ischemic brain parenchyma (deleterious effect).
Matrix metalloproteinase-9 is a member of the metalloproteinase family, which has been directly implicated in the pathophysiology of cerebral ischemia (Rosenberg et al, 1996). One of the main sources of MMP-9 is activated microglial cells. During cerebral ischemia, there is an increase in MMP-9 activity in the ischemic tissue associated with the development of cerebral edema (Asahi et al, 2000, 2001; Yang et al, 2007). There is considerable debate about the sources of MMP-9 after the onset of ischemic insult. Indeed, it has been postulated that inflammatory cells, glia, and endothelial cells are the main source of MMP-9 in the ischemic brain. In this study, we show that one of the main mechanisms of increased MMP-9 activity in the ischemic brain is triggered by the interaction between tPA and microglial LRP1. However, other mechanisms whereby the interaction between tPA and LRP1 may lead to increased MMP-9 activity in the ischemic brain include augmentation of the permeability of the NVU with passage of MMP-9-bearing inflammatory cells into the ischemic tissue, as well as the induction of an nuclear factor-κB-mediated cell signaling event in perivascular astrocytes and endothelial cells.
A growing body of evidence indicates that tPA induces MMP-9 actvity in the ischemic brain through a plasminogen-independent mechanism. Indeed, it has been shown that the increase in MMP-9 activity observed in Wt mice in response to the ischemic insult is significantly attenuated in tPA−/− mice (Yepes et al, 2003). Similarly, incubation of glial cells with tPA (Lee et al, 2007) or the intravenous administration of tPA in an animal model of embolic stroke (Lapchak et al, 2000) induces a significant increase in MMP-9 activity. It has been shown in in vitro culture systems that under normoxic conditions, LRP1 functions as an endocytic receptor for MMP-9 (Hahn-Dantona et al, 2001). Accordingly, either the inhibition of LRP1's cleavage (Rozanov et al, 2004) or the incubation with the receptor associated-protein (Hahn-Dantona et al, 2001) results in a significant increase in MMP-9 activity in the media. In this study, we postulate that during ischemic conditions microglial LRP1 functions as a cell signaling receptor, which on its interaction with tPA leads to an increase in MMP-9 activity in the ischemic tissue. At present, it is unknown whether the interaction between tPA and LRP1 has a direct effect on the expression of MMP-3, an activator of pro-MMP-9 present in microglial cells. However, our results indicate that the interaction between tPA and LRP1 has a direct effect on the expression of MMP-9 mRNA, which subsequently may be activated by MMP-3.
One of the main functions of the NVU is the regulation of the passage of substances from the intravascular space into the brain (del Zoppo and Mabuchi, 2003). Early after the onset of ischemic insult, there is an increase in tPA activity around the NVU associated with the development of cerebral edema (Yepes et al, 2003). Importantly, our previous work has indicated that cerebral ischemia also induces an increase in LRP1 expression in the abluminal side of the NVU (Polavarapu et al, 2007), and that the deleterious effect of tPA on the permeability of the NVU is mediated by its interaction with LRP1 (Yepes et al, 2003; Polavarapu et al, 2007). The data presented in this study indicate that the interaction between tPA and microglial LRP1 leads to an increase in MMP-9 expression and activity associated with the development of ischemic edema. The integrity of tight junction proteins between endothelial cells is one of the most important determinants of the barrier function of the NVU. Indeed, the onset of ischemic insult is associated with a progressive degradation of these proteins associated with an increase in the permeability of the NVU and development of cerebral edema. Our results show that the interaction between tPA and microglial LRP1 leads to degradation of the tight junction protein claudin-5, which has been previously described as a substrate for MMP-9 activity during cerebral ischemia (Yang et al, 2007; McColl et al, 2008). In summary, in this study, we propose a model in which in response to the ischemic insult there is an increase in tPA activity in the ischemic tissue, originated either in the intravascular space or in the ischemic parenchyma, or in both. This tPA interacts with LRP1 in microglial cells leading to the activation of a cell signaling event that results in an increase in MMP-9 activity in the NVU with degradation of the tight junction protein claudin-5 and the development of cerebral edema. This is a novel mechanism for the development of ischemic edema and a potential target for the treatment of acute ischemic stroke patients.
Footnotes
The authors declare no conflict of interest.