
Abstract
The metabolic demands of the brain are met by oxygen and glucose, supplied by a complex hierarchical network of microvessels (arterioles, capillaries, and venules). Transient changes in neural activity are accommodated by local dilation of arterioles or capillaries to increase cerebral blood flow and hence nutrient availability. Transport and communication between the circulation and the brain is regulated by the brain microvascular endothelial cells that form the blood–brain barrier. Under homeostatic conditions, there is very little turnover in brain microvascular endothelial cells, and the cerebrovascular architecture is largely static. However, changes in the brain microenvironment, due to environmental factors, disease, or trauma, can result in additive or subtractive changes in cerebrovascular architecture. Additions occur by angiogenesis or vasculogenesis, whereas subtractions occur by vascular pruning, injury, or endothelial cell death. Here we review the various processes that lead to changes in the cerebrovascular architecture, including sustained changes in the brain microenvironment, development and aging, and injury, disease, and repair.
Keywords
Introduction
The brain has no moving parts and yet it is one of the most energy-expensive organs in the body, consuming 15–20 W.1,2 To power the adult human brain, nutrients are supplied to the 100 billion neurons via a 600 km network of capillaries and microvessels. 3 Since the brain does not have significant capacity to store metabolic nutrients, cerebral blood flow (CBF) is generally proportional to cerebral metabolic rate, 4 and the cell bodies of neurons are typically 10–20 µm from the nearest capillary. 2 Therefore, the cerebrovasculature is crucial to the maintenance of normal brain function.
The cerebrovasculature, in addition to supplying nutrients and other essential molecules, is a key component of the blood–brain barrier (BBB) which maintains tight control of the brain microenvironment by regulating fluctuations in chemistry, transport of immune cells, and the entry of toxins and pathogens.2,5 Under homeostatic conditions, the turnover of brain microvascular endothelial cells (BMECs) is very low, and the cerebrovascular architecture is considered to be static. However, due to the precise neurovascular coupling between energy supply and demand, perturbations to the cerebrovasculature and the neuronal architecture are closely related. The cerebrovascular architecture displays profound plasticity during development, aging, injury, and disease (Figure 1).
Cerebrovascular architecture displays profound plasticity during the human lifespan. A summary of changes during: (a) development, (b) homeostasis, and (c) aging, injury, and disease. (d) Microvascular density increases via angiogenesis in the brain. Shown is the mouse postnatal cortex, arrowheads denote endothelial sprouts.
85
(e) The blood–brain barrier (BBB) displays functionally low permeability during early stages of brain development; here, a guinea pig embryo injected with trypan blue demonstrates restriction of dye entry into CNS.
199
(f) Neurovascular coupling is the process by which increases in neural activity create changes in local cerebral blood flow and cerebral consumption of oxygen; here, we display a change in local total hemoglobin concentration (dark red) in response to electrical stimulation in the somatosensory cortical surface of a rat.
14
(g) Chronic hypoxia results in increased microvascular density in the mouse motor cortex.
63
(h) TBI results in a rapid decrease in microvascular density in rats.
141
(i) During Alzheimer’s disease, the most common neurodegenerative disease, amyloid-beta accumulates around human microvessels and BBB integrity is compromised (downregulation of the tight junction protein claudin-5) among other cerebrovascular changes.
200
Images were cropped from original and have labels added.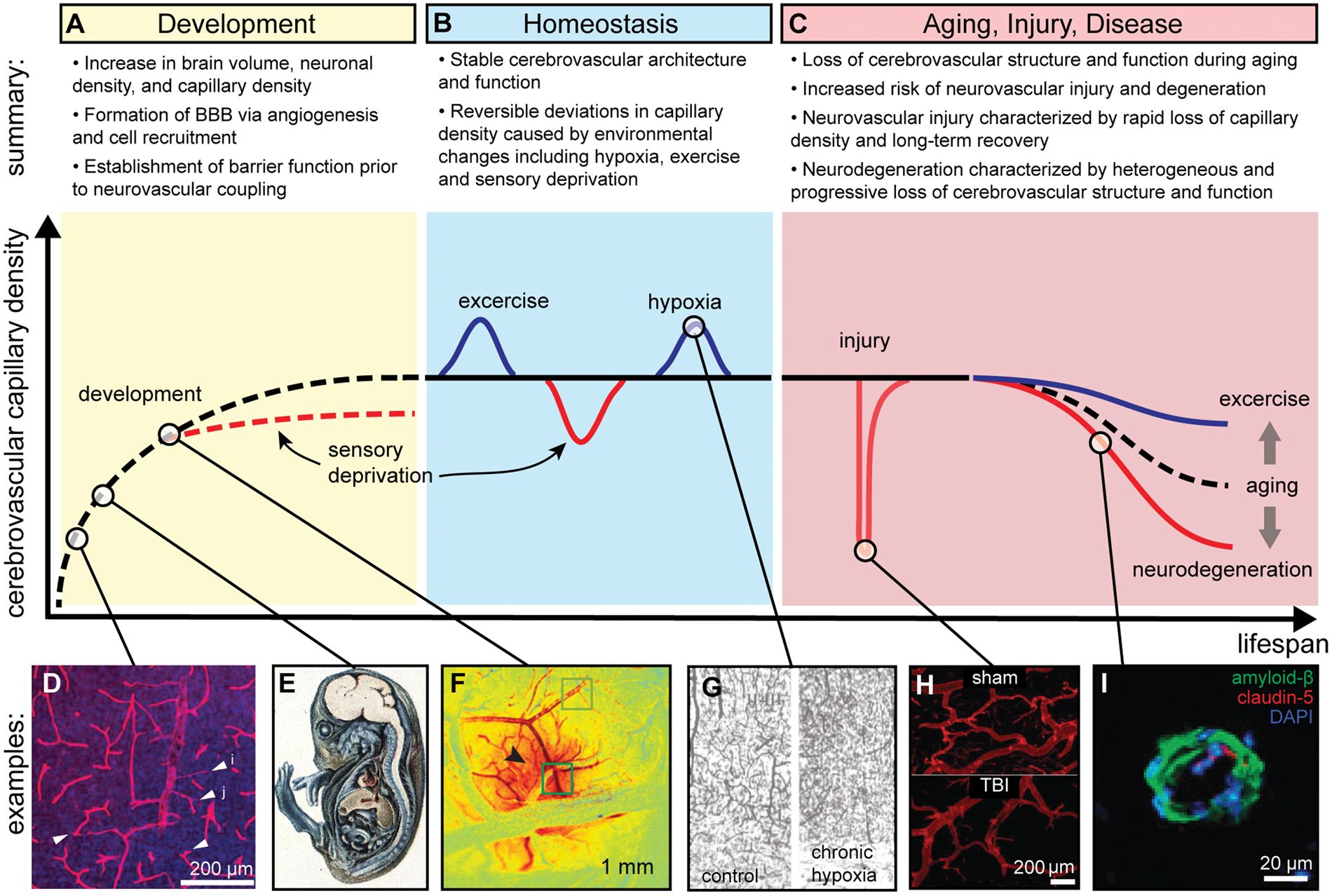
Transient changes in neural activity can be accommodated by local dilation or contraction of arterioles and capillaries. However, sustained changes in the global brain microenvironment, for example, due to sensory deprivation, hypoxia, or aerobic exercise, can lead to robust remodeling of the cerebrovascular architecture. At the cellular level, changes in the cerebrovascular architecture involve changes in the rates of proliferation and loss of BMECs that are associated with addition or subtraction of capillary segments. As a result of neurovascular coupling, these changes are usually closely associated with local gain or loss of neurons and may ultimately be associated with changes in cognitive function. Although cerebrovascular plasticity is observed at all hierarchical levels, here we focus on the microvasculature (arterioles, capillaries, and venules).
The time scales of these processes span a wide range. For example, changes in CBF in response to increases in neural activity occur with a time constant on the order of seconds. Changes in cerebrovascular architecture, due to sustained sensory deprivation, aerobic exercise, or hypoxia, occur with time constants of several weeks. The cerebrovasculature displays elevated plasticity during development, while during aging, plasticity declines. The time scale of changes during development and aging is on the order of months to years, where the rate often depends on mitigating factors such as exercise (in the case of aging). Injury can result in rapid damage to cerebrovascular and neuronal architecture, followed by partial or complete repair over weeks to months. In contrast, neurodegenerative disease is characterized by highly heterogeneous changes that occur over months to years. Here, we review processes that lead to changes in cerebrovascular architecture.
Cerebral blood supply and metabolism
Cerebral blood supply
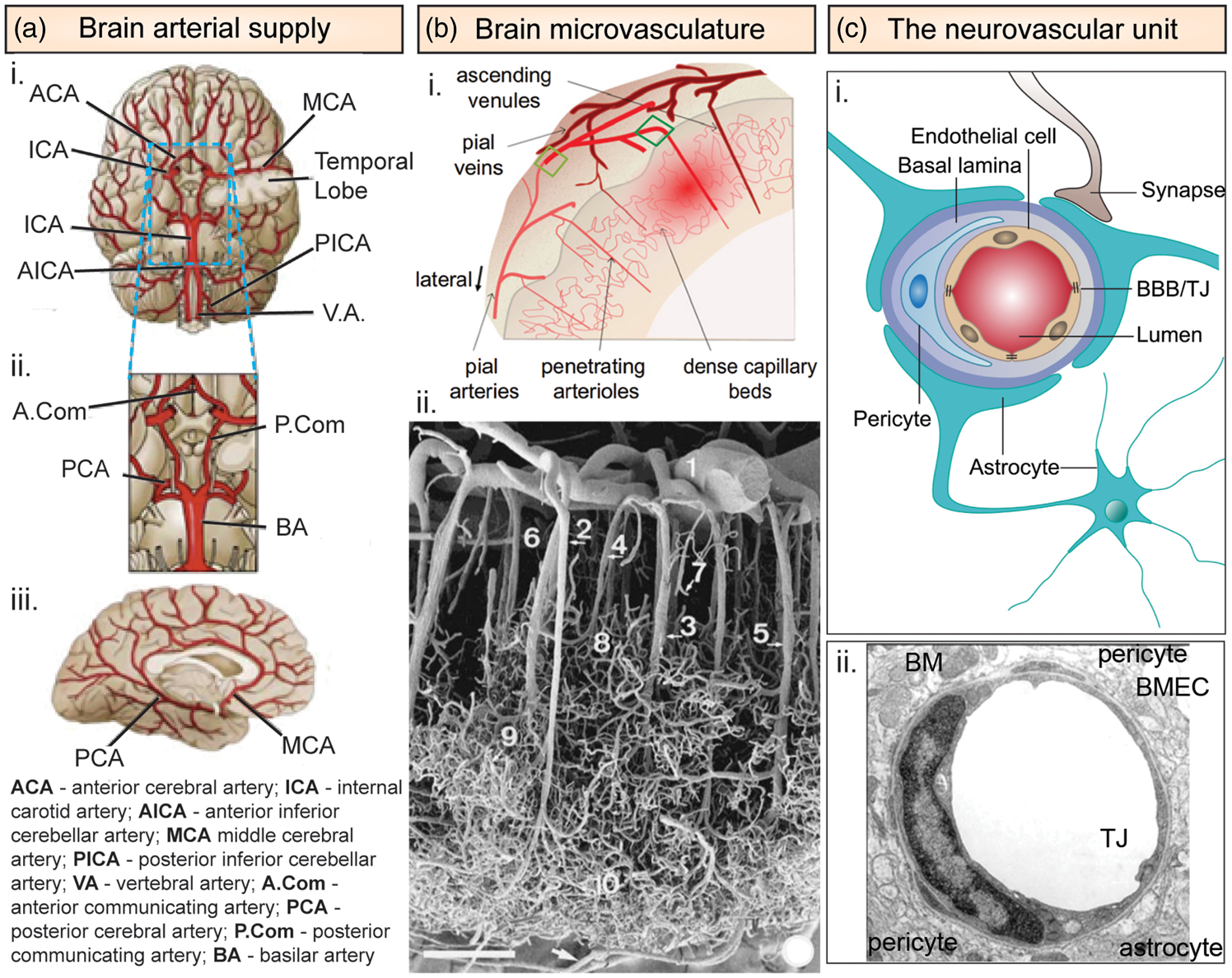
The brain receives blood supply from the cerebral branches of the internal carotid (which then branch into the anterior and middle cerebral arteries) and the vertebral arteries (that later merge to form the basilar artery; Figure 2a).
6
The internal carotid arteries and the vertebral arteries are interconnected in the cranial cavity to produce the Circle of Willis, which creates collateral circulation. If any part of the Circle of Willis or supplying arteries becomes blocked or narrowed, blood flow remains at least partially preserved by the other supplying vessels, reducing the likelihood of ischemia. Anastomoses also occur between the branches of the three cerebral arteries on the surface of the brain. In adults, if one of the four arteries delivering blood to the brain is blocked, the remaining three are not usually capable of providing adequate circulation, resulting in ischemic stroke.
7
Additionally, cerebral blood supply reflects the metabolic demands of specific brain regions and defines their susceptibility to vascular damage. For example, regions that are supplied by a single major artery and have a continuous capillary network with weak collateral flow are at higher risk for hypoxic injury and stroke.
8
Cerebrovascular architecture. (a) Arterial architecture: (i) inferior view of the base of the brain with cerebral arterial Circle of Willis; (ii) magnified view of the Circle of Willis; and (iii) right lateral view of the right hemisphere. (b) Microvasculature ultrastructure in the cerebral cortex: (i) schematic illustration of the vasculature in the cerebral cortex showing both arterial and venous systems. Pial arteries located on the surface penetrate deep into the cerebral cortex as penetrating arterioles, which branch into capillary beds that then reemerge from the cortex as ascending venules.
14
(ii) Scanning electron micrograph of a corrosion cast showing the vasculature of the temporal lobe of the human cerebral cortex.
16
(Scale bar = 375 µm): (1) pial artery, (2) long cortical artery, (3) middle cortical artery, (4) short cortical artery, (5) cortical vein, (6) subpial zone, (7) precapillary vessel with blind ending, (8) superficial capillary zone, (9) middle capillary zone, and (10) deep capillary zone. (c) Neurovascular unit. (i) Schematic illustration of the neurovascular unit comprised of brain microvascular endothelial cells surrounded by pericytes and astrocytes.
85
(ii) Electron microscope cross section of a capillary from the rat frontoparietal cortex.
163
Cerebral blood flow
The average CBF is determined by the Poiseuille equation for laminar flow:
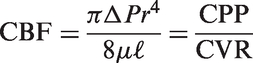
CBF is maintained at a relatively constant value over a range of CPPs (50–150 mm Hg) by autoregulation: changes in CPP are compensated by changes in artery diameter (vasodilation or vasoconstriction).6,10 The lower limit corresponds to the point of maximum dilation, and at lower pressures, the CBF decreases with decreasing pressure due to vessel collapse. The upper limit corresponds to maximum constriction, and higher pressures result in disruption of the BBB. 9 Autoregulation is maintained through multiple mechanisms including metabolic (e.g. CO2 levels), myogenic (e.g. nitric oxide (NO)), and neurogenic (e.g. sympathetic innervation) regulation. 10
The average cerebral blood volume (CBV) is 3.5–4.5 mL per 100 g tissue (about 50 mL for an adult brain). 11 A large fraction of this volume is contained in the venous sinuses and pial veins. Although the CBV is a small fraction of the brain volume (around 4%), it plays an important role in regulating ICP since it can be modulated very quickly by changing the blood flow. Since CBV ∝ r2 and CBF ∝ r 4 (see above), it is predicted that CBV ∝ CBFn where n = 0.5. 12 Studies in humans and animals have reported exponents of 0.3–0.4. 12
Cerebral metabolism
The brain is one of the most energy-expensive organs in the human body. Overall, the brain accounts for 15–20% of the base metabolic rate, consuming 15–20% of oxygen, leaving the heart, and 15–20% of the glucose consumed daily.1,2 Since the brain does not have significant capacity to store metabolic nutrients, fuel to power the brain is provided on-demand by the lungs and gastrointestinal system, which transfer oxygen and glucose, respectively, to the vascular system. Glucose is transported into the brain by glucose transporter 1 (GLUT1) localized to BMECs, while oxygen passively diffuses across endothelial cells. 2
Microvascular architecture
Microvascular ultrastructure
Neuronal architecture, and thus cerebrovascular architecture, is spatially heterogeneous.6,13 In the cerebral cortex, pial arteries on the brain surface descend to become parenchymal arterioles to supply microvascular beds of cortical gray matter (Figure 2b). 14 At the interface between gray and white matter, the microvascular density decreases substantially along with the density of neurons and other supporting cells. In white matter, capillaries are generally arranged in parallel with axons, and microvascular density is roughly 10% of that in the gray matter (consistent with the lower blood flow).6,13 Pathological and physiological stresses can promote increases or decreases in microvascular density via angiogenesis or apoptosis, as addressed in subsequent sections.
In mammals, capillaries have a diameter that is slightly larger than red blood cells (RBCs), 15 which in humans corresponds to 8–10 µm in diameter. In the human brain, capillaries have branch points approximately every 30 µm and an inter-capillary spacing of ∼50 µm. 16 Venular capillaries in the mouse cortex are up to 20 branches away from penetrating arterioles, 17 and the average path length of RBCs through a single capillary network in the cortex (where flow can be visualized in real time by two-photon microscopy) is 150–500 µm. 18 Wall shear stress in the capillaries ranges from 20 to 40 dyne cm–2 and is regulated by capillary tone via neurovascular coupling. 19 Downstream from capillaries, post-capillary venules (PCVs) have a perivascular space that serves as a preferential site for extravasation of leukocytes, tumor cells, and parasites. 20 – 22 PCVs have an average wall shear stress from 1 to 4 dyne cm–2, and smaller cardiac cycle-based fluctuations compared to arterioles. 19
The neurovascular unit
The neurovascular unit, which comprises the BBB, embodies the system that regulates neurovascular coupling. The cellular components of the BBB are BMECs, pericytes, and astrocytes, and implicitly include neurons in the context of coupling (Figure 2c).5,19 Highly specialized BMECs provide the physical barrier between the vasculature and brain parenchyma. Pericytes are mural cells with a round nucleus and long processes that extend along the abluminal endothelial wall. A basement membrane (BM), composed of collagen IV, laminin, nidogen, and heparan sulfate proteoglycans, surrounds BMECs and pericytes. 2 Astrocytes are star-shaped glial cells with small cell bodies and long radial processes that contact both synapses and the BM. In capillaries, endothelial cells, pericytes, and BM are completely surrounded by astrocytic end-feet. 23 Astrocytes serve multiple functions in the brain, including spatiotemporal modulation of local blood flow, by relaying signals from neurons to BMECs via their end-feet to induce dilation and contraction.23,24
Cerebrovascular and neuronal plasticity
Under homeostasis, the cerebrovascular architecture is assumed to be fixed, with very low cell turnover. Direct evidence for cerebrovascular plasticity comes from two-photon microscopy studies in animal models. 25 – 28 In the somatosensory and motor cortex of mice, increases in capillary length and the number of branch points have been observed up to post-natal day 25. However, in adult mice, there was negligible change in capillary segment diameter, capillary segment length, or the position of branch points over about 30 days.25,26 These results imply that there was no turnover in BMECs during the imaging period. The subtraction or pruning of redundant capillary sections in short vascular loops occurred once every 6.25 days per mm 3 in the motor cortex. 25 Subtraction events may occur in response to disturbances in shear flow and could contribute to pathological processes, such as vascular atrophy, rarefaction, wall degeneration, and the neurodegeneration observed in aged animals.25,29
Increases in microvascular density occur by formation of capillaries from existing blood vessels (angiogenesis) or organization of proliferating precursor cells into new blood vessels (vasculogenesis). Angiogenesis involves endothelial cell activation, proteolytic degradation of BM and matrix, migration, alignment, proliferation, tube formation, and anastomosis with other vessels. During maturation, capillaries develop tight intracellular junctions, produce BM, and recruit pericytes and astrocytic end-feet. Arterioles recruit a layer of smooth muscle cells (SMCs), which allows control of vessel tone. Vascular endothelial growth factors (VEGFs) and their respective receptor tyrosine kinases (VEGFR) are key regulators of angiogenesis and are inducible in many cell types, including astrocytes, pericytes, and endothelial cells. 23 VEGF-A also supports neurogenesis, nerve migration, axonal guidance, and neuronal survival. 30 During adulthood, brain angiogenesis is observed in response to stimuli such as hypoxia, injury, or neurodegenerative disease, as discussed in subsequent sections. Additionally, vasculogenesis can occur in response to stimuli such as trauma or tissue hypoxia as endothelial progenitor cells (EPCs) are mobilized from bone marrow to participate in endothelial cell repair/regeneration and tissue neovascularization processes. 31 For example, a higher concentration of circulating EPCs during the first week after stroke onset is associated with improved patient outcomes. 32 Cerebral arteries also display profound plasticity; for example, during hypertension (elevated blood pressure) extensive remodeling of arterial walls occurs to reduce lumen diameter. 33 In subsequent sections, we consider the cerebrovascular plasticity of microvessels (arterioles, capillaries, and venules).
Neuronal plasticity is distinct from cerebrovascular plasticity in terms of location. In adulthood, neurogenesis occurs in only two regions of the brain: the subgranular zone of the dentate gyrus in the hippocampus and the subventricular zone. 34 These specialized regions are often referred to as neurovascular niches since the vasculature has been identified as a crucial component of the stem cell environment. 35 In the subventricular zone, neural stem cells directly contact blood vessels in homeostasis as well as regeneration, 36 a feature unique to this region of the brain. It has also been shown that new neurons are born in close proximity to blood vessels in the hippocampus. 37
Factors that influence cerebrovascular plasticity
Neural activity
A key feature of brain metabolism is the tight coupling between energy supply and demand, commonly known as neurovascular coupling or functional hyperemia. 24 Local increases in neural activity are followed by dilation of local capillaries and upstream arterioles, increasing CBF, and increasing cerebral metabolic rate of oxygen consumption (CMRO2).9,14 This response is modulated by secretion of vasodilators, such as NO, vasoactive intestinal polypeptide, and prostaglandin by endothelial cells, neurons, and other glial cells.24,38 Recently, extracellular K+, a byproduct of neural activity, has been shown to induce a retrograde hyperpolarization signal from capillaries to arterioles, resulting in dilation. 39 These chemical factors typically act by promoting relaxation of SMCs surrounding upstream arterioles. There have been contradictory reports on the role of pericytes in dilation and contraction of capillaries. 40 – 43 Increased metabolism, which follows an increase in neural activity, is quantifiable though the measured increase in CMRO2.44,45 The response to increased neuronal activity is relatively fast with changes in CBF occurring within a few seconds. 46
Neuronal activity results in increases in CBF and CMRO2, with CBF increasing two- to four-fold more than CMRO2.45,47 Local changes in oxygen consumption result in downstream changes in the concentration of deoxygenated hemoglobin, which is the basis for blood oxygenation level dependent (BOLD) contrast functional magnetic resonance imaging (fMRI). 48 – 50 Changes in the concentration of deoxygenated hemoglobin are most prevalent in the venous tree following consumption of oxygenated hemoglobin in capillaries. 51
Cerebrovascular architecture is maintained during neurovascular coupling. However, persistent changes in neural activity or nutrient supply can lead to adaptions that include remodeling of cerebrovascular architecture. Examples of such changes are sensory deprivation, hypoxia, and aerobic exercise.
Sensory deprivation
Evidence for coupling between neural activity and cerebrovascular architecture comes from sensory deprivation studies in rats and studies of the effect of single-eye vision depravation in cats. 52 – 55 Juvenile rats raised in stimulating, complex environments for 30 days were found to have increased density of capillary branch points, increased capillary surface area per unit tissue volume, and increased number of branch points per unit of capillary surface area within the striate cortex when compared to rats in non-complex environments.52,53 Similar studies have shown further increases in microvascular density in the cerebellar cortex of adult rats, while learning tasks increased the number of synapses per unit area. 54 Adult rats raised in complex environments showed increased microvascular density and number of branch points. New microvessels were observed after 10 days with full capillary neovascularization occurring between 30 and 60 days compared to animals housed in individual cages. Juvenile rats raised in the dark for up to 60 days showed a lower density of neurons, significantly lower cortical thickness, and lower vessel density in the striatum.56,57
Increases in neural activity are associated with increased growth rate in the somatosensory cortex in rat models. 58 Increased neural activity induced by whisker stimulation for one week, from post-natal day 14–21, resulted in increased microvascular density and the number of branch points in the somatosensory cortex. 59 A reduction in sensory neural activity induced by whisker plucking from post-natal day zero to five resulted in decreased microvascular density and the density of branch points at post-natal day 14. 59 These results indicate relatively fast changes in cerebrovascular architecture in juvenile mice.
Hypoxia
Since oxygen is essential for normal brain function, transient or sustained decreases in oxygen supply can lead to hypoxia and result in changes in the cerebrovascular architecture. In adulthood, local hypoxia is usually associated with injury or disease. For example, ischemic stroke or circulating tumor microemboli can cause local hypoxic regions downstream from occlusion. In contrast, changes in the oxygen partial pressure of the environment can lead to global hypoxia in the brain. At sea level where oxygen comprises 21% of the atmosphere, the arterial oxygen partial pressure (PaO2) is 75–100 mm Hg. Mild hypoxia usually refers to arterial oxygen partial pressures down to 50 mm Hg, corresponding to an atmospheric oxygen concentration of about 10% or equivalent to an altitude of about 5000 m. 60 While mild hypoxia results in numerous adaptations including angiogenesis, moderate hypoxia (PaO2 = 35–50 mm Hg) can result in cognitive deficits, and severe hypoxia (PaO2 < 35 mm Hg) can result in loss of consciousness.
Acute exposure to mild hypoxia results in an increase in CBV due to vasodilation and an increase (up to two-fold) in CBF, which returns to baseline after a few days. 60 The renormalization in CBF is associated with an increase in RBC volume and hemoglobin concentration, a well-known response to high altitude training in the field of human performance. 61 Rats subjected to chronic mild hypoxia display regional specifies increases in microvascular density; a three-fold increase in density in the hippocampus and striatum was observed over 3–4 weeks, while other brain regions display more modest changes.62,63 Increases in microvascular density are driven by angiogenesis through expression of hypoxia-inducible factor-1α (HIF-1α) and subsequent upregulation of VEGF, while declines are achieved via BMEC apoptosis following return to normoxia. 60
Physical activity
Physical activity is associated with increased neuronal plasticity, cognitive function, and cerebrovascular plasticity. The interplay between physical activity and cognitive function is emerging as an important area of research in human performance, and the benefits of exercise in restoring health and preventing disease are well established. 64 – 66 In particular, exercise is a practical therapeutic intervention to enhance cognitive function and prevent or slow cognitive decline.
Aerobic physical exercise can improve cognitive function in childhood, young adults, and older individuals.64,65 Numerous studies in humans and animals have also shown that aerobic exercise increases neuronal plasticity, providing evidence for the link between exercise and cognitive function. 66 Greater aerobic fitness has been correlated with increased gray matter volume, and evidence suggests that the benefits of exercise are dose dependent.64,65 In mouse models, acute aerobic activity results in a selective increase in neuronal activity, primarily in the hippocampus, and an increase in proliferation, survival, and differentiation of neurons in the dentate gyrus of the hippocampus, the region of the brain responsible for memory, spatial orientation, and navigation. 64 Extensive studies of older individuals have shown that exercise can delay or slow down age-related cognitive decline, enhance brain plasticity and learning, and improve aspects of cognitive function, particularly related to learning, memory, and executive function.64,65
Cerebrovascular plasticity
Since aerobic exercise can promote neuronal plasticity, it is not surprising that it also promotes cerebrovascular plasticity. 67 Several studies in mouse models have shown exercise-induced increases in microvascular density in the motor cortex and striatum. 68 Mice housed with a running wheel for about six weeks showed exercise-induced neurogenesis in the dentate gyrus and striatum, along with increased microvascular density in these regions of the hippocampus. 69 In a similar study, mice housed with a running wheel showed increased neurogenesis and microvascular density in the dentate gyrus after three days and an increase in microvascular density of around 20% after 10 days, however, microvascular density and neurogenesis returned to baseline after the running wheel was removed. 70 In contrast, an intravital microscopy study of adult mice housed with a running wheel showed no change in capillary segment length, diameter, or branching in the somatosensory and motor cortex over 30 days. 25 In mouse models of stroke, exercise resulted in an increased density of perfused microvessels and improved outcome after four weeks.71,72 Evidence suggests that this is related to increased endothelial nitric oxide synthase (eNOS) activity and an increase in circulating EPCs. 71
Exercise and CBF
As described previously, autoregulation maintains CBF at about 750 mL min–1 in the adult brain. However, studies in animals and humans show that CBF increases during exercise. 68 Blood flow in the external carotid artery, the common carotid artery, and the vertebral artery increases with exercise intensity in a dose-dependent manner. 68 Results from animal studies show increases in neuronal activity in regions of the brain associated with running, including the somatosensory cortex and hippocampus. 68
Studies in humans have shown an increase in CBV in the hippocampal dentate gyrus in healthy adults following aerobic exercise training for three months. 73 In similar experiments in mice, increased CBV in the dentate gyrus was correlated with neurogenesis, 73 implying increased angiogenesis. Basal levels of CBF decline with aging, but individuals who engage in regular aerobic exercise have higher rates of CBF than age-matched sedentary individuals. 74 In related studies, three months aerobic exercise decreased endothelin-1-mediated vasoconstriction in sedentary older individuals. 75 Increased endothelin-1 leads to vasoconstriction, which is associated with many cardiovascular diseases. 76
Neurotrophic coupling
Relationship between aerobic activity, neurogenesis, and changes in the cerebrovascular architecture.
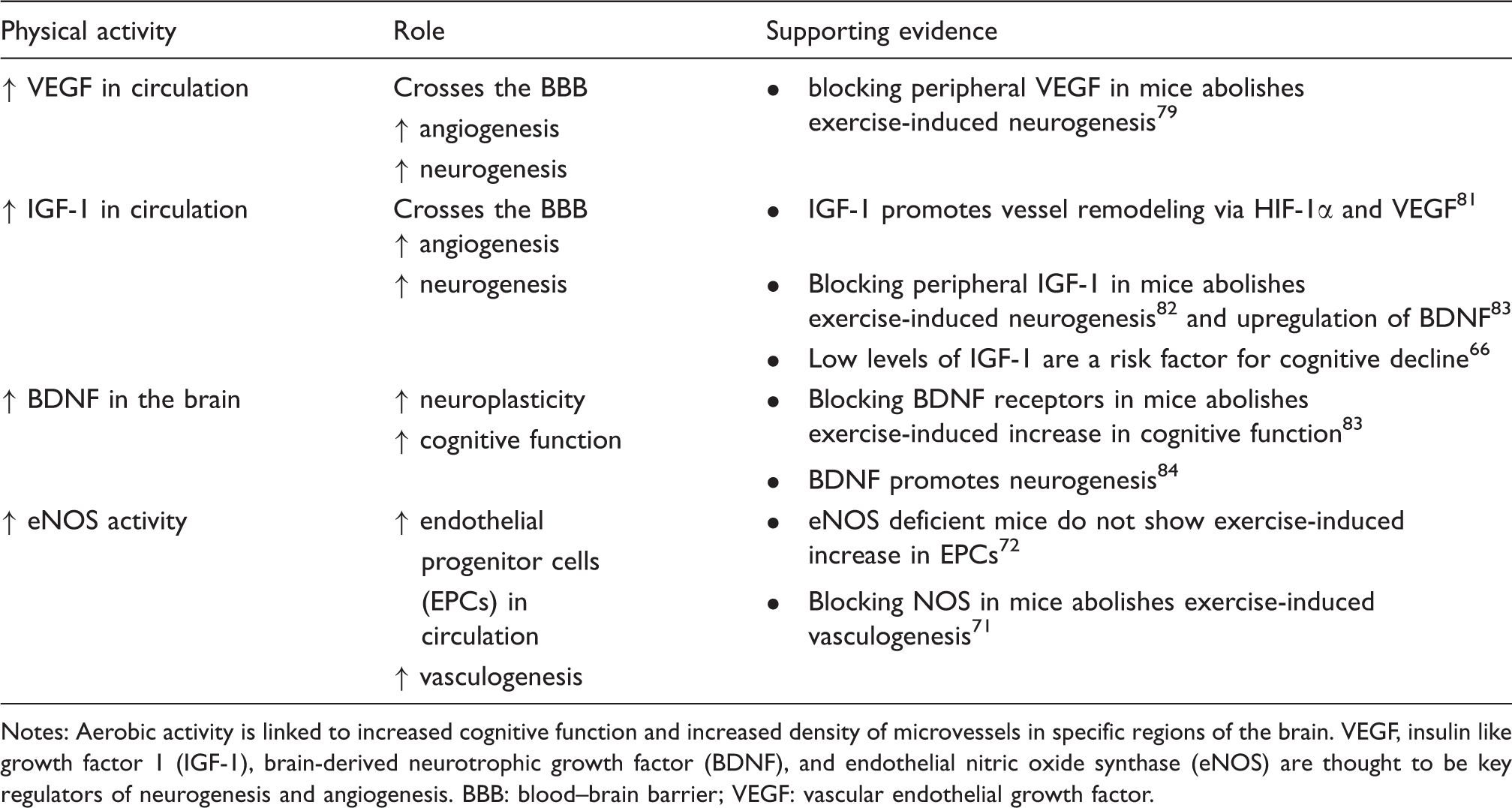
Notes: Aerobic activity is linked to increased cognitive function and increased density of microvessels in specific regions of the brain. VEGF, insulin like growth factor 1 (IGF-1), brain-derived neurotrophic growth factor (BDNF), and endothelial nitric oxide synthase (eNOS) are thought to be key regulators of neurogenesis and angiogenesis.
BBB: blood–brain barrier; VEGF: vascular endothelial growth factor.
The concentration of IGF-1 in circulation is increased with aerobic exercise and is associated with improvements in cognitive function.68,77 Acute aerobic exercise results in a dose-dependent increase in BDNF and IGF-1 levels in circulation, with values returning to baseline after about one hour. 66 Evidence suggests that IGF-1 plays a role in regulating neurogenesis and angiogenesis in the hippocampus.80,81 For example, blocking IGF-1 in circulation abolishes exercise-induced neurogenesis in the hippocampus, 82 and low levels of IGF-1 are risk factors for cognitive decline and aging. 66
BDNF is expressed in the brain and is associated with adaption to environmental changes, response to injury and disease, and modification of brain circuitry. 66 Exercise upregulates expression of BDNF in the brain, particularly in the hippocampus, where production in neurons can remain elevated for weeks with sustained exercise.66,77 Support for the role of exercise-induced BDNF on neural plasticity comes from functional blocking studies in animal models, where blocking the tropomyosin receptor kinase B (TrkB) receptor abolishes exercise-induced improvements in cognitive function. 83 Evidence suggests that both IGF-1 and BDNF are required in exercise-induced neuroplasticity: (1) blocking IGF-1 signaling in animals abolishes upregulation of BDNF in the hippocampus; (2) in cell culture, IGF-1 increases expression of the TrkB receptor, providing a mechanism for increasing BDNF signaling; and (3) BDNF activates a pathway associated with neuroplasticity. 77
Development and aging
Development
During development, the cerebrovasculature is dynamic in structure and function. Many signaling pathways converge to facilitate cerebrovascular plasticity.5,85 VEGF-A, released by the developing neural tube, initiates formation of the perineural vascular plexus (PNVP) via vasculogenesis. Next, BMECs invade the brain parenchyma from the PNVP via sprouting angiogenesis. Non-CNS-specific signaling pathways (e.g. VEGF-A/VEGFR2) and CNS-specific signaling pathways (e.g. Wnt7a/b) guide angiogenesis as well as formation of the BBB (barriogenesis).5,85 Cues in the microenvironment that guide angiogenesis and barriogenesis display unique temporal and spatial expression profiles. For example, VEGF-A expression by neurons guides early angiogenesis, whereas postnatally, astrocytes surrounding the microvasculature are the predominate source of VEGF-A; chronic developmental hypoxia disrupts this expression pattern and results in hypervascularization. 86 Wnt/β-catenin signaling is uniquely required for CNS angiogenesis and barriogenesis; neural progenitor cells release Wnt ligands (Wnt7a and Wnt7b) that guide sprouting of BMECs and induce BBB formation via expression of GLUT1. 87
The timing of angiogenesis and barriogenesis during brain development is controversial. However, accumulating physiological and molecular evidence indicates that the developing brain displays barrier properties appropriate for each developmental stage. 88 Studies in zebrafish suggest that angiogenesis and barriogenesis are temporally indistinguishable, 89 while histological studies in human embryos show that during brain angiogenesis, capillaries are positive for the tight junction protein claudin-5 and already restrict transport of plasma proteins. 90 Postnatally, vascular remodeling continues but is not well understood; however, VEGF-A and axonal growth inhibitor, NOGO-A, have been implicated. 85
As BMECs invade the CNS, they interact with neurons, neural stem cells, glial precursors, and pericytes. Sprouting BMECs release platelet-derived growth factor β to recruit pericytes and initiate BBB formation during embryogenesis. 91 Astrocytes are not present in the developing brain when it is initially vascularized, however, astrocytic-derived sonic hedgehog ligands are critical in promoting BBB integrity and immune quiescence postnatally. 92 Glial–endothelial interactions also guide spatial patterning during brain development; radial glia cells provide a scaffold for BMEC migration, whereas BMECs later provide a scaffold for oligodendrocytes precursor migration.85,93
Aging
Aging is associated with heterogeneous decreases in both microvascular and neuronal density in the brain. Although these decreases can be linked, evidence suggests that cerebrovascular plasticity and neurovascular coupling are also impaired during aging.29,94 Histological analysis of post-mortem tissue in humans frequently shows a decrease in microvascular density (typically 10–30%) during normal aging, particularly in the prefrontal cortex and hippocampus.29,94,95 Aging is also associated with thickening of the BM, pericyte degeneration/loss, and swelling of astrocytic end-feet.94,96 Age-associated functional changes include increased tight junction defects and increased permeability.97,98 Similar age-associated changes have been observed in brain arterioles in the cerebral cortex, including decreased density (∼40%), loss of SMCs and elastin, and gain of BM and collagen. 94
Evidence from two-photon microscopy of the cerebral cortex in mouse models showed BMEC senescence, reduced turnover, and diminished hypoxia-induced angiogenesis compared to younger mice. 26 Similar to other vascular beds, impaired angiogenesis is thought to be related to HIF1-α hyporesponsiveness, growth factor downregulation, matrix metalloproteinase inhibition, eNOS inactivity, reduced bioavailability of NO, impairment of endothelial proliferation, reduced recruitment of EPCs, and pericyte dysfunction. 99 Enriched environments or administration of VEGF viral vectors, which elicit increases in brain microvascular density in juvenile and adult rodents, result in reduced changes in aged rodent brains.100,101 Age-associated failure of angiogenic signaling is implicated as VEGFR-2 is not properly unregulated upon stimulation, 101 and brain region-specific changes in angiogenesis-related genes are found in aged mice. 95
Numerous brain-imaging studies have shown age-associated decreases in CBF and CMRO2 in many regions of the cerebral cortex, in particular, in regions vulnerable to neurodegeneration. 102 In the cortex, decreases of 0.38–0.76% per year have been reported.11,102– 104 Studies of healthy brains indicate that decreases in CBF are largely distinct from gray-matter atrophy, suggesting that age-associated changes in ultrastructure and hemodynamics are independent. 103 CBF reductions may result from a loss of arterial supply to the brain (mediated by decreased IGF-180), a hypercontractile arteriole phenotype, and impaired neurovascular coupling.
Neurovascular coupling is impaired during aging in humans and animal models. 105 – 108 Dysfunctional neurovascular coupling causes a mismatch between supply and demand of oxygen/metabolites for neurons, likely contributing to cognitive decline. Interestingly, pharmacologically induced cerebrovascular uncoupling in mice mimics the aging phenotype (e.g. impaired spatial working memory, recognition memory, and motor coordination) without changing basal CBF. 109 The contributions of IGF-1 deficiency, oxidative stress, endothelial dysfunction, and astrocyte dysfunction to cerebrovascular uncoupling during normal aging have been reviewed elsewhere. 108 Pericyte degeneration has also been implicated in cerebrovascular uncoupling via reductions in oxygen delivery and increased metabolic stress. 41 BOLD fMRI studies of neural activity during aging are complicated by cerebrovascular changes including the loss of neurovascular coupling. 110 Contrary to other reports, a recent study utilizing a deconvolution technique found no significant change in BOLD neurovascular coupling during visual and auditory task with normal aging. 111 Thus it remains unclear if age-associated declines in neural activity precede cerebrovascular changes or result from impaired cerebral hemodynamics.
Response to injury and disease
Changes in cerebrovascular structure, plasticity, and coupling occur in AD, PD, HD, and ALS.

Notes: For each disease, neuropathological hallmarks as well as cerebrovascular changes are summarized. Across the four most common neurodegenerative disease there are reported reductions in cerebral blood flow (CBF) and increases in blood–brain barrier (BBB) permeability. Overexpression (AD, PD, and HD) or underexpression (ALS) of vascular endothelial growth factor, which drives brain angiogenesis, may underlie ultrastructural changes. While vascular degeneration is widely reported in AD and PD (characterized by decreased microvessel density), elevated microvessel density may occur in early-stage disease and contribute to BBB permeability increases also associated with these diseases. Changes are brain region specific.
VEGF: vascular endothelial growth factor; hiPSC: human-induced pluripotent stem cell; BMEC: brain microvascular endothelial cell.
Ischemic stroke
Stroke is the most common form of cerebrovascular injury and second leading cause of death worldwide. 117 Each year, roughly 15 million individuals experience stroke worldwide. Of these, one-third do not survive, while another third become permanently disabled. The most common site of ischemic stroke in humans is the middle cerebral artery, which affects the posterior frontal, lateral, and parietal lobes. 118 During a typical ischemic stroke (80% of all stroke cases), the human brain ages 3.6 years for each hour without treatment; this translates to loss of about 120 million neurons and 830 billion synapses per hour. 119 Cerebrovascular plasticity is equally dynamic; middle cerebral artery occlusion in mice results in rarefication (75% loss) of brain capillaries within the microinfarction core, while larger microvessels are unaffected. 120
The induced occlusion of a single cortical penetrating arteriole in rats results in a cylindrical microinfarction core about 500 µm in diameter and extending about 1 mm into the cortex. Neuronal activity is completely diminished within 2 h, and conditions become severely hypoxic (PaO2 < 10 mm Hg) in 6 h. 121 However, occlusion of microvessels, two or more branch, points away from cortical penetrating arterioles (i.e. capillaries) does not lead to detectable microinfarctions, as flow in interconnected microvascular networks can reverse to compensate for the occlusion. 121 Thus, the severity of ischemic stroke is heavily dependent on the location of the occlusion and presence of collateral circulation.
The normal progression of ischemic stroke involves three phases. In the acute phase (up to 48 h after onset), hypoxia leads to release of neurotransmitter glutamate, which results in widespread excitotoxicity and subsequent neuronal injury. 122 When blood flow is restored to the infarction site, either naturally or by intervention, reperfusion leads to further damage known as reperfusion injury. Activated glial cells, damaged neurons, and other cells of the neurovascular unit release growth factors, 123 matrix metalloproteinases, 124 reactive oxygen species, 125 glutamate, 126 NO, 127 and cytokines. 128 These compounds are implicated in diverse roles post-injury including angiogenesis, BBB breakdown, BM degradation, glial activation, and neurotoxicity.113,129,130 Additionally, peripheral immune cells enter the brain with unique temporal dynamics, which in tandem with innate glial activation, generates a proinflammatory environment. 131
The sub-acute phase of ischemic stroke, occurring between two days and six weeks, is associated with the initiation of repair. In this phase, cerebrovascular plasticity is at its highest, and many of the mediators of vascular injury begin to take on neuroprotective and regenerative roles. VEGF plays a critical role in post-ischemic neurovascular remodeling. However, the route and timing of VEGF administration following ischemic stroke modulate its therapeutic effects; for example, systemic VEGF delivery during the acute phase promotes BBB disruption and worsens brain injury, while local modulation of VEGF or systemic delivery during the sub-acute phase may reduce ischemic damage.132,133 Following occlusion, BMECs begin proliferating and generating sprouts within 12–24 h, leading to formation of new capillaries within three days after ischemic injury. 134 Additionally, brain pericytes play various roles in promoting microvessel stabilization and neuroprotection following stroke. 135 An important characteristic of the sub-acute phase is the regenerative neurovascular niche, in which angiogenic blood vessels signal to neural progenitor cells to mediate neurogenesis. This model is supported by the presence of newly formed neurons near remodeled vessels in the rodent striatum and cerebral cortex following stroke, 136 most likely due to recruitment by BMECs that form a niche for neural stem cells. 35 During the chronic phase (after approximately three months following stroke), endogenous plasticity declines while vascular repair, angiogenesis, and behavioral improvement continue (although at a slower rate). 137
Traumatic brain injury
TBI has diverse causes including blast-waves (i.e. improvised explosive devices), blunt trauma (i.e. contact sports), and penetration trauma (i.e. firearms). Cerebrovascular changes during TBI occur in two distinct phases: primary and secondary. During primary injury, direct biomechanical damage to cerebral tissue results in loss of cerebrovasculature architecture, CBF abnormalities, BBB disruption, and often fatal hemorrhaging and edema, while during secondary injury, cellular and molecular responses promote repair via angiogenesis, vasculogenesis, and neurogenesis.138,139 The timescale and severity of responses in these phases is dependent on the injury modality, severity, and location, among other factors.
TBI results in rapid and significant decreases in microvascular density.140,141 In an animal model of fluid percussion trauma, the cerebrovascular density of the ipsilateral cerebral hemisphere decreased by approximately 50% within 24 h, while recovery was dependent on the severity of the injury and associated with establishment of a proangiogenic environment. 140 Other ultrastructural changes following TBI include BMEC swelling, vessel thinning, BM thickening, and pericyte degeneration. 114 In TBI patients, reduced CBF is observed within 12 h after injury, while neurovascular uncoupling is thought to occur during the secondary phase. 142 Additionally, BBB disruption is widely observed following TBI and is associated with worse long-term outcomes, and may persist for years after injury contributing to risk of neurodegenerative disease.143,144 Angiogenesis and vasculogenesis are initiated during the secondary injury phase, resulting in increases in microvascular density over several weeks.114,140,145 Local hypoxia is thought to be the driver of neovascularization with HIF-1α and VEGF acting as critical molecular regulators. 145
Neurodegeneration
Cerebrovascular structure, plasticity, and coupling are altered during neurodegenerative disease, beyond the impairments observed during normal aging.146,147 Whether cerebrovascular changes precede, follow, or occur concurrently with neurodegeneration is not yet fully understood, however, accumulating evidence suggests that cerebrovascular changes can occur prior to the presentation of symptoms and promote neurodegeneration. 148 – 157
Neurodegenerative disease is characterized by progressive loss of functional neurons and compromised human brain function. Age is the greatest risk factor for neurodegenerative disease. 158 Neurodegeneration and normal brain aging share common pathologic origins including oxidative stress, mitochondrial dysfunction, and proteotoxicity.158,159 However, neurodegenerative diseases are also highly heterogeneous with distinct risk factors (environmental and genetic), histopathological hallmarks (neuronal loss in specific brain regions and formation of protein aggregates), and clinical manifestations. Despite the pathogenic differences, several themes connect cerebrovascular changes during neurodegeneration: (1) BBB dysfunction;5,96,146 (2) cerebral hypoperfusion and glucose hypometabolism; 160 and (3) overexpression (Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD)) or underexpression (amyotrophic lateral sclerosis (ALS)) of VEGF (Table 2). 30
Alzheimer’s disease
Cerebrovascular changes associated with Alzheimer’s disease include vascular degeneration, altered angiogenesis, reduced CBF, increased BBB permeability, and neurovascular uncoupling.146,149 The amyloid hypothesis states that the formation of amyloid-β (predominantly the Aβ-42 isoform) within the brain parenchyma induces gain of toxic function and neurodegeneration. However, AD is also closely associated with cerebral amyloid angiopathy (CAA), which is characterized by perivascular accumulation of predominantly the Aβ-40 isoform in the BM of leptomeningeal and cortical vessels of the cerebrum and cerebellum. 161 The epidemiological link between cerebrovascular risk factors and AD pathogenesis is well established and has been reviewed elsewhere. 162
Post-mortem analysis of AD brain reveals capillary loss, as well as atrophied BMECs and pericytes, swollen astrocytic end-feet, hypercontractile SMCs, BM thickening, and BM deposits.163,164 In severe CAA, additional pathologies include SMC loss, duplicated lumens, fibrinoid necrosis, and hyaline degeneration. 161 Despite capillary loss, AD is associated with increased concentrations of VEGF.165,166 In post-mortem human AD tissue, an increase in the number of angiogenic vessels (integrin αvβ3 expression) was correlated with several AD pathologies including neurofibrillary tangles and Aβ load. 167 Transgenic AD animal models corroborate these findings; in aged Tg2576 mice, disruption of tight junctions was linked to increased microvascular density, suggesting that amyloid-β initiates hypervascularization and BBB disruption in early-stage AD, while microvascular degeneration occurs in last-stage AD. 168 However, the effects of amyloid-β on brain angiogenesis remain convoluted as amyloid-β has been reported to both promote and suppress angiogenesis both in vitro and in vivo.169,170
Reduced CBF, increased BBB permeability, and reduced glucose metabolism are found to precede AD neurodegeneration in animal models and human studies. 43 Reduced CBF is found in patients at high risk of developing AD (APOE ɛ4 allele) and patients with mild cognitive impairment.171,172 These reductions persist during AD progression and appear to correlate with the degree of cognitive decline and AD pathology. 163 Recently, neutrophil adhesion in brain capillaries has been shown to reduce cortical blood flow in AD mouse models. 173 Additionally, BBB permeability increases before plaque formation in an AD mouse model, while MRI studies indicate that increased BBB permeability of mild cognitive impairment patients correlates with AD cognitive decline.148,150 Positron emission tomography imaging studies find reduced glucose metabolism in patients at high risk of AD, which precedes brain atrophy.146,174 CBF, metabolic rate, and BBB permeability changes contribute to ischemic/hypoxic damage of the neurovascular unit and initiate and/or exacerbate AD pathology.43,146
AD patients, APOE ɛ4 allele carriers, and transgenic AD mice exhibit impaired neurovascular coupling responses, while therapies that rescue neurovascular coupling are associated with cognitive improvements.108,151,175 AD-associated changes in vascular reactivity factors (i.e. eNOS and endothelin-1) may underlie neurovascular uncoupling and chronic hypoperfusion. 176 Interestingly, partial eNOS deficiency in mice results in CAA-like pathologies and stroke in brain regions associated with hypoperfusion in preclinical AD patients. 177
Parkinson’s disease
Cerebrovascular changes associated with PD include both vascular degeneration and angiogenesis. PD is histopathologically characterized by dopaminergic neuronal loss within the substantia nigra in the brainstem. 178 Additionally, Lewy bodies, aggregates of predominately α-synuclein, form in the cytoplasm of specific neurons in widespread brain regions. Impairment of α-synuclein degradation during aging may contribute to dopaminergic neuronal loss and downstream motor deficiency (bradykinesia) observed in PD patients. 159
PD-associated vascular degeneration resembles that of AD; morphological changes include BMEC degeneration and decreases in microvascular density.164,179 However, other studies find increased angiogenic vessels (integrin αvβ3 expression) in PD-associated brain regions, without overt changes in microvascular density. 180 Thus, it is possible that a proangiogenic environment is established in PD patients, as evidenced by heightened angiogenic biomarkers. 181 Integrin αvβ3 expression is co-localized with BBB leakage in animal models of PD (6-hydroxydopamine mouse model), while the striatum in post-mortem PD sections demonstrates signs of BBB disruption, suggesting that angiogenesis disrupts the BBB, thereby contributing to vascular degradation in late-stage disease.182,183 Additionally, hypoperfusion of specific brain regions of PD patients is correlated with motor dysfunction. 184
Elevated microvascular plasticity in PD patients treated with deep brain stimulation (DBS) has recently been reported. 185 Reductions in microvascular density, endothelial tight junction protein expression, and endothelial VEGF expression were found in PD patients compared to age-matched controls. However, in PD patients treated with subthalamic nucleus DBS, these cerebrovascular changes were reversed. Increased neural activity promoted by DBS rapidly modulates production of neurotrophic and angiogenic factors, such as VEGF and BDNF. 186
Huntington’s disease
HD is associated with cerebrovascular alterations including increased microvascular density, BBB disruption, and altered cerebral hemodynamics. HD is caused by expansion of cytosine–adenine–guanine repeats in the huntingtin gene. 187 Mutant huntingtin (mHTT) protein, produced by neurons and astrocytes, aggregates within the brain parenchyma, BM of microvessels, and cells of the NVU. 188 Loss-of-function of normal HTT and gain-of-function of mHTT contributes to progressive loss of neurons in the striatum, cortex, and hippocampus, among other regions, accompanied by declines in cognitive and motor functions. 187
In HD patients and mouse models of HD, microvascular density is increased up to two-fold compared to age-matched healthy controls.188,189 BBB disruption has been observed in HD mouse models, brain imaging of mild-to-late stage HD patients, and post-mortem studies of HD tissue. 188 Changes in tight junction expression that underlie increased BBB permeability were recently found to precede disease presentation in a mouse model (R6/2). 152
Astrocytes have been implicated as a mediator of microvascular changes; increased astrocytic VEGF in a HD mouse model (R6/2) promoted angiogenesis and loss of pericytes contributing to abnormal cerebral autoregulation. 190 Recent work suggests that BMECs of HD patients are autonomously dysfunctional; human-induced pluripotent stem cells derived into BMECs from HD patients exhibit elevated angiogenic potential and deficient BBB properties compared to healthy cell lines. 153 Additionally, reductions in cortical CBF and impaired neurometabolic coupling have been reported in early stage HD patients and the R6/2 mouse model, respectively.154,155
Amyotrophic lateral sclerosis
During ALS, progressive degeneration of motor neurons in the cortex, brainstem, and spinal cord lead to loss of voluntary muscle control and generalized paralysis. Many molecular mechanisms have been implicated in ALS pathogenesis, including failure of proteostasis, mutations of RNA-binding proteins, axonal transport dysfunction, cytoskeletal disarrangement, and dysfunction of glial and neurovascular cell types.191,192 ALS-associated neurovascular unit dysfunction, including BBB breakdown, has been reviewed elsewhere. 193 In SOD-1 mutant mouse models, disruption of the blood–spinal cord barrier was observed prior to the development of symptoms of the disease and prior to an inflammatory response, 156 suggesting that vascular changes can precede symptomatic disease.
Genetic alterations in expression of hypoxia-inducible genes are heavily implicated in ALS pathogenesis. Mice with reduced VEGF levels develop progressive motor neuron degeneration resembling human ALS, and reduced VEGF accelerates motor neuron degeneration in familial ALS mouse models.194,195 Humans with genetic variations of the VEGF promoter that confer low levels of circulating VEGF are at heightened risk to develop ALS. 195 Interestingly, early ALS patients demonstrated reduced CSF VEGF levels independent of VEGF promoter polymorphisms. 157 Insufficient VEGF likely contributes to ALS progression by both depriving neurons of neurotrophic signals and by contributing to BBB dysfunction and hypoperfusion/ischemia. 30
Summary
During normal brain function, local changes in neural activity are accompanied by local dilation/contraction of microvessels to meet current metabolic demands. Under these conditions, the cerebrovascular architecture is generally thought to be fixed and, at the cellular level, the turnover of BMECs is negligible. However, subtraction and pruning of redundant loops suggest that the cerebrovasculature is not completely static during homeostasis. During development, neuronal and glial interactions drive additive changes in the cerebrovasculature, while the loss of neurons associated with aging is accompanied by heterogeneous subtractive changes. Injury and disease can also result in subtractive loss and induce subsequent repair processes. Sustained changes in the global brain microenvironment due to sensory deprivation, aerobic exercise, or hypoxia, can also lead to vascular remodeling and additive or subtractive changes in the cerebrovascular architecture. A common factor in all additive changes in the cerebrovasculature is VEGF, a protein involved in vasculogenesis and angiogenesis, and expressed and recognized by BMECs, glial cells, and neurons
Model systems to study cerebrovascular plasticity are continually advancing. Multiphoton and intravital microscopy support studies of cerebrovascular plasticity in living animal models. 25 – 28 In humans, neuroimaging modalities (i.e. fMRI) provide real-time metrics of cerebrovascular function, while post-mortem histological analyses provide information on cerebrovascular structure. There remains a need for techniques to study human cerebrovascular plasticity with high temporal and spatial resolution. In vitro models of cerebrovascular plasticity may satisfy this need. However, current models fail to recapitulate in vivo structure, function, or plasticity. For example, while transwell assays (comprised of BMECs cultured on a permeable membrane) can achieve physiological permeability, it does not mimic physiological hierarchy or geometry. 19 Without three-dimensional structure, models of cerebrovascular plasticity will fail as they cannot recapitulate additive or subtractive changes of capillaries. Recent advances in stem cell technology, microfluidics, and tissue engineering have led to three-dimensional cerebrovascular models with increasing complexity.196,197 However, their ability to achieve characteristic stability of cerebrovascular architecture during homeostasis and plasticity during perturbation has yet to be shown. Development of stable three-dimensional models of the cerebrovasculature will support studies of brain plasticity, as animal models do not always accurately model human homeostasis or pathogenesis.
Authors’ contributions
MIB, JGD, RML, ADW, and PCS wrote and edited the paper.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors gratefully acknowledge support from DTRA (HDTRA1-15-1-0046) and NIH (R01NS106008). JGD acknowledges support from the Nanotechnology for Cancer Research training program. RML acknowledges a National Science Foundation Graduate Research Fellowship (grant no. DGE1746891).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.