
Abstract
The inflammatory response triggered by stroke has been viewed as harmful, focusing on the influx and migration of blood-borne leukocytes, neutrophils, and macrophages. This review hypothesizes that the brain and meninges have their own resident cells that are capable of fast host response, which are well known to mediate immediate reactions such as anaphylaxis, known as mast cells (MCs). We discuss novel research suggesting that by acting rapidly on the cerebral vessels, this cell type has a potentially deleterious role in the very early phase of acute cerebral ischemia and hemorrhage. Mast cells should be recognized as a potent inflammatory cell that, already at the outset of ischemia, is resident within the cerebral microvasculature. By releasing their cytoplasmic granules, which contain a host of vasoactive mediators such as tumor necrosis factor-α, histamine, heparin, and proteases, MCs act on the basal membrane, thus promoting blood–brain barrier (BBB) damage, brain edema, prolonged extravasation, and hemorrhage. This makes them a candidate for a new pharmacological target in attempts to even out the inflammatory responses of the neurovascular unit, and to stabilize the BBB after acute stroke.
Introduction
Inflammatory reaction, a host response to noxious stimuli, has been the focus of research on ischemic brain injury for several decades. It has been viewed as a detrimental process with a wide array of mechanisms, and approaches to harness it are still considered as promising avenues for future stroke treatment (Hallenbeck et al, 2006). The purpose of this review is to provide new evidence supporting a hypothesis that mast cells (MCs), which are resident in several brain structures and meninges, act as early regulators of the immediate blood–brain barrier (BBB) changes and ensuing vasogenic edema that characterize sudden cerebral ischemia. By releasing potent preformed vasoactive mediators that then act on the cerebral vessels, MCs could promote the breakdown of the BBB, edema, extravasation, and hemorrhage.
We review the following in this paper:
Existing data on MC activation and their role in immediate host response;
the multitude of released vasoactive mediators of MC granule contents;
antagonism of inflammatory cell responses as a strategy for clinical stroke therapy;
recent experimental ‘proof-of-concept’ studies suggesting that MC stabilization might provide a basis for effective novel therapies in ischemic or hemorrhagic stroke in the future;
evidence on how MCs could participate in a step-by-step degradation of the BBB and promote secondary inflammatory responses in relation to other well-known regulators in this cascade, such as cytokines, matrix metalloproteinases (MMPs), and cathepsins;
the putative interrelationships between MCs and components of the vascular wall, perivascular inflammatory, neuronal, and glial cells assembled to form an integral neurovascular unit (NVU); and
the potential relevance of the reviewed evidence to therapeutic attempts to harness ischemia-dependent inflammation, expansive brain edema, and cerebral hemorrhages.
Mast cells and host response in the brain
The brain has long been considered an immunologically privileged organ, and recent findings have implied an involvement of immune mechanisms in neurologic disease and illness, including ischemic and hemorrhagic stroke. The presence of the BBB and the lack of lymphatic vessels alluded that immunologic host response within the central nervous system (CNS) is limited. However, at present, considerable literature attests the innate participation of several resident brain cells in immune surveillance, e.g., by astrocytes, oligodendrocytes, microglia, various perivascular cells (endothelial cells, smooth muscle cells, pericytes, perivascular macrophages, MCs), and even neurons during health and disease (Griffiths et al, 2009; Price et al, 2003). Although resident cerebral cells do not characteristically express proteins of major histocompatibility complex, microglia and astrocytes as so-called nonprofessional antigen-presenting cells can present novel antigens to naive helper T cells that will infiltrate into the neuropil on appropriate stimuli (Griffiths et al, 2009; Price et al, 2003).
Although inhibition of the inflammatory cascade of blood-borne neutrophil and phagocyte infiltration after cerebral insults has been the subject of serious attempts to establish a new therapeutic avenue in stroke, as reviewed previously (Emerich et al, 2002; Frijns and Kappelle, 2002), recent research on ischemia has not been focused on resident brain cell types that are capable of mounting immediate host responses in the brain and meninges, namely the MCs. Although MCs are present in the brain from the time of birth and earlier investigators had noticed the presence of perivascular MCs in the brain and meninges (Figure 1) (Dropp, 1976; Florenzano and Bentivoglio, 2000; Goldschmidt et al, 1984; Hough, 1988; Manning et al, 1994; Silver et al, 1996; Theoharides, 1990), they have thereafter received insufficient attention.
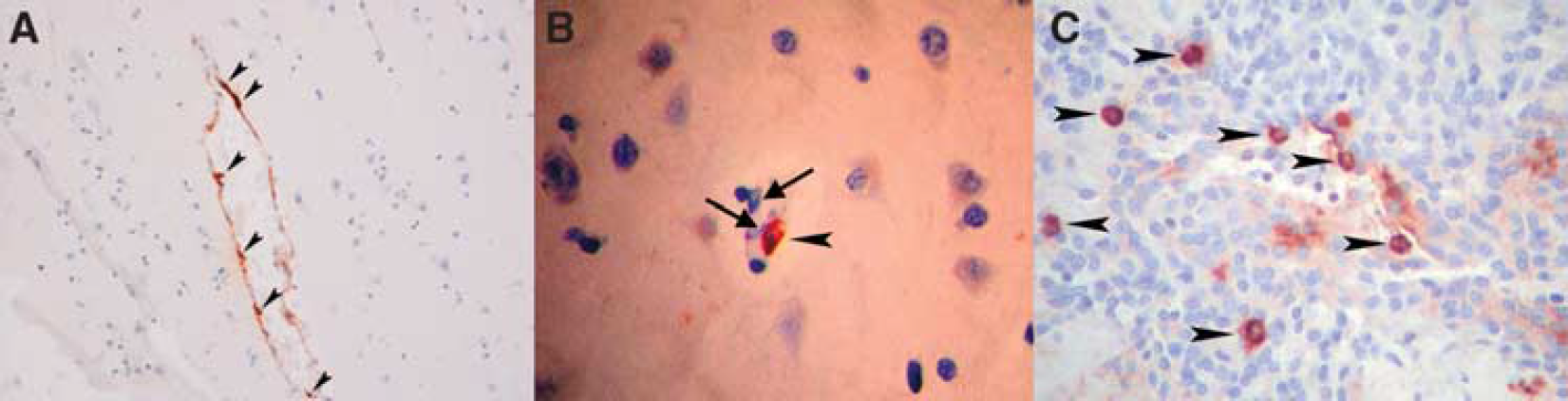
Mast cells in human cerebral and meningeal tissues. (
Mast cells enter the CNS during development through penetrating blood vessels, with which they remain associated (Lambracht-Hall et al, 1990), as shown by ultrastructural studies showing the predominantly perivascular location (Figures 1 and 2) of MCs (Dimitriadou et al, 1990; Letourneau et al, 2003). Even during development, MCs occur in two locations: the pia and the brain parenchyma. Although the pial MC population in rodents declines rapidly and reaches adult levels by postnatal week 30, the thalamic population does increase during the same period (Asarian et al, 2002; Khalil et al, 2007). Almost 97% of MCs lie on the abluminal (brain) side of the blood vessels (Khalil et al, 2007). In the body, MCs are typically arranged in the proximity of outer layers and barriers, such as the epithelia, mucosal membranes, and vascular walls, where they can protect organs against noxious environmental or blood-borne stimuli. In the CNS, corresponding tissues are the meninges and the blood vessels forming the BBB.
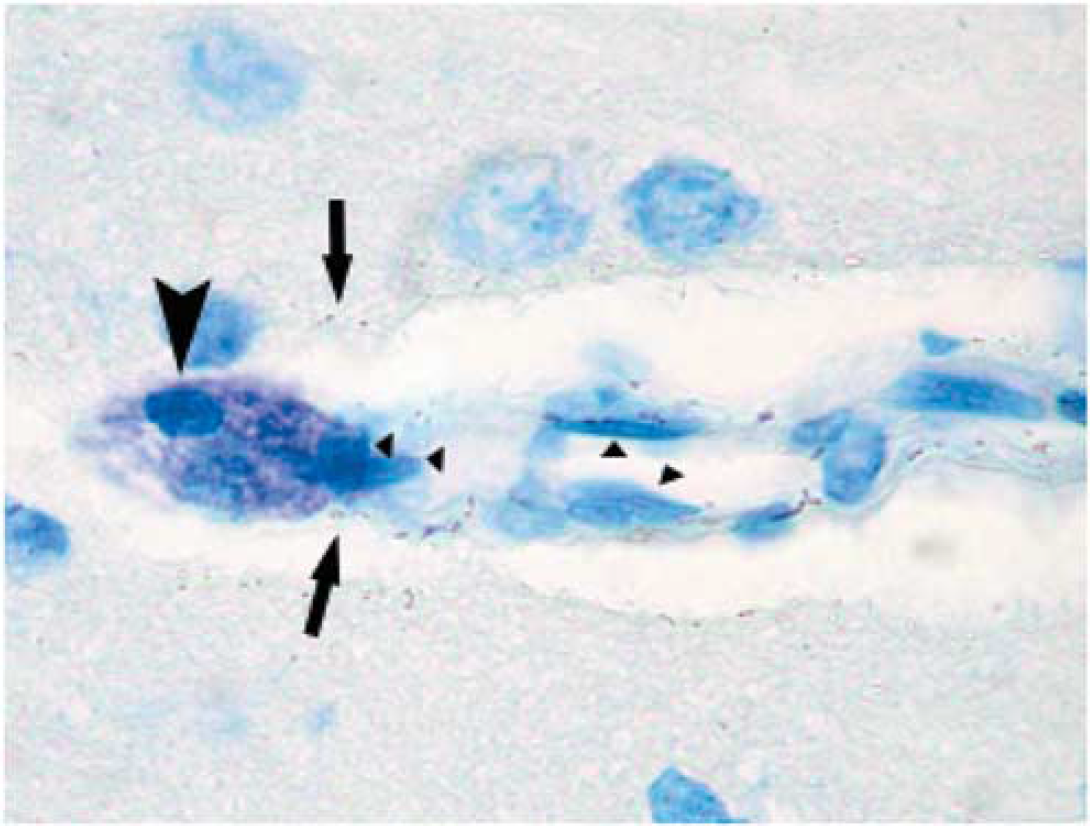
Toluidine blue staining of rat brain sections. Toluidine blue staining of ischemic rat brain sections highlighting the metachromatic (deep violet) heparin-containing cytoplasmic granules associated solely with MCs in an intensely stained round/oval MC (arrows). The typical perivascular position along a penetrating cortical artery must be noted. Arrows point to exocytosed MC granules, and triangles highlight endothelial cells.
Mechanisms of action of mast cells
Mast cells have been recognized as major cellular mediators of fulminant type-I hypersensitivity responses and systemic anaphylaxis. These responses have also long been known to be biphasic, the immediate phase occurring within minutes and the late phase within several hours. The former is caused by the release of MC granule contents (such as histamine, tumor necrosis factor (TNF)-α, chymase, tryptase, cathepsin-G, heparin, and various chemoattractants). The second phase is characterized by
Should acute cerebral insults trigger an analogous biphasic MC-driven phlogistic response, the predicted result would be an acute increase in vascular permeability, followed by the gradual accumulation of circulating PMNLs facilitated by the released histamine, chemotactic agents, and morphologic alteration of the BBB. Indeed, earlier data support the observation that edematous changes in the brain may be regulated by histamine and its receptors (Abbott, 2000; Esposito et al, 2001; Joo et al, 1994; Yong et al, 1994; Zhuang et al, 1996), as histamine is a main granule constituent of MCs. Even earlier experiments had shown that MC activation by potent enzymes such as phospholipase-A could induce edema in the peripheral vascular bed (Brain et al, 1977). Recent results obtained from experimental models summarized in detail below have provided more data supporting the existence of such an immediate hypersensitivity-type response that opens the BBB and leads to very acute expansive brain edema and secondary PMNL infiltration (Strbian et al, 2006, 2007
Inflammatory cell inhibition as a strategy in acute stroke therapy
There have been three major attempts to test the clinical value of limiting inflammatory cell-dependent injury in ischemic stroke (Becker, 2001; ICOS; Investigators, 2001; Krams et al, 2003). These have targeted the cascade of tissue entry of leukocytes that is well elucidated in various experimental ischemia–reperfusion injury models and in a multitude of organs. Typically, such approaches have targeted adhesion, transmigration, and infiltration of PMNLs that start within hours after the onset of tissue ischemia (Frijns and Kappelle, 2002). Antibodies have been developed to bind to adhesion molecules on the circulating leukocyte membranes (e.g., CD11/CD18) (Rieu et al, 1994) or on their counterpart molecules on the luminal side of the vascular endothelium (e.g., intercellular adhesion molecule-1) (Rothlein et al, 1993).
The first medicament to reach clinical development targeted intercellular adhesion molecule-1, and was shown to be beneficial in several preclinical experimental models and to be tolerated in humans (Bowes et al, 1993, 1995; Schneider et al, 1998). Unfortunately, it led to paradoxical neutrophil activation and hyperthermia. The antibody was found to elicit host antibodies and paradoxically activated neutrophils in later laboratory examinations (Vuorte et al, 1999; Furuya et al, 2001). It activated blood complement (Vuorte et al, 1999), which is common for this isotype of murine IgG2a. However, the trial was completed with the inclusion of 625 patients and resulted in an excess of CNS-related deaths and neurologic worsening in the active treatment arm (Investigators, 2001; Jiang et al, 1998).
Another clinical investigation used the antibody UK-279,276 (neutrophil inhibitory factor), a recombinant glycoprotein with selective binding to the CD11b integrin of CD11b/CD18 (Mac-1), which reduced neutrophil infiltration and infarct volume in transient (2-h) rat MCAo (middle cerebral artery occlusion) models when administered within 4 h after the onset of ischemia (Jiang et al, 1998) and was shown to be tolerated in patients with acute stroke (Lees et al, 2003). The results of this study (
The hypothesis of PMNL-targeted therapies has probably not yet been adequately tested in clinical trials. However, PMNLs as the early wave of phagocytic cells arrive at the scene only after the most immediate ischemic damage has already occurred. It may well be that the tissue volume to be salvaged by blocking the activity of PMNLs is very limited and impossible to detect in clinical outcome measures (Emerich et al, 2002). To continue the debate, translation of preclinical data to clinical trials is considered to be not straightforward (Dirnagl et al, 2007). A key issue in this regard is the need to devote more attention to validating the nominal effect of the interaction between the drug candidate and its molecular target, and its relationship to pharmacodynamic treatment end points. It is equally important to prove that clinical outcomes are linked to the alleged mechanism of action of the compound (Feuerstein et al, 2008).
As the trials targeted to PMNLs have been subjected to criticisms possibly because of being rather late in responding to inflammatory cells in the face of ischemic brain injury (Emerich et al, 2002), more proximal effector cells inciting the inflammation driven by ischemia–reperfusion injury should be sought as pharmacological targets. Immediate hypersensitivity reactions that are well-established physiologic responses to either environmental exogenous or endogenous or direct physico-chemical noxious stimuli constitute an archaic but powerful host defense mechanism. These immediate reactions within various end organs are performed by resident immune cells that are already present in the exterior aspects (such as the epithelia, mucosae, blood vessels) of target organs—-tissue MCs—-which can also produce an abrupt systemic anaphylactic reaction. This is in contrast to circulating immune cells, which must first be invited by locally released chemotactic substances and anchored by endothelial adhesion molecules before they gradually infiltrate the organs to carry out their purpose.
Mast cells are present and active in the brain
Detection of Tissue Mast Cells
Mast cells are not easily visible in standard histopathological stainings such as hematoxylin and eosin, and therefore they need to be sought after using few specialized stainings. Mast cells arise from a CD34 +/CD117 + progenitor cell population (Kirshenbaum et al, 1999) and reside in the bone marrow and peripheral organs, often associated with blood vessels (Figures 1 and 2), nerves, glandular ducts, stromal connective tissue, or mucosa. In contrast to basophils, they characteristically have a round or elongated nucleus and finer but dense cytoplasmic granules ∼1 μm in diameter. Mast cell morphology varies from round or oval to spindle (Figures 1A and 1B). Their abundant cytoplasmic heparin sulfate-containing granules can be shown by metachromatic dyes such as toluidine blue (Figure 2), azures A and B, thionin, and methylene blue, which induce purple or violet staining of the granules. Toluidine blue and Giemsa are the most commonly used staining procedures. Fluorophoreconjugated egg white avidin also binds to heparin and has been used as well. With enzyme cytochemistry, MCs can be detected by the demonstration of Naphthol AS-D chloroacetate esterase activity (Leder staining) (Figure 3), which is lacking in basophils (Horny et al, 2008; Leder, 1964). Safranin and alcian blue stains have been used for showing the immaturity of MCs.
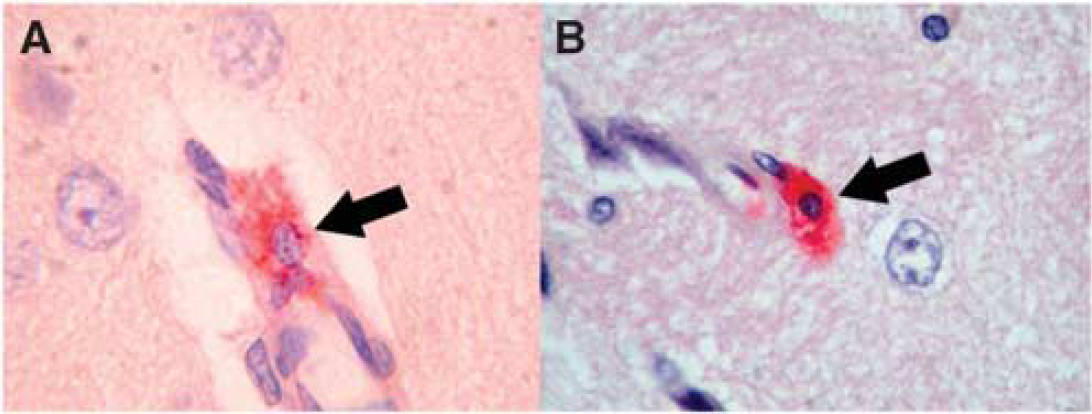
Chloroacetate esterase (Leder)-stained mast cells in ischemic rat brain. (
In addition to CD117, MCs express various myelomonocytic antigens, including CD68, CD11c, CD 33, CD43, CD45, and C5aR. They can also be immunohistochemically detected by preformed granule-associated mediators such as histamine, heparin, chymase, and tryptase (Figure 1A). In practice, the most sensitive and specific antigen for immunohistochemistry is tryptase; CD117 (Figure 1B) is highly sensitive but is not as specific (Horny et al, 2008).
Mast Cell Trafficking into The Brain
Alterations do appear in the abundance of MCs in the brain, which may be caused by changes in marker-dependent detection, by the rate of both entry and proliferation of precursor cells and the differentiation activity of MCs (Zhuang et al, 1999). It is generally accepted that MCs circulate as devoted precursors rather than as mature cells (Galli et al, 1992; Kitamura et al, 1993). However, what is also possible is that mature MCs translocate from peripheral sources into the CNS (Silverman et al, 2000). In the latter study, migrated donor MCs represent 2% to 20% of the total MC population in the analyzed brain region 1 h after injection, suggesting fast crossing of the BBB. Reconstructions of confocal images showed that MCs were localized deep within the basal lamina, in nests of glial processes. Furthermore, electron microscopic analysis showed that MCs indeed migrate into the CNS (Silverman et al, 1994).
Although fully differentiated MCs and their precursors have been considered to be able to divide (Dvorak et al, 1976), no evidence of MC division was found through 5-bromo-2-deoxyuridine labeling (Silverman et al, 2002), supporting the hypothesis that an increase in end-organ MC population is apparently due to migration from the periphery or entry of new precursors from the circulation. As mature cells MCs do not circulate in the blood, it is suggested that the source of the augmented population is likely to be through the pial sheath of the thalamic blood vessels (Lambracht-Hall et al, 1990; Manning et al, 1994).
Mast Cell Activation in Cerebral Ischemia
In the brain, MCs are typically perivascular (Figures 1 and 2), have substantial granule content, and are frequently observed siding cortical penetrating arterioles (Figure 2). After ischemia, MCs are found more abundantly in the cerebral tissue, where they release their granules, thus leading to permeability change and perivascular edema, as documented in our laboratory in the MCAo model in the Wistar rat (Figure 3A) (Karjalainen-Lindsberg et al, 2001; Strbian et al, 2006). These findings have been confirmed by other laboratories (Hu et al, 2004; Jin et al, 2007). More recently, the rapid recruitment and activation of TNF-α-positive MCs within 1 h after generalized hypoxic–ischemic (HI) challenge has been shown in the neonate rat brain (Jin et al, 2009), although such a rapid MC accumulation has not been documented in other ischemia models. Mast cells recruited into the brain tissue are observed along elongating blood vessels, some of which express the GLUT1 isoform of the glucose transporter protein, indicative of vessels within the BBB (Jin et al, 2007). The potential regulatory role of MCs in vascular permeability is not a novel concept, as this property has been known for several decades (Valent, 2000) and was also shown in the vessels of the CNS by previous studies (Abbott, 2000; Esposito et al, 2001; Yong et al, 1994; Zhuang et al, 1996). However, this literature has focused more on inflammatory, endocrinological, and stress-related disorders in quite distinct animal models.
The Effect of Mast Cells in Focal Ischemic and Hemorrhagic Brain Damage
The finding of the increased presence of MCs and their rapid degranulation in association with morphologic changes of ischemic, edematous brain injury (Figure 3) led us to interventive, ‘proof-of-concept’ studies on the role of MCs in the deleterious sequelae of ischemic and hemorrhagic stroke. In the MCAo model, pharmacological MC stabilization with sodium cromoglycate yielded significantly reduced ischemic brain swelling by 40% and BBB leakage by 50%, whereas abrupt MC degranulation induced with a secretagogue compound 48/80 increased these rates by 90 and 50%, respectively (Strbian et al, 2006). These effects probably share pathophysiological mechanisms with traumatic brain injury and its MC-dependent edematous sequelae, which have also been described previously (Lozada et al, 2005; Stokely and Orr, 2008).
The specificity of the above-described effects was supported by investigations using rats born with no MCs, which is a trait induced by a mutation of the
These results provide compelling evidence of the involvement of MCs in augmenting postischemic BBB leakage and vasogenic edema formation, and also in the regulation of the latter after intracerebral hemorrhage. In the past, the pathophysiology of these phenomena was extensively studied by many research groups in an effort to identify the underlying molecular mechanisms. However, the reviewed data suggested that MCs represent a cell type that might set forth a multitude of downstream pathways. Thus, there are several pathobiologic rationales that would explain a critical role for MCs in these cascades.
Ischemic blood–brain barrier opening—magic bullets or shotgun fire?
Although a prevailing strategy in research devoted to cerebral ischemia has been focused on showing pivotal roles for particular single molecules or receptors in producing direct or delayed vasculopathic brain injury, identification of the cellular sources of molecules such as MMPs and substances that cleave pro-MMPs needs careful clarification. Clearly, astrocytes, macrophages, and pericytes have been identified as dominant sources or activators of several members of the MMP family (Cunningham et al, 2005; Rosenberg et al, 1998). However, instead of dissecting isolated activities of specific molecular mediators in a ‘seamless web of intricately interwoven relationships’ (Ratnoff, 1969) produced in ischemic brain injury, a compound effect mounted by an immune cell capable of releasing a multitude of deleterious, vasoactive, and proteolytic mediators might lead us to develop more fruitful therapeutic concepts. In other words, rather than considering brain edema and secondary hemorrhages to be a result of excessive production of single proteins (such as MMP-2 and MMP-9), we believe that a composite effect of biphasic MC activation could regulate a multitude of those vasoactive molecular responses that have been viewed to be central in ischemia–reperfusion injury. To this end, in the following paragraphs, we review data that place MCs within the context of well-known vasculopathic consequences of acute cerebral ischemia.
Mast Cell Proteases and Integrity of the Basal Lamina
Several investigations have linked inflammation to the production of MMPs and other proteases capable of degrading major proteins of the basal lamina (laminin, collagen type IV, and fibronectin), which loses its integrity rapidly after the onset of ischemia (Hamann et al, 1995; Wagner et al, 1997). At present, other investigators have already provided us with ample evidence showing that MMPs (especially MMP-9 and MMP-2) have a pivotal role in BBB damage and acute dissolution of the basal lamina (Asahi et al, 2001; Hamann et al, 1995, 1996; Heo et al, 1999; Lo et al, 2002; Rosenberg et al, 1998; Rosenberg, 2002; Sumii and Lo, 2002). Later research by other investigators has implicated MMP-9 as a biomarker of ischemic brain injury that leads to BBB failure and hemorrhagic transformation (Montaner et al, 2003; Rosell et al, 2006; Tejima et al, 2007). In addition to MMPs, other types of proteases, e.g., cathepsins (Fukuda et al, 2004), were suggested to have a role.
How can MCs disrupt the BBB and degrade components of the basal lamina? Several possible mechanisms are involved, in which the vasoactive and matrix-degrading components of MCs might act in concert. These proposed mechanisms are outlined in Figure 4. Mast cell activation produces a potent vasodilatory mediator, histamine, and contributes up to 90% of its content in the thalamus and up to 50% in the whole brain (Goldschmidt et al, 1985). Hence, of the resident cells in the brain and meninges, only MCs acutely release massive amounts of preformed vasodilatory mediators, such as histamine. Mast cell-borne heparin markedly accelerates the generation of another potent vasoactive substance, bradykinin (Reddigari et al, 1995). Under experimental conditions
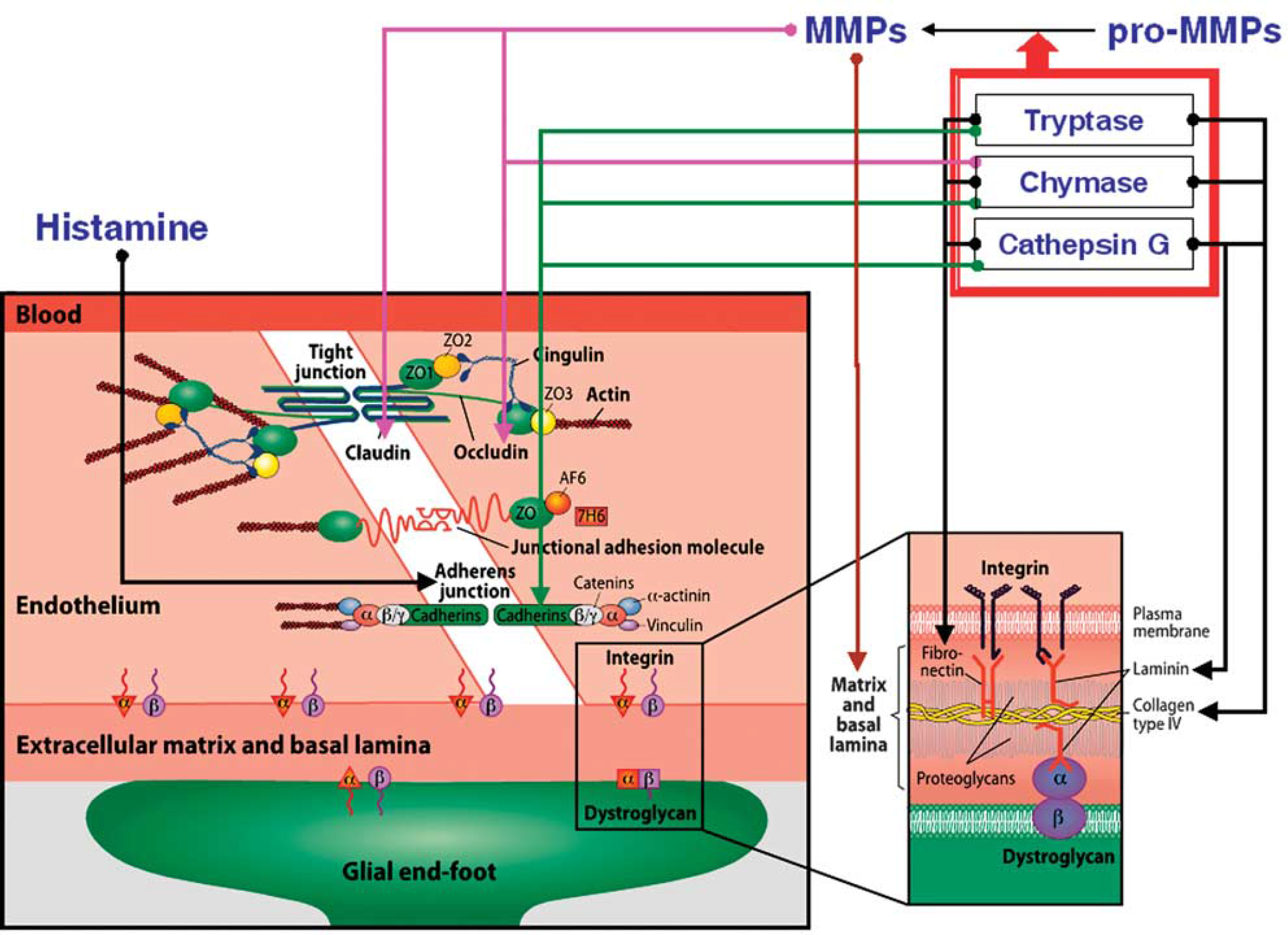
Illustration of the components of the blood–brain barrier (left) and the basal lamina (right). Arrows represent the possible involvement of MC-derived mediators in blood–brain barrier and basal lamina damage. ECM, extracellular matrix; ZO, zona occludens; AF6, afadin; 7H6, tight junction-associated phosphoprotein; MMP, matrix metalloproteinase.
Importantly, MCs themselves can release gelatinase A (MMP-2) and gelatinase B (MMP-9) (Fang et al, 1999). Cathepsin G, a neutral protease present in some MCs, cleaves many components of the extracellular matrix and pericellular matrix, including fibronectin and vitronectin (Helske et al, 2006; Mäyränpää et al, 2006). The role of cathepsins in microvascular matrix degradation after focal cerebral ischemia has been shown (Fukuda et al, 2004). Finally, apart from proteases, MCs can release TNF-α instantly from its preformed granules and interleukin (IL)-1; both of these cytokines are involved in BBB failure and in ischemic brain edema formation (Kim et al, 1992; Yamasaki et al, 1992). Indeed, MCs do possess a potent armamentarium for targeting the key constituents of the BBB and basal lamina (Figure 4) shortly after their activation, whereas
Mast Cells and Fibrinolysis
Hemorrhage formation, either spontaneous or iatrogenic (in association with thrombolytic therapy with tPA), can devastate the outcome after successful vessel recanalization. Investigators searching for improved control of unwanted fibrinolysis during blood-clotlysing therapy may have to take a closer look at MCs, which have been considered essentially as a fibrinolytic cell type (Valent, 2000). To this end,
The mechanism of postthrombolytic hemorrhage formation still awaits full understanding. It is interesting to note that it occurs with any of the fibrinolytic substances used clinically, namely tPA, streptokinase, prourokinase, and reteplase (Furlan et al, 1999; Qureshi et al, 2001). They all are potent serine proteases, which, in addition to their local plasminogen-activating effect to dissolve fibrin locally, also induce systemic plasminemia (Sobel, 1994). Plasmin, again, has an independent proinflammatory activity that causes direct brain tissue damage (Asahi et al, 2000; Xue and Del Bigio, 2001) and, in addition, by activating MMPs it degrades a range of extracellular matrix proteins (Castellino, 1998). Interestingly, inhibition of MMPs reduced tPA-mediated mortality in experimental ischemia–reperfusion (Pfefferkorn and Rosenberg, 2003).
Perhaps even more important in the setting of thrombolytic therapy of ischemic stroke is the observation that the development of a hemorrhage may be further augmented and prolonged by heparin. This mediator is acutely released by activated MCs, which are the only source of this strong anticoagulant (Lane and Björk, 1992; Rosenberg and Bauer, 1994). Heparin release from perivascularly positioned MCs might impair hemostasis and prevent plugging of BBB breaches. In theory, endogenously released heparin may accelerate erythrocyte extravasation and contribute to the formation of frank hemorrhages. In dogs bearing MC tumors, intense activation of the fibrinolytic system has been observed after injection of minute amounts of the MC-degranulating agent 48/80 (Ende and Auditore, 1964). In addition to the fact that besides heparin, tPA also belongs to the palette of MC-derived anticoagulant mediators (Valent, 2000), these data confirm the antithrombotic and fibrinolytic potential of MCs. We believe that the reviewed data support a hypothesis that MC activation could participate in the formation of vicious, space-occupying intracerebral hematomas (spontaneous or tPA-mediated), as well as less dangerous hemorrhagic transformations.
Mast cells and brain protection
Effects in the Immature Brain
In the rodent brain with excitotoxic lesions mimicking those described in human HI brain injury (cerebral palsy), IL-9 pretreatment exacerbated brain damage produced by intracerebral injections of the glutamatergic analog ibotenate (Dommergues et al, 2000). Among its different cell targets, the Th2 cytokine IL-9 is an MC growth and differentiation factor that can cause MC degranulation. Interleukin-9 pretreatment had no significant effect on ibotenate-induced excitotoxic brain lesions in genotypically MC-deficient mice, whereas IL-9 exacerbated these lesions in wild-type littermates. Cromoglycate or antihistamine drugs significantly reduced ibotenate-induced brain lesions in IL-9-treated mice (Patkai et al, 2001). In a retrospective study of children with cerebral palsy, higher levels of several perinatal circulating cytokines, including IL-9, were detected (Nelson et al, 1998).
Hypoxic–ischemic brain damage in immature rats led to a rapid increase in the cerebral population of MCs along with their activation (Jin et al, 2007). In that study, activated MCs were shown in the pia, brain parenchyma, and, importantly, in regions showing neuronal loss. Mast cell stabilization with cromoglycate reduced the MC count in the latter region and limited brain damage by more than 50% compared with controls. In the same study, it was noted that only HI—but not hypoxia alone—led to brain damage, suggesting that a tissue-level metabolic or inflammatory challenge is required. Another study (Biran et al, 2008) showed the contribution of MC-derived histamine to ischemia-induced neuronal death in P7 newborn rats. Another important finding was that MC-associated genes were upregulated after HI brain damage (Hedtjarn et al, 2004). Most recently, cerebral MC counts and their activation were shown to be elevated immediately after HI before detection of cleaved caspase-3 in neurons (NeuN +; 2 h after HI), astroglial activation (GFAP + with swollen cell body, 4 h after HI), or microglial activation (OX42 +, 4 h after HI) (Jin et al, 2009). Tumor necrosis factor-α-positive MCs were present in a subpopulation of MCs in control animals, and the fraction of TNF-α-positive MCs increased dramatically ipsilaterally immediately after HI. Microglial TNF-α was evident at 4 h, but endothelial cells had no detectable TNF-α until 48 h after HI. Tumor necrosis factor-α was implicated in the generation of early inflammatory and neurotoxic effects. Cromoglycate prevented MC migration, reduced brain damage/neuronal loss, glial activation, and brain atrophy through 4 weeks of recovery (Jin et al, 2009). In summary, these data support the observation that MC activation may have a significant detrimental effect during neonatal HI brain damage. In these models, MCs seem to be early responders with their preformed TNF-α pools, and MC stabilizing treatments seem a promising novel neuroprotective avenue to prevent neonatal brain injuries.
Effects in the Adult Brain
Although the features of cerebral palsy share the morphologic characteristics of ischemic and HI cortical damage (Gressens et al, 1997; Marret et al, 1995), similar evidence of neuroprotection by MC stabilization in ischemic brain damage is scarce. Sodium cromoglycate probably does not possess any intrinsic neuroprotective activity. Any neuroprotective net effect would probably be mediated by prevention of the release of neurotoxic mediators, such as TNF-α, and reduced inflammation and vasculopathic effects, such as BBB damage and edema. Importantly, inhibition of tPA-mediated hemorrhage formation with this drug led to better neurologic outcomes and reduced mortality up to 24 h after MCAo (Strbian et al, 2006 2007
To reconcile the above evidence, we believe that there are differences between immature and adult rodent brain in vulnerability to ischemic or HI damage, and in their responsiveness to MC stabilization. Furthermore, intraperitoneal cromoglycate administration was shown to be neuroprotective in the immature mice brain (Jin et al, 2007, 2009), whereas in adult rats, the drug had to be administered intraventricularly to be efficacious. An exception was the model of intracerebral hemorrhage, in which also the intravenous route of administration was effective in reducing edema, mortality, and neurologic recovery (Strbian et al, 2007b). Obviously, immature BBB and permeability to circulating macromolecules in the immature brain can account for this difference. Future studies should be directed to the therapeutic potential of MC stabilization, regardless of whether it is dominantly dependent on direct neuroprotection or secondary brain protection through reduction of microcirculatory failure and expansive edema.
Mast cells in cerebral micromilieu
Interplay and Crosstalk With Endothelial and Perivascular Cells
A conceptual framework, ‘the neurovascular unit’ (Figure 5), has been proposed, which consists of vascular and perivascular structures and cells; microvessels (endothelial cells—-basal lamina matrix—-astrocyte end-feet and pericytes), astrocytes, neurons and their axons, in addition to other supporting cells (e.g., microglia and oligodendroglia) (del Zoppo, 2009) (Figure 5). The endothelial cell is viewed central to the NVU, but its function is regulated by crosstalk between neighboring astrocyte end-feet, neurons, and pericytes (del Zoppo, 2009; Simard et al, 2003). The extracellular matrix and the matrix-degrading enzymes and their inhibitors have a key role at the basal lamina and the cell surface in the regulation of cell signaling (Cunningham et al, 2005).
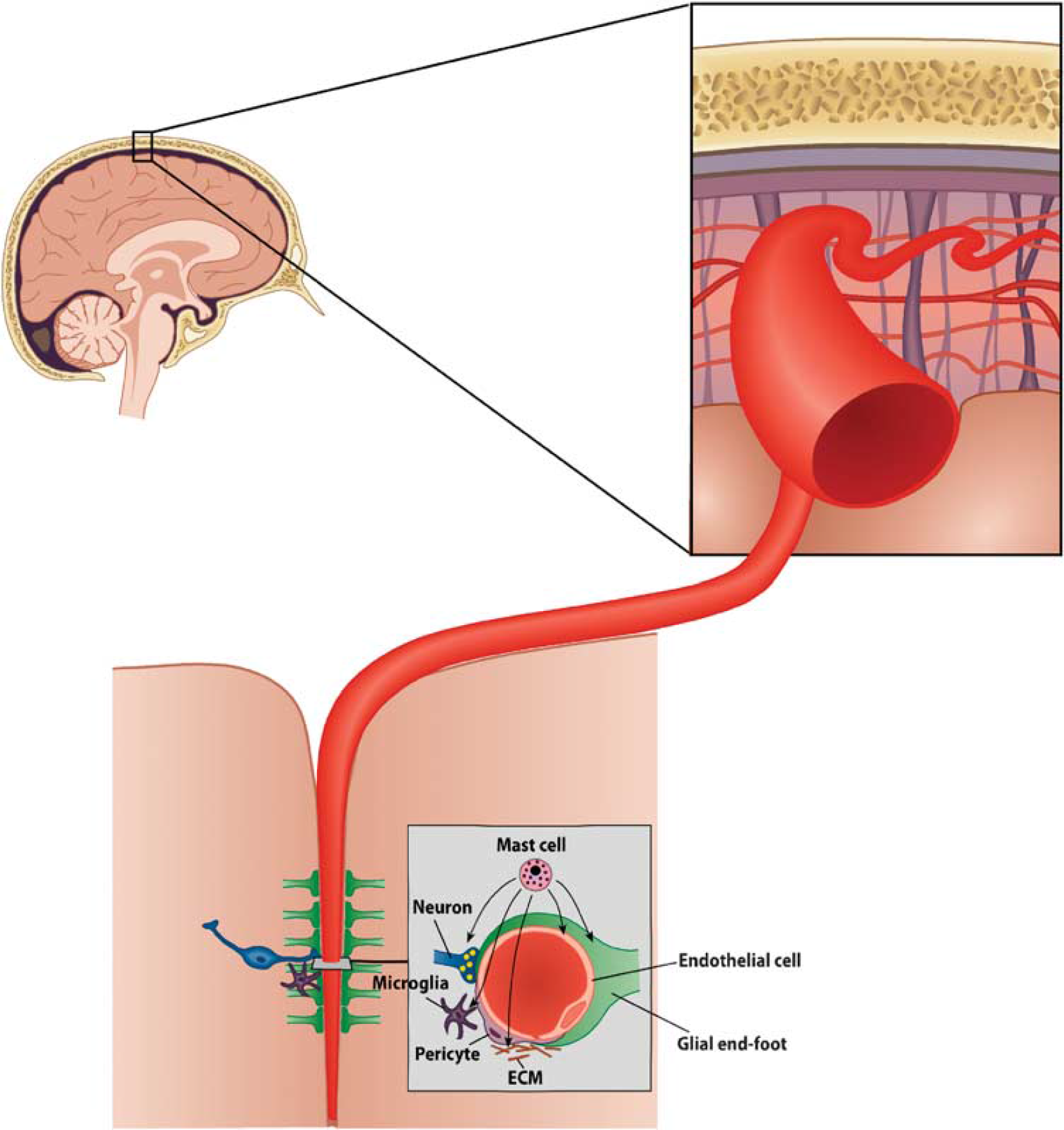
Schematic illustration of the cellular elements of the neurovascular unit.
In addition, MCs have recently been viewed to be involved in several types of interactions within the NVU (del Zoppo, 2009), the various components of which together with their complex crosstalk secure the integrity and homeostasis of the microvasculature. Mast cells possess a palette of mediators that could participate in the fine-tuning of the microcirculatory and metabolic milieu nurtured within an NVU—histamine in regulating the degree of vasodilation and bidirectional permeability to circulating or extracellular substances—heparin and tPA in regulating the balance between hemostasis and fibrinolysis—and TNF-α in regulating inflammatory changes, such as expression of adhesion molecules and chemotactic signaling. Early release of chemotactic signals and facilitation of BBB permeation could pave the way for circulating phagocytic cells necessary for clearance of noxious substances and cell debris. Theories such as these need to be addressed in studies yet to come, but in the following paragraphs, we review shortly what is presently known of MCs and their cellular interactions with dominant cells within the NVU (Figure 5).
Astrocytes
During development, association of yet undifferentiated MCs with the vascular bed (preferentially at branching points) is dependent on the contact of the blood vessel wall with astroglial processes, which involves α4-integrins expressed by MCs (Khalil et al, 2007). Adjacent astrocytes can influence the differentiation, the eventual tissue-specific phenotype, and the migration of MCs (Dimitriadou et al, 1996), presumably by synthesizing MC growth factors such as IL-3 (Farrar et al, 1989; Frei et al, 1985) and nerve growth factor (Carman-Krzan et al, 1991). Furthermore, mature MCs can be grown on astrocytes (Johnson and Krenger, 1992; Shalit et al, 1993), and astroglial processes elongate in close proximity to MCs (Shanas et al, 1998).
Neurons
Mast cell products enter neurons in at least three ways (transgranulation) in the dove brain: (Wilhelm et al, 2005) (1) direct fusion of the granule and plasma membranes of both MCs and neurons, (2) engulfment of MC processes containing granules or capture of released granule remnants, and (3) receptor-mediated endocytosis. Interestingly, the frequency of transgranulation events is related to the activity status of the MCs.
Endothelial Cells
Changes in endothelial cell–matrix interactions may be influenced by TNF-α and IL-1β (del Zoppo and Mabuchi, 2003), most likely by the downregulation of integrin receptors of the β1 subfamily (Defilippi et al, 1992). Accordingly, TNF-α reduces the integrin α1β1 expression of ECs, leading to decreased adhesion to laminin (Defilippi et al, 1992), whereas IL-1β contributes to early ischemic brain edema, presumably by altering β1 expression (Yamasaki et al, 1992). Both cytokines can be released by different cells, but are included among MC mediators as well.
Basal Lamina and Extracellular Matrix
Mast cells can attach to and migrate on laminin- and fibronectin-coated surfaces (Thompson et al, 1993). Furthermore, surface receptors of MCs (one of them for laminin) regulate MC trafficking and distribution by engaging extracellular matrix components, including the classical integrin receptors (Metcalfe, 1995).
These data suggest that biologically and pathobiologically meaningful crosstalk exists between MCs and the NVU. Finally, one should recognize the large pool of MCs resident within the meninges (largely pial in the developing CNS and dural in the adult). The effects of MCs within this compartment might be dominant in the early phase of catastrophic space-occupying or lacerating ischemic, hemorrhagic, and traumatic brain injuries.
What Triggers Mast Cell Responses in the Ischemic Brain?
Ischemic and hemorrhagic stroke are catastrophic situations to be managed merely locally by the NVU (Figure 5), which subserves its ‘neurosphere’ under physiologic conditions. We raise the possibility that the sudden cessation of blood circulation and rapid accumulation of waste products and reduction of pH probably trigger MCs to degranulate, and they act as a ‘fast response force’ to tackle the noxious, nonhomeostatic micromilieu within the NVU. Mast cells are well known to respond and degranulate rather stereotypically on a multitude of physico-chemical challenges, such as ionizing irradiation, trauma, toxic substances, allergens, complement activation, thermal challenges, hyperosmolality, and pH changes. Therefore, sudden dense ischemia, the release of, e.g., toxic-free hemoglobin or activated serum complement proteins, such as anaphylatoxins (C3a, C5a) into the abluminal interstitial space, could well represent one candidate for sufficient noxious stimuli to which MCs in the affected brain area respond by rapid degranulation. This is an interesting subject for further study.
Although brain tissues are known to possess MCs scattered within the cerebral cortex and basal ganglia, they are found much more numerously in the meningeal tissues (Figure 1C). This pool of MCs has been shown to lead to edematous cerebral changes in models of traumatic brain injury (Stokely and Orr, 2008). Less disrupting metabolic or irritating challenges such as stress and migraine-type models are associated with spontaneous MC degranulation within leptomeningeal or cerebral tissue (Esposito et al, 2001; Reuter et al, 2001). We find it plausible that, after a major stroke or hemorrhage, early activation and degranulation of dural and pial MCs ensue and participate in the process occurring within the intracranial vault, leading to aggravation of BBB damage and expansive brain edema. Collateral circulation from the leptomeninges could pass MC-derived mediators humorally into the brain tissue proper to exert the deleterious effects described above. However, at present, these suggestions remain exciting research areas for future investigations.
Conclusions
The main aim of this review is to shed light on the potentially significant, yet not sufficiently realized, role of MCs in mounting rapid host responses within different structures of the CNS, especially the cerebral cortex, basal ganglia, and the meninges. Through several decades, similar responses have been amply described in other organs, and we are aware of the potent, acute effects of MC degranulation and activation apart from the CNS. We have now reviewed a body of recent literature that provides rather compelling yet largely experimental evidence for hypothesizing a role for cerebral MCs in influencing BBB permeability, space-occupying brain edema, neuroprotection, and important outcome variables such as preservation of neurologic function and survival after HI, ischemic, and hemorrhagic brain damage. However, the bulk of the reviewed material remains at the level of ‘proof-of-concept’ study.
Future studies on this subject should include a more detailed dissection of the pathobiological mechanisms underlying MC-mediated BBB disruption, as well as more rigorous elucidation of potential beneficial effects of MC-stabilizing agents given in therapeutic, postinsult protocols. Given the promiscuity of MC-dependent responses in allergic, inflammatory, and additional hypersensitivity reactions throughout the body, vast literature has been accumulated to support the stabilization of MCs in helping various therapeutic regimens. Stabilization of MCs is obviously amenable also for novel purposes within the CNS using familiar drugs used for several decades, such as sodium cromoglycate. Perhaps the most immediate area of further work in stroke research would be the development of pharmacological compositions to be entered in clinical assessment of fibrinolytic therapies to minimize the risk of threatening hemorrhagic complications. In the field of basic research, a more thorough understanding of MC-related phenomena in the crosstalk with components of the NVU will probably reveal exciting novel perspectives on physiologic and pathobiologic regulation of the microvascular milieu in the CNS.
Footnotes
Acknowledgements
Financial support has been received from the Finnish Academy (PJL), Sigrid Juselius foundation (PJL) and the Helsinki University Hospital District (EVO) funds (PJL).
The authors declare no conflict of interest.