
Abstract
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) is a member of the tumor necrosis factor superfamily. TWEAK acts via binding to a cell surface receptor named Fn14. To study the role of this cytokine in the regulation of the permeability of the neurovascular unit (NVU) during cerebral ischemia, TWEAK activity was inhibited in wild-type mice with a soluble Fn14-Fc decoy receptor administered either immediately or 1 h after middle cerebral artery occlusion (MCAO). Administration of Fn14-Fc decoy resulted in faster recovery of motor function and a 66.4%±10% decrease in Evans blue dye extravasation when treatment was administered immediately after MCAO and a 46.1%±13.1% decrease when animals were treated 1 h later (n=4, P<0.05). Genetic deficiency of Fn14 resulted in a 60%±12.8% decrease in the volume of the ischemic lesion (n=6, P<0.05), and a 87%±22% inhibition in Evans blue dye extravasation 48 h after the onset of the ischemic insult (n=6, P<0.005). Compared with control animals, treatment with Fn14-Fc decoy or genetic deficiency of Fn14 also resulted in a significant inhibition of nuclear factor-κB pathway activation, matrix metalloproteinase-9 activation and basement membrane laminin degradation after MCAO. These findings show that the cytokine TWEAK plays a role in the disruption of the structure of the NVU during cerebral ischemia and that TWEAK antagonism is a potential therapeutic strategy for acute cerebral ischemia.
Introduction
Stroke causes 5.5 million deaths in the world every year. In the United States, stroke is the third leading cause of mortality after cancer and coronary artery disease (The World Health Organization, 1999). During cerebral ischemia, disruption of the architecture of the neurovascular unit (NVU) results in increase in the permeability of the blood—brain barrier (BBB) with the development of cerebral edema (Garcia et al, 1978), which is a major cause of mortality among patients with acute stroke. The NVU is a dynamic structure consisting of endothelial cells, the basal lamina, astrocytic end-feet processes, pericytes, and neurons (del Zoppo and Mabuchi, 2003). The permeability of the NVU is determined not only by the integrity of the interendothelial tight junctions but also by the composition of the basal lamina (del Zoppo and Mabuchi, 2003) and the interaction between astrocytes, endothelial cells, and the extracellular matrix (Rieckmann and Engelhardt, 2003). It has been reported that proinflammatory cytokines released in response to the ischemic signal act directly on elements of the NVU with resultant increases in BBB permeability. Also, the nuclear factor-κB (NF-κB) family of transcription factors and the secreted protease matrix metalloproteinase-9 (MMP-9) have been implicated in disruption of the NVU and increases in BBB permeability in response to ischemic injury (Rosenberg et al, 1996; Asahi et al, 2000, 2001).
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK), a member of the tumor necrosis factor superfamily of cytokines (Chicheportiche et al, 1997), binds to a cell surface receptor named Fn14 (Wiley et al, 2001) and promotes various biologic responses, depending on the cell type analyzed (Wiley and Winkles, 2003). Furthermore, TWEAK is an angiogenic factor (Lynch et al, 1999; Ho et al, 2004) and can induce the expression of various gene products commonly associated with the inflammatory response (Campbell et al, 2004). TWEAK binding to Fn14 has been shown to activate the NF-κB signal transduction pathway in multiple cell types (Brown et al, 2003; Donohue et al, 2003; Potrovita et al, 2004; Polavarapu et al, 2005; Campbell et al, 2006). We have recently reported that under nonischemic conditions, the intracerebral injection of TWEAK results in NF-κB and MMP-9 activation, disruption of the structure of the NVU, and increase in the permeability of the BBB (Polavarapu et al, 2005).
Three recent studies have indicated that TWEAK may be involved in the pathophysiologic process leading to cell death and edema during cerebral ischemia (Potrovita et al, 2004; Yepes et al, 2005; Polavarapu et al, 2005). These studies showed that TWEAK and Fn14 expression increases in the area of ischemic penumbra after middle cerebral artery occlusion (MCAO) in the mouse (Potrovita et al, 2004; Yepes et al, 2005) and that inhibition of TWEAK activity either with a soluble Fn14-Fc decoy receptor (Polavarapu et al, 2005) or with neutralizing anti-TWEAK antibodies (Potrovita et al, 2004) is neuroprotective.
Here, we show that during cerebral ischemia TWEAK—Fn14 interactions play a role in NF-κB pathway activation, MMP-9 activation, laminin degradation, and increased permeability of the NVU. We also show that inhibition of endogenous TWEAK activity either with a soluble Fn14-Fc decoy receptor or by genetic deficiency of Fn14 after MCAO results in preservation of the integrity of the NVU with attenuation of ischemia-induced cerebral edema.
Materials and methods
Animal Model
Male C57BL/6J wild-type mice or Fn14−/− mice (Jakubowski et al, 2005) backcrossed at least 10 generations on a C57BL/6J background were used. All procedures were approved by the Emory University Institutional Animal Care and Use Committee. Mice were anesthetized with 4% chloral hydrate (400 mg/kg, intraperitoneally). The rectal and masseter muscle temperatures were controlled at 37°C with a homeothermic blanket. The middle cerebral artery was exposed and occluded with a 10 to 0 suture as described elsewhere (Nagai et al, 1999). Cerebral perfusion in the distribution of the middle cerebral artery (laser Doppler; Perimed Inc., North Royalton, OH, USA), and changes in pH, PO2, and PCO2 were monitored 10 mins before, throughout the surgical procedure and 10 mins after MCAO (Table 1). After MCAO, wild-type animals were placed on a stereotactic frame and 2 μL of either Fn14-Fc decoy (1 μg/μL) or Fc protein (1 μg/μL) purified as described previously (Donohue et al, 2003; Yepes et al, 2005) were injected into the third ventricle at bregma: −2; mediolateral: 0; dorsoventral: 2. Forty-eight hours later, 1 mm brain sections were stained with 2,3,5 triphenyltetrazolium chloride and the volume of the ischemic lesion was measured as described (Nagai et al, 1999). Statistical analysis was performed with the Student's t-test.
Physiologic variables in wild-type and Fn14−/− mice
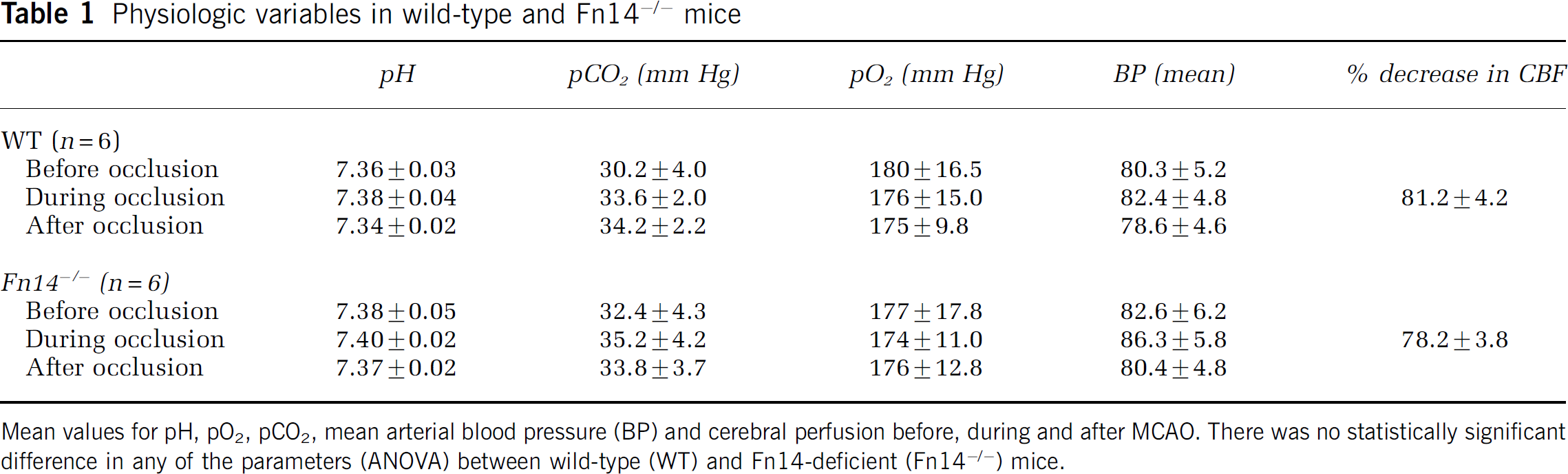
Mean values for pH, pO2, pCO2, mean arterial blood pressure (BP) and cerebral perfusion before, during and after MCAO. There was no statistically significant difference in any of the parameters (ANOVA) between wild-type (WT) and Fn14-deficient (Fn14−/−) mice.
Studies of Motor Activity and Evaluation of the Permeability of the Neurovascular Unit
After MCAO and the intraventricular injection of Fn14-Fc decoy or Fc, a subgroup of wild-type and Fn14−/− animals was intravenously injected with 2 μl of 2% Evans blue dye (Sigma-Aldrich, St Louis, MO, USA). Six hours before MCAO and 6, 24, and 48 h thereafter, animals were placed in individual acrylic chambers for 2 h and horizontal and vertical motor activity were recorded with a Computerized Activity Monitor (Omnitech Instruments, Inc., Columbus, OH, USA). The number and extent of horizontal and vertical movements was determined by breaks in photo beams and converted into locomotor activity counts. Results for each time point were compared with the baseline evaluation obtained 6 h before MCAO. After the last evaluation, Evans blue dye extravasation was quantified as described elsewhere (Yepes et al, 2003). Statistical analysis was performed with the Student's t-test.
Immunogold Electron Microscopy
Fn14−/− mice and wild-type littermate controls (n=4) were deeply anesthetized 6 h after MCAO. Brains were fixed by transcardial perfusion and processed as described elsewhere (Polavarapu et al, 2005), followed by dissection of the areas of interests (fronto-temporal, temporo-parietal, and subcortical surrounding the necrotic core) from flat embedded vibrating microtome sections. Cuts were then re-embedded and 60 nm cuts were stained and examined with a Hitachi H-7500 transmission electron microscope.
Western Blot Analysis
Polyclonal antibodies to phospho-p65, total p65, phospho-IKKβ, and total IKKβ were purchased from Cell Signaling Technology (Beverly, MA, USA). Polyclonal antibodies to pan-laminin and β-actin were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Wild-type and Fn14−/− mice underwent MCAO. Wild-type animals were injected with either Fc protein or Fn14-Fc decoy into the third ventricle immediately after MCAO as described above. Brains were extracted at 1, 3, or 6 h after MCAO for analysis of NF-κB activation, and 24 h after MCAO for studies of laminin degradation. Tissue was processed and gels were loaded as described (Polavarapu et al, 2005). A total of four observations were made for each time point. The density of the bands was analyzed using the NIH Image Analyzer System as described elsewhere (Polavarapu et al, 2005). Statistical analysis was performed with the Student's t-test.
Immunofluorescence
Wild-type and Fn14−/− mice underwent MCAO followed by intraventricular treatment with either Fc protein or Fn14-Fc decoy in wild-type animals as described above. Twenty-four hours after MCAO, 10 μm frozen sections were processed as described (Yepes et al, 2005) and incubated with an anti-pan-laminin antibody, 1:1,000 dilution (Sigma-Aldrich). After three washes with phosphate-buffered saline a 1:500 dilution of goat anti-rabbit secondary antibody conjugated to Alexa 488 (Molecular Probes, Eugene, OR, USA) was applied for 1 h at room temperature. The number of vascular structures with intact perivascular laminin staining was counted in the above-described areas of interest in the ipsilateral (ischemic) and contralateral (non ischemic) hemispheres in cuts obtained in each brain at the level of the putamen (+1 mm bregma), third ventricle (0.00 mm bregma), and hippocampus (−2.0 mm bregma). In each case, results were expressed as a percentage of the number of vascular structures per mm2 of brain with intact laminin staining in the contralateral, nonischemic hemisphere. Statistical analysis was performed with the Student's t-test.
Matrix Metalloproteinase Activity Analysis
For analysis of MMP-9 activity, brains of wild-type animals treated either with Fn14-Fc or Fc (control), and Fn14−/− mice, were removed, divided into ipsilateral and contralateral hemispheres, and homogenized in a 2:1 volume-to-weight ratio of lysis buffer containing 0.5 mol/L NaCl, 1% Triton X-100 (Sigma-Aldrich), 0.05% Tween 20 (Sigma-Aldrich), and 50 mmol/L sodium acetate. The samples were centrifuged at 10,000g for 10 min and the supernatant was added to Gelatin Sepharose 4B beads (Amersham Biosciences, Piscataway, NJ, USA). After 3 h incubation at room temperature, the beads were washed three times in phosphate-buffered saline, incubated in sample buffer for 30 min and centrifuged. The supernatant was then loaded in a gelatin zymography gel (Invitrogen, Carlsbad, CA, USA). After electrophoresis, the gel was washed twice during 45 min with 2.5% Triton X-100 (Sigma-Aldrich), followed by a 30 min wash with Tris-buffered saline containing 5 mmol/L CaCl2, then incubated overnight at 37°C in the same buffer after which the gel was stained with 0.5% Coomassie Brilliant Blue R-250 (Amersham Biosciences) for 30 min and then destained. Purified murine MMP-9 from Chemicon (Temecula, CA, USA) was used as a marker for MMP-9. In a different subset of experiments, wild-type mice underwent MCAO followed by treatment with increasing concentrations of Fn14-Fc decoy (0 to 2 μg). Matrix metalloproteinase-9 activity was analyzed 6 h after MCAO and the volume of the ischemic lesion was measured at 48 h as described above. Each experiment was performed four times and the density of the band in each observation was analyzed using the NIH Image Analyzer System. Statistical analysis was performed with Student's t-test.
Electrophoretic Mobility Shift Assay
Wild-type and Fn14−/− animals underwent MCAO followed by treatment of wild-type animals with either Fn14-Fc decoy (2 μg) or vehicle control. Brain tissue was suspended in buffer A at 10 mL/g (10 mmol/L N-2-hydroxyl piperazine-N′-2-ehane sulfonic acid, pH 7.9, 25 mmol/L KCl, 5 mmol/L MgCl2, 250 mmol/L sucrose, 0.6% NP-40, and protease inhibitors), homogenized for 25 to 30 sec, and centrifuged at 2,000 r.p.m. for 5 min. The pellet was resuspended in 1 mL buffer B (10 mmol/L N-2-hydroxyl piperazine-N′-2-ehane sulfonic acid, pH 7.9, 10 mmol/L KCl, 10 mmol/L MgCl2, 250 mmol/L sucrose, and protease inhibitors), centrifuged at 2,000 r.p.m. for 5 min and then washed and resuspended in 150 μL electrophoretic mobility shift assay buffer (20 mmol/L N-2-hydroxyl piperazine-N′-2-ehane sulfonic acid, pH 7.9, 80 mmol/L KCl, 5 mmol/L MgCl2, 0.2 mmol/L dithiothreitol, 0.1 mmol/L ethylene diaminetetraacetic acid, 5% glycerol, 0.1% NP-40, 0.04 μg of polydeoxyinosinic-deoxycytidylic acid). Nuclear extracts were recovered after centrifugation for 10 min at maximum speed. Protein concentrations were determined with Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). The NF-κB consensus sense (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and antisense (5′-GCCTGGGAAAGTCCCCTCAACT-3′) oligonucleotides were obtained from Operon (Huntsville, AL, USA). They were labeled with [32P]-γATP using T4 polynucleotide kinase (Invitrogen) to produce double-stranded DNA probes. For binding reactions, 8 μg of nuclear extract was incubated with poly (dI-dC) and the 32P-labeled double-strand oligonucleotide (1 ng, 1 × 105 cpm). The reaction products were fractioned on a nondenaturing 6% polyacrylamide gel, which was then dried and subjected to autoradiography. As control, binding reactions were also conducted in the presence of an excess concentration of cold double-stranded probe.
Results
Effect of Inhibition of TWEAK Activity on Motor Recovery and Development of Cerebral Edema After Middle Cerebral Artery Occlusion
To study the effect of endogenous TWEAK on the permeability of the BBB and the recovery in motor activity after cerebral ischemia, wild-type mice underwent MCAO followed by the intravenous administration of Evans blue dye and the intraventricular administration of either the Fn14-Fc or Fc protein either immediately after the onset of the ischemic insult or 1 h later. In previous studies, we found that treatment with either Fc protein or phosphate-buffered saline has no effect on the volume of the ischemic lesion or in the permeability of the BBB after MCAO (Yepes et al, 2005; Polavarapu et al, 2005); therefore, in the present experiments we used only one of these negative controls (Fc injection). The mean locomotor activity and Evans blue dye extravasation were evaluated as described in Materials and methods. We found that inhibition of TWEAK activity with Fn14-Fc decoy results in an accelerated recovery in motor function and in a significant decrease in the magnitude of cerebral edema 48 h after MCAO (Figure 1A). The effect of Fn14-Fc decoy on motor activity was not observed 7 days after MCAO; however, at that time we did detect a significant difference in Evans blue dye extravasation between Fn14-Fc-treated mice and controls (data not shown). Additionally, the protective effect of TWEAK inhibition was also observed when mice were treated with Fn14-Fc decoy 1 h after MCAO (Figure 1B). In contrast, we did not find any difference in motor activity or Evans blue dye extravasation when Fn14-Fc decoy was administered 3 h after MCAO (data not shown). These results indicate that endogenously expressed TWEAK has a direct effect on the permeability of the BBB after MCAO.
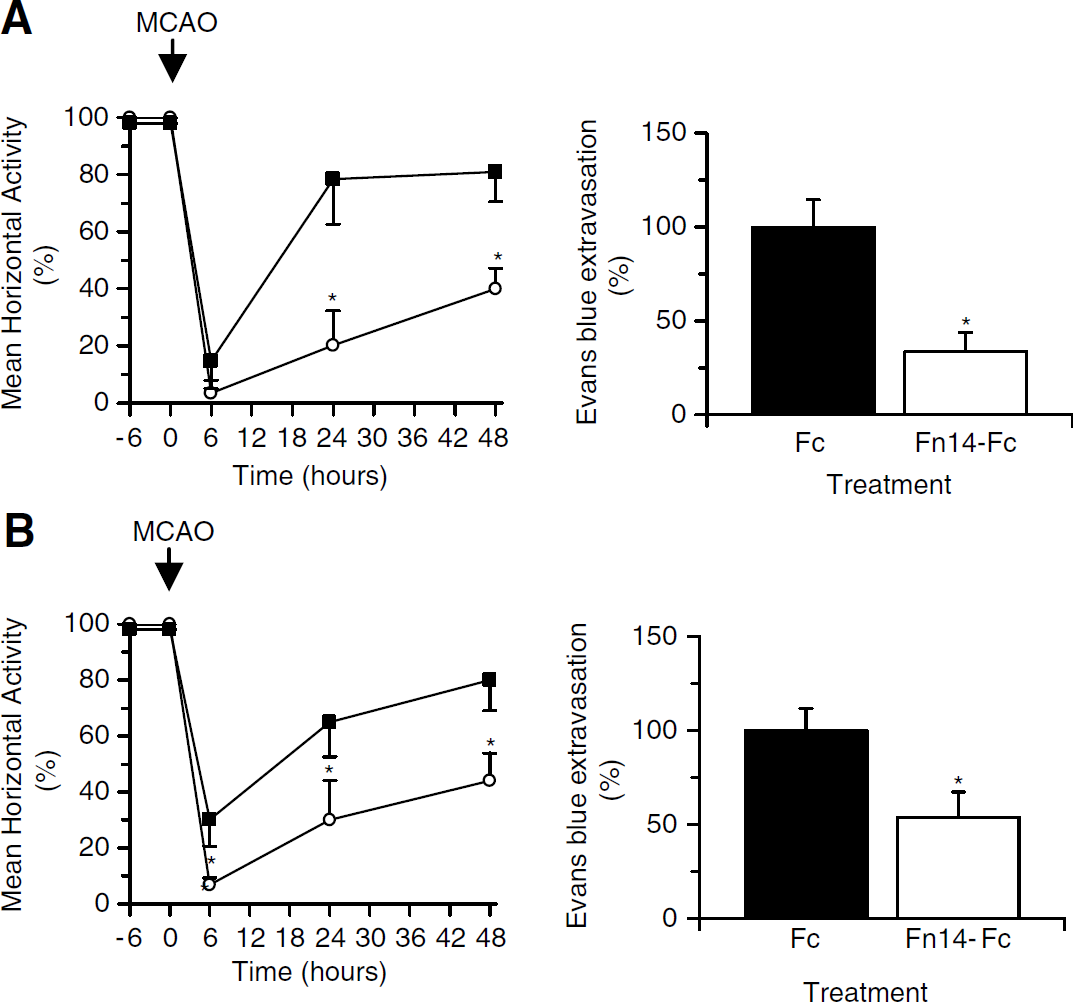
Inhibition of TWEAK function improves motor activity and decreases cerebral edema after MCAO. Mean locomotor activity at 6, 24, and 48 h and Evans blue dye extravasation at 48 h after MCAO after treatment with either Fc protein (white circles) or Fn14-Fc decoy (black squares), either immediately after (
Effect of Genetic Deficiency of Fn14 on the Volume of the Ischemic Lesion After Middle Cerebral Artery Occlusion
Fn14 gene-deficient mice (Fn14−/− mice) have been characterized to some extent (Jakubowski et al, 2005) but their response to cerebral ischemic injury has not been reported. Therefore, we measured the volume of the ischemic lesion in Fn14−/− mice and their wild-type littermate controls 48 h after MCAO. A summary of the physiologic parameters before, during, and after the surgical procedure is included in Table 1. We found that the volume of the ischemic lesion decreased from 15.6±1.08 mm3 in wild-type littermate controls to 6.25±1.05 mm3 in Fn14−/− mice (60% reduction; P<0.05) (Figure 2). This observation shows that genetic deficiency of Fn14 has a protective effect during cerebral ischemia.
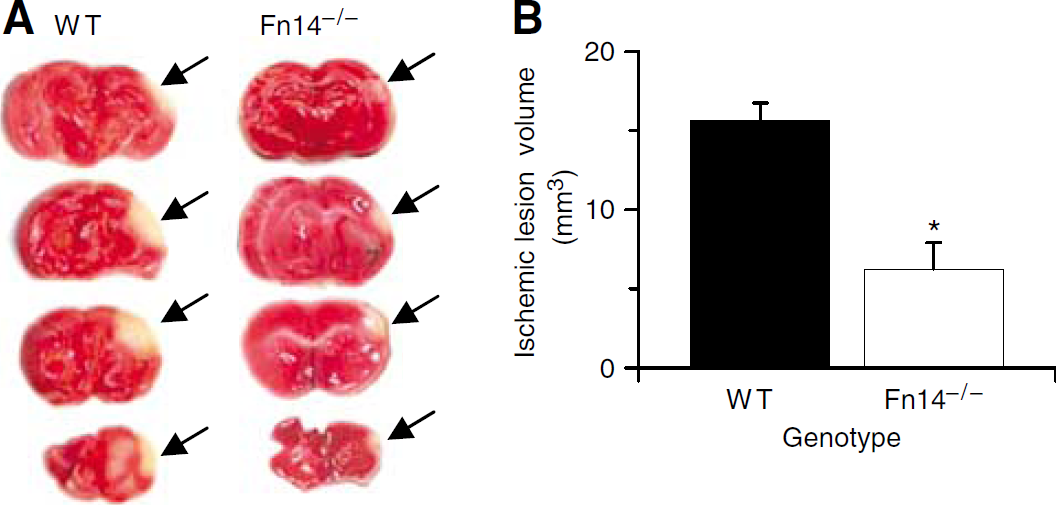
Genetic deficiency of Fn14 decreases the volume of the ischemic lesion after MCAO. (
Effect of Genetic Deficiency of Fn14 on Motor Recovery, Development of Cerebral Edema, and Disruption of the Architecture of the Neurovascular Unit After Middle Cerebral Artery Occlusion
To better characterize the protective effect of Fn14 deficiency during cerebral ischemia, locomotor activity and Evans blue dye extravasation were examined in Fn14−/− mice and their wild-type littermate controls after MCAO. A subset of mice was euthanized 6 h after MCAO and brains were processed for electron microscopic analysis of NVU structure. We found that compared with wild-type mice, Fn14−/− mice exhibit a faster recovery in motor activity (Figure 3A) and a significant decrease in the extent of cerebral edema (Figure 3B) after MCAO. Evans blue dye extravasation was 0.003±0.012 units (measured as optical density units at 620 nm/g of brain tissue) in sham-operated animals, 2.192±0.283 units in wild-type mice, and 0.304±0.271 units in Fn14−/− mice after MCAO. This represents a 87% decrease in Evans blue dye extravasation in Fn14−/− mice (Figure 3B). Electron microscopy analysis showed preservation of astrocytic end-feet processes-basement membrane contacts, and significant decrease in the development of perivascular edema 6 h after MCAO in Fn14−/− mice (Figure 3C). Together our results show that genetic deficiency of Fn14 results in protection of the integrity of the NVU and reduced permeability of the BBB during cerebral ischemia.
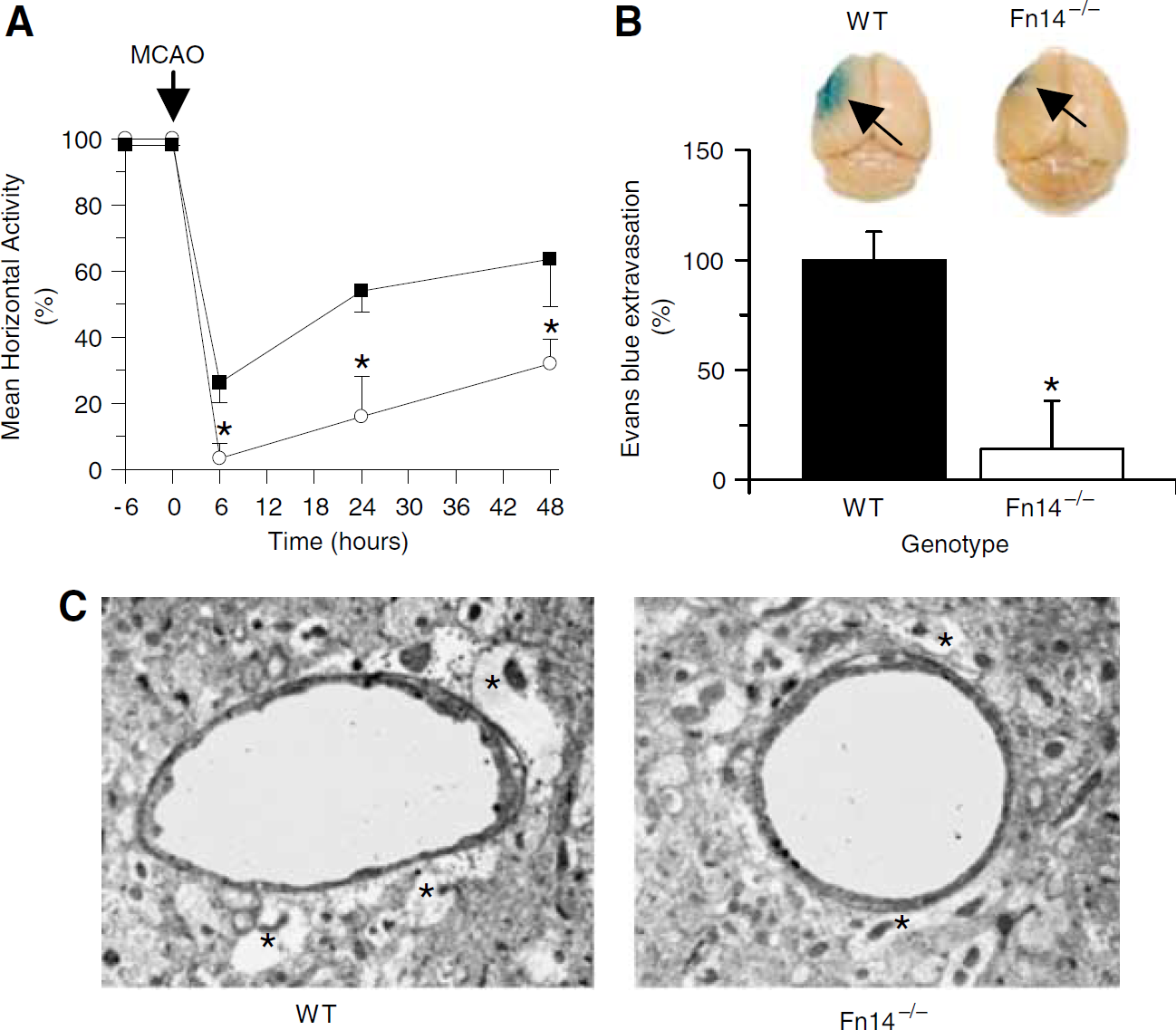
Genetic deficiency of Fn14 improves motor activity, decreases cerebral edema, and protects the integrity of the neurovascular unit after MCAO. (
TWEAK Contributes to Cerebral Ischemia-Induced Nuclear Factor-κB Pathway Activation
Because TWEAK induces activation of the NF-κB pathway when added to various cell types (Brown et al, 2003; Donohue et al, 2003; Kim et al, 2004), and when injected directly into the brain (Polavarapu et al, 2005), we investigated the effect of endogenous TWEAK on cerebral ischemia-induced NF-κB pathway activation. The IκB kinase (IKK) complex is composed of two catalytic subunits (α and β) and one regulatory subunit (γ). Several kinases can phosphorylate IKKβ and stimulate its activity. Phosphorylated IKKβ, in turn, induces IκBα phosphorylation and resultant NF-κB activation. Serine phosphorylation of the NF-κB p65 subunit also occurs during NF-κB pathway activation. Therefore, in our experiments, we first monitored NF-κB pathway activation by assaying for IKKβ and p65 phosphorylation.
Wild-type and Fn14−/− mice underwent MCAO followed by the intraventricular administration of either Fn14-Fc decoy or Fc protein in wild-type animals. Brains were harvested at 0, 1, 3, or 6 h later and Western blot analysis for phospho-p65, total p65, phospho-IKKβ, and total IKKβ was performed. We observed a rapid phosphorylation of the IKKβ protein (Figure 4A) and the p65 protein (Figure 4B) 1 h after MCAO in wild-type mice treated with Fc protein. In contrast, inhibition of TWEAK activity by Fn14-Fc decoy or genetic deficiency of Fn14 resulted in a significant inhibition of cerebral ischemia-induced IKKβ and p65 phosphorylation (Figures 4A and 4B).
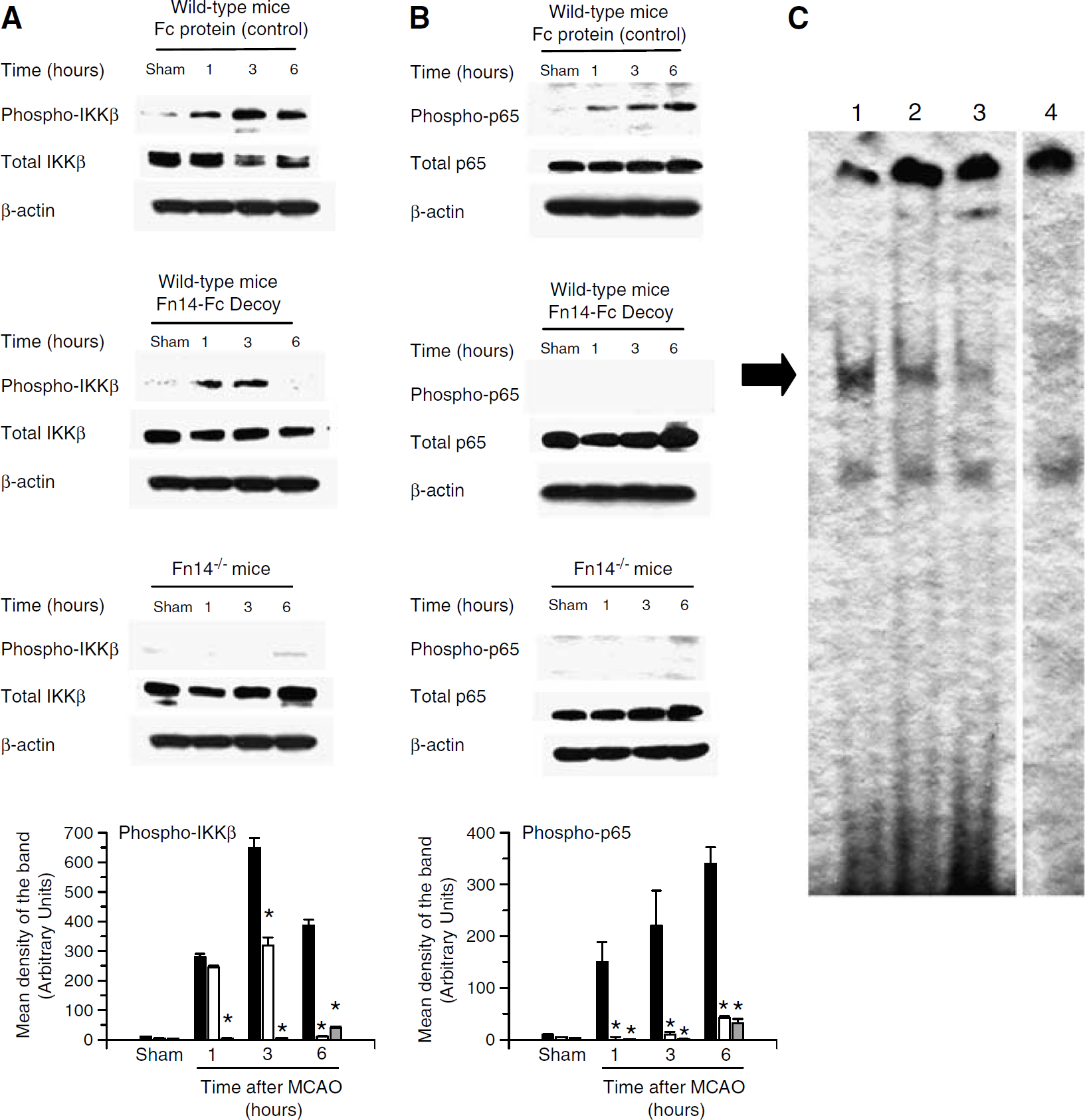
TWEAK contributes to NF-κB pathway activation after MCAO. Western blot analysis of phospho-IKKβ and total IKKβ (
To further confirm these results, we also performed an electrophoretic mobility shift assay using brain extracts from wild-type mice, wild-type mice treated with Fn14-Fc decoy, and Fn14−/− mice 6 h after MCAO. We found that MCAO induces a significant change in NF-κB activation 6 h after the onset of the ischemic insult (black arrow, Figure 4C, lane 1). In contrast, treatment with Fn14-Fc decoy (lane 2) or genetic deficiency of Fn14 (Fn14−/− mice, lane 3) resulted in a significant decrease in the NF-κB DNA-binding activity after MCAO. Together, our data show that inhibition of TWEAK activity either with Fn14-Fc decoy or by genetic deficiency of Fn14 results in a significant blockade of MCAO-induced NF-κB pathway activation.
TWEAK Contributes to Cerebral Ischemia-Induced Matrix Metalloproteinase-9 Activation
TWEAK can induce MMP-9 activation when added to macrophages (Kim et al, 2004), and astrocytes cultured in vitro, and when injected directly into the mouse brain (Polavarapu et al, 2005). Because it has been proposed that MMP-9 plays an important role in the proteolytic degradation of the NVU during cerebral ischemia (Asahi et al, 2000, 2001), we studied the effect of endogenous TWEAK on MMP-9 activity after MCAO. Wild-type and Fn14−/− mice underwent MCAO followed by the intraventricular administration of either Fn14-Fc decoy or Fc protein in wild-type animals. Brains were harvested 6 h later and gelatin zymography was performed to detect MMP-9 activity. We observed a significant increase in MMP-9 activity in wild-type mice treated with Fc protein 6 h after MCAO (Figure 5A, lane 3). In contrast, the increase in MMP-9 activity in either wild-type mice treated with Fn14-Fc decoy or in Fn14−/− mice was minimal (Figure 5A, lanes 4 and 5). Additional experiments using various Fn14-Fc decoy concentrations showed that the effect on MMP-9 activation was dose-dependent and correlated with the final volume of the ischemic lesion (Figure 6). These results show that endogenous TWEAK stimulates MMP-9 activity during cerebral ischemia.
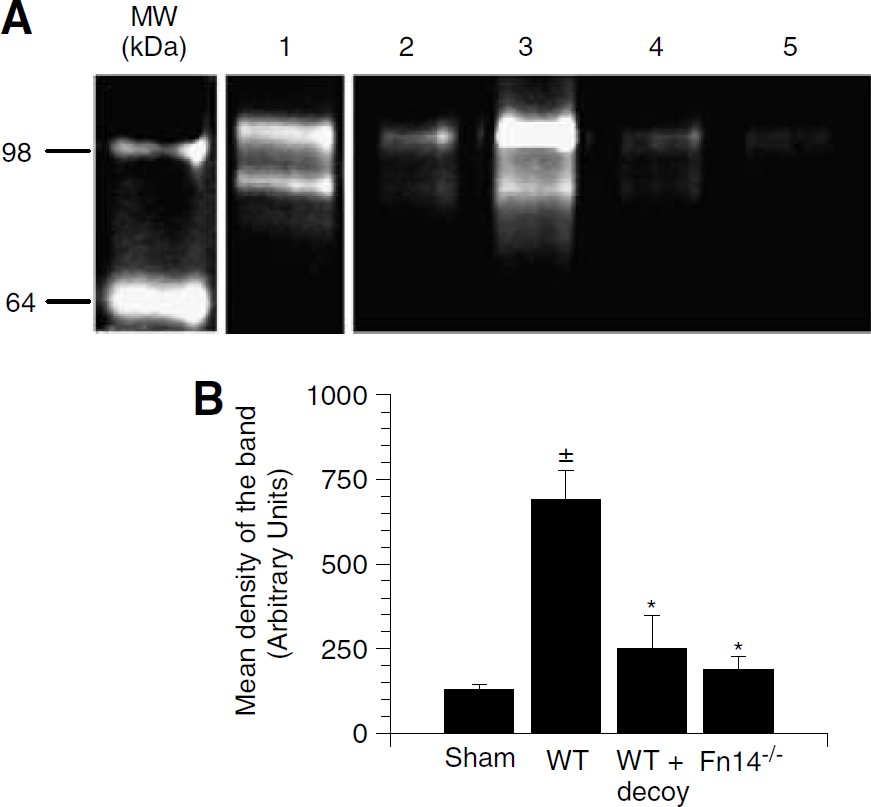
TWEAK contributes to MMP-9 activation after MCAO. (
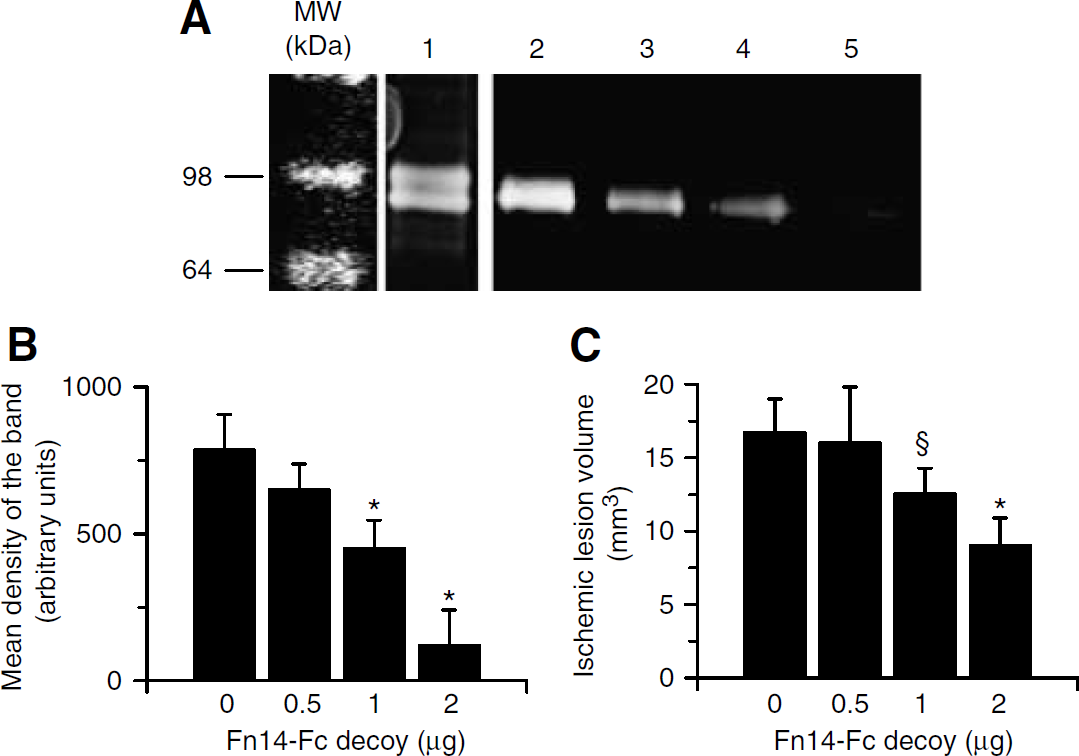
Fn14-Fc decoy induces a dose-dependent decrease in MMP-9 activation and ischemic lesion volume after MCAO. (
Tweak Contributes to Cerebral Ischemia-Induced Degradation of the Basement Membrane
To examine whether TWEAK could be involved in cerebral ischemia-induced laminin degradation, Fn14−/− mice and wild-type littermate controls underwent MCAO followed by intraventricular administration in wild-type mice of either Fc protein or Fn14-Fc decoy. Brains were harvested 24 h later and Western blot analysis for laminin was performed using an anti-pan-laminin antibody. We observed laminin proteolysis, especially degradation of the 360 and 170 kDa subunits to 51 and 39 kDa fragments, in wild-type animals treated with Fc protein (Figure 7A, lane 5). In contrast, either treatment with Fn14-Fc decoy or genetic deficiency of Fn14 resulted in significant inhibition of laminin degradation (Figure 7A, lanes 3 and 4). To further confirm these observations, we quantified the number of vascular structures with intact laminin per mm2 in the areas of interest of the ischemic and nonischemic hemispheres of the brains of wild-type animals treated with either Fc protein or Fn14-Fc decoy, and in Fn14−/− mice. We observed a significant decrease in the percentage of vascular structures/mm2 with intact laminin from 56.2%±11.8% in wild-type mice treated with Fc protein, to 22.39%±7.6% in wild-type animals treated with Fn14-Fc decoy, to 10.5%±7.2% in Fn14−/− mice (P<0.05; Figures 7B and 7C). Together, our results show that endogenous TWEAK is involved in cerebral ischemia-induced degradation of laminin, and this may be one mechanism responsible for the effect of TWEAK on the integrity of the NVU and the permeability of the BBB after the onset of the ischemic insult.
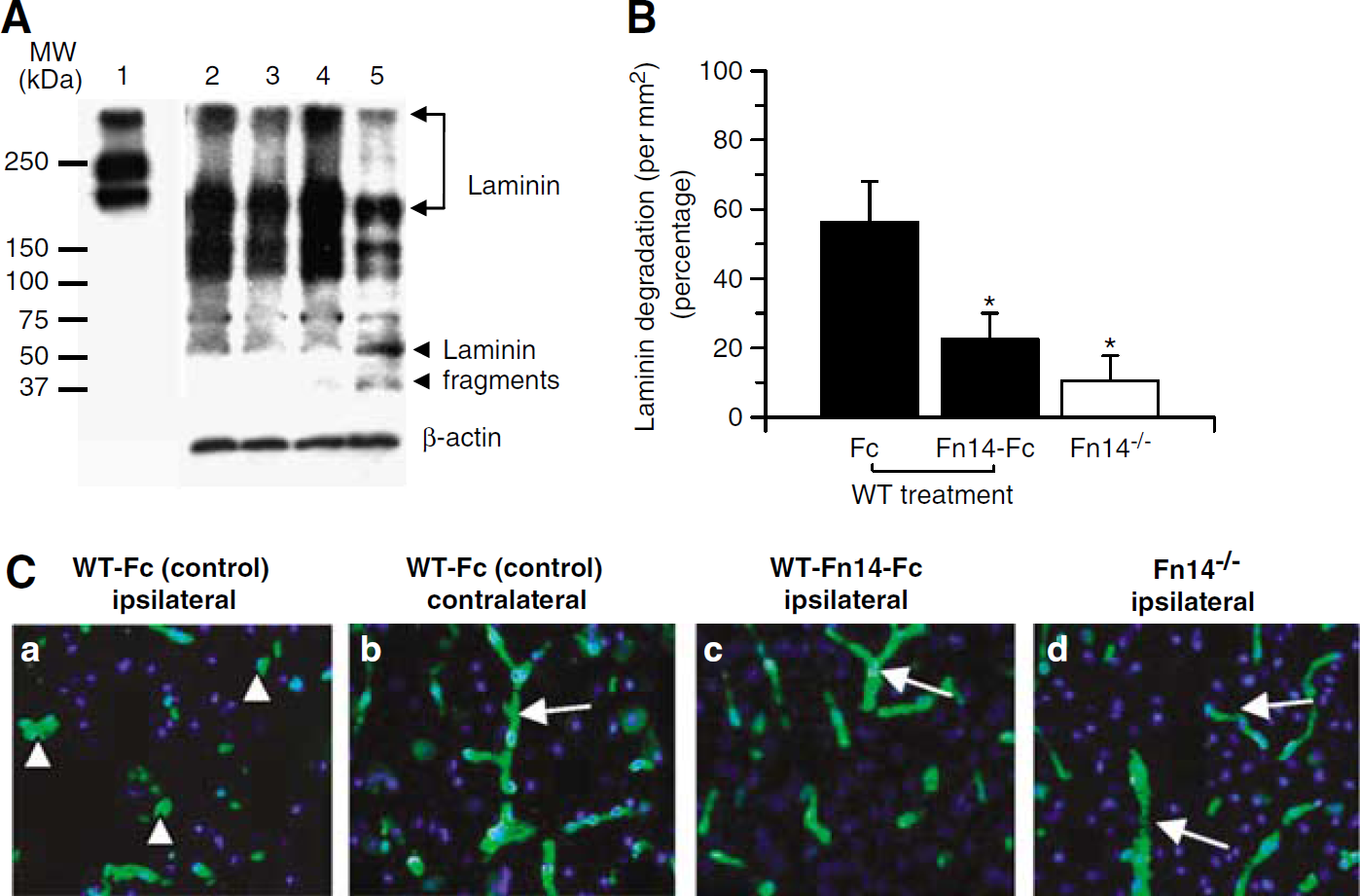
TWEAK contributes to laminin degradation after MCAO. (
Discussion
The NVU is assembled from endothelial cells, the extracellular matrix of the basal lamina, the astrocytic end-feet processes that surround the microvessels, and adjacent neurons (del Zoppo and Mabuchi, 2003). One of the main functions of the NVU is the regulation of the supply of nutrients to the brain and prevention of the passage of harmful substances from the intravascular space into the central nervous system. The onset of cerebral ischemia modifies the association between the cellular and noncellular components of the NVU. This results in disruption of the architecture of this structure with increase in the permeability of the BBB and development of cerebral edema (Garcia et al, 1978; Yepes et al, 2003), which is a major cause of mortality in acute stroke patients. In our previous report, we showed that treatment of nonischemic brains with recombinant TWEAK results in disruption of the astrocytic end-feet processes—basement membrane contacts with a dose-dependent increase in the permeability of the BBB by a tumor necrosis factor-α-independent mechanism (Polavarapu et al, 2005). The results presented here show that inhibition of endogenous TWEAK activity by treatment with Fn14-Fc decoy or genetic deficiency of Fn14 result in a significant decrease in the extent of cerebral edema after MCAO, and in protection of the structure of the NVU in the early phases of the ischemic insult. The lack of efficacy on motor activity and BBB permeability when the decoy was administered 3 h after the onset of the ischemic insult may reflect that the dose of the decoy is still too low, or that most of the deleterious effect of TWEAK occurs relatively soon after the onset of the ischemic insult.
The precise mechanism responsible for TWEAK-mediated increase in BBB permeability after MCAO is not known. Astrocytes induce many of the properties of the NVU including the low permeability of endothelial tight junctions (Wolburg et al, 1994). In previous studies, we showed that TWEAK and Fn14 were expressed in astrocytes (Yepes et al, 2005). Also, it has been shown that TWEAK treatment of astrocytes cultured in vitro results in the release of proinflammatory cytokines and metalloproteinases (Saas et al, 2000; Polavarapu et al, 2005). Therefore, TWEAK may bind the Fn14 receptors on astrocytes within the NVU and activate the NF-κB pathway. Cytokine and MMP production may then promote increased BBB permeability and cell death. This possibility is supported by evidence demonstrating that loss of the interaction between the cellular components of the NVU and the extracellular matrix results in cell death. Alternatively, the interaction of TWEAK with neuronal and glial cell Fn14 may directly lead to cell death. Indeed, it has been shown that exposure to recombinant TWEAK induces apoptotic cell death in primary neuronal cultures under oxygen-glucose deprivation conditions (Potrovita et al, 2004) and that inhibition of TWEAK activity after MCAO results in a decrease in the number of cells with apoptotic features in the area of ischemic penumbra (Yepes et al, 2005).
Cerebral ischemia induces a rapid activation of the NF-κB pathway in the brain (Schneider et al, 1999; Song et al, 2005). In vitro studies have showed that activation of NF-κB in glial cells induces cell death whereas activation of this pathway in neurons increases their survival after exposure to hypoxic conditions (Yu et al, 1999). In contrast, studies in experimental animal models of focal cerebral ischemia have showed that NF-κB enhances ischemic cell death (Schneider et al, 1999), and that this effect decreases when neuronal but not astrocytic NF-κB is inhibited (Zhang et al, 2005). It has been shown that TWEAK binding to its receptor Fn14 results in activation of the NF-κB pathway in vitro (Han et al, 2003; Saitoh et al, 2003; Donohue et al, 2003; Xu et al, 2004; Kim et al, 2004; Jin et al, 2004; Tran et al, 2005; Campbell et al, 2006) and in vivo (Polavarapu et al, 2005). To study whether TWEAK plays a role in activation of the NF-κB pathway during cerebral ischemia we evaluated the effect of either inhibition of TWEAK activity or genetic deficiency of Fn14 on cerebral ischemia-induced NF-κB activation. Our results show a rapid activation of the NF-κB pathway in the ischemic brain of control animals that is ameliorated by either Fn14-Fc decoy injection or by genetic deficiency of Fn14.
It is known that MMP-9 is an NF-κB-regulated gene (Bond et al, 1998). This metalloproteinase is upregulated after the onset of the ischemic insult (Rosenberg et al, 1996) and its activation has been associated with degradation of components of the NVU (Asahi et al, 2001; Gu et al, 2005) and increased BBB permeability (Heo et al, 1999). The cause of increased MMP-9 activity during cerebral ischemia is unknown. However, elevated cytokines may significantly upregulate matrix metalloproteinases (Rosenberg et al, 1995). It has been shown that TWEAK can induce MMP-9 activation when added to astrocytes in culture and when injected directly into the nonischemic brain (Polavarapu et al, 2005). To study the relationship between endogenous TWEAK activity and MMP-9 function during cerebral ischemia, we performed gelatin zymography assays using brain extracts from wild-type and Fn14−/− mice subjected to MCAO. We found that 6 h after the onset of the ischemic insult there was an increase in MMP-9 activity that is inhibited by either treatment with Fn14-Fc decoy or by genetic deficiency of Fn14.
The basal lamina is a specialized part of the extracellular matrix that is formed by type IV collagen, laminin, fibronectin, entactin, thrombospondin, proteoglycans, and heparan sulfates (del Zoppo and Mabuchi, 2003). After the onset of cerebral ischemia there is a progressive loss of the astrocytic end-feet processes—basement membrane contacts (Wagner et al, 1997) that is associated with loss of laminin in the extracellular matrix and increase in BBB permeability (Hamann et al, 1996). It has been proposed that, in the ischemic brain, laminin is a substrate for MMP-9 (Gu et al, 2005). Thus, we examined whether endogenous TWEAK played a role in cerebral ischemia-induced degradation of laminin in the ischemic brain. We found that either inhibition of TWEAK activity or deficiency of Fn14 results in a significant decrease in cerebral ischemia-induced laminin degradation.
In summary, our results show that TWEAK has a detrimental effect on the structure of the NVU and the permeability of the BBB in the early stages of cerebral ischemia. We postulate a model in which in response to the ischemic signal, there is release of TWEAK from perivascular astrocytes. This TWEAK interacts with Fn14 on the astrocyte cell surface, with activation of the NF-κB pathway and induction of MMP-9 expression. TWEAK-induced increase in MMP-9 activity results in degradation of laminin in the extracellular matrix with detachment of astrocytic end-feet processes from the basement membrane and increase in the permeability of the NVU. Alternatively, the interaction between TWEAK and Fn14 on pial vessels could also result in increase in cerebrovascular permeability. These findings indicate that inhibition of TWEAK activity with Fn14-Fc decoy is a potential therapeutic strategy aimed at preserving the integrity of the NVU during cerebral ischemia.
Footnotes
Acknowledgements
We thank Ms Hong Yi from the Emory University School of Medicine Electron Microscopy Core Facility for her assistance with the electron microscopy studies.