
Abstract
Preconditioning describes the ischemic stimulus that triggers an endogenous, neuroprotective response that protects the brain during a subsequent severe ischemic injury, a phenomenon known as ‘tolerance’. Ischemic tolerance requires new protein synthesis, leads to genomic reprogramming of the brain's response to subsequent ischemia, and is transient. MicroRNAs (miRNAs) regulate posttranscriptional gene expression by exerting direct effects on messenger RNA (mRNA) translation. We examined miRNA expression in mouse cortex in response to preconditioning, ischemic injury, and tolerance. The results of our microarray analysis revealed that miRNA expression is consistently altered within each group, but that preconditioning was the foremost regulator of miRNAs. Our bioinformatic analysis results predicted that preconditioning-regulated miRNAs most prominently target mRNAs that encode transcriptional regulators; methyl-CpG binding protein 2 (MeCP2) was the most prominent target. No studies have linked MeCP2 to preconditioning or tolerance, yet miR-132, which regulates MeCP2 expression, is decreased in preconditioned cortex. Downregulation of miR-132 is consistent with our finding that preconditioning ischemia induces a rapid increase in MeCP2 protein, but not mRNA, in mouse cortex. These studies reveal that ischemic preconditioning regulates expression of miRNAs and their predicted targets in mouse brain cortex, and further suggest that miRNAs and MeCP2 could serve as effectors of ischemic preconditioning-induced tolerance.
Introduction
Ischemic brain injuries are among the most common and important causes of disability and death worldwide. However, a sublethal duration of ischemia, ischemic preconditioning, triggers endogenous responses that protect the brain against a subsequent severe ischemic insult, a phenomenon known as ‘tolerance’ (Dirnagl et al, 2009). The mechanisms of preconditioning-induced tolerance are not well known, but are characterized by three key features. Ischemic tolerance requires
MicroRNAs (miRNAs) regulate posttranscriptional gene expression in plants, animals, and viruses (Ambros, 2004; Bartel, 2004; Chen and Meister, 2005) and are integral components of RNA-induced silencing complexes, which repress translation by directly interacting with messenger RNAs (mRNAs). In animals, miRNAs regulate mRNA translation through an imperfect pairing with nucleotide sequences within the 3′ untranslated region (3′ UTR) of targets, and repressed translation is enhanced for those mRNAs targeted by multiple miRNAs (Doench and Sharp, 2004). MiRNAs repressing translation are sequestered and localized in processing bodies (Pillai et al, 2005; Sheth and Parker, 2003); release of miRNA-targeted mRNAs sequestered in processing bodies can occur without affecting mRNA stability (Liu et al, 2005; Pillai et al, 2005; Sen and Blau, 2005). Cellular stress can cause the release of mRNA from processing bodies, leading to recruitment of translating ribosomes and protein synthesis (Bhattacharyya et al, 2006). MiRNAs expressed within dendrites regulate translation of proteins mediating dendritic growth (Schratt et al, 2006). MiRNAs that regulate synaptic plasticity can target, and be targeted by, plasticity mediators such as cAMP response element binding protein (CREB), fragile X mental retardation protein, and MeCP2 (methyl-CpG binding protein 2) (Smalheiser and Lugli, 2009).
We propose that ischemic preconditioning could regulate miRNA expression and thus serve as novel effectors of altered protein expression that leads to ischemic tolerance. Accordingly, we show that ischemia does result in significant changes in miRNA expression in preconditioned, ischemic, and tolerant cortices, relative to sham. Target prediction analysis revealed MeCP2 as the most prominent target of miRNAs regulated in preconditioned cortex; thus, we further propose that the preconditioning-regulated miRNAs serve, at least in part, to regulate protein expression of transcriptional regulators required for ischemic tolerance.
MeCP2 has been considered as a global transcriptional repressor because of its methyl-DNA binding and transcription repression domains. For example, in neurons, MeCP2 bound to the brain-derived neurotrophic factor promoter is released on membrane depolarization, resulting in transcription of brain-derived neurotrophic factor mRNA (Chen et al, 2003). However, results of new studies indicate that MeCP2 is a global regulator of transcription; MeCP2 can repress transcription when complexed with histone deacetylase, or activate transcription when complexed with CREB1 (Chahrour et al, 2008). Further studies have established MeCP2 as a multifunctional nuclear protein with roles in chromatin architecture, regulation of RNA splicing, and transcriptional activation (Hite et al, 2009). Together, these studies show a complex role for MeCP2 in brain function and synaptic plasticity; as such, MeCP2 is associated with epigenetic regulation of the nervous system (MacDonald and Roskams, 2009).
The results of our studies show that preconditioning regulates miRNA expression, and target prediction of the preconditioning-regulated miRNAs identified novel proteins that could serve as effectors governing the epigenetic changes that mediate preconditioning-induced tolerance. Among these novel proteins, expression of MeCP2 protein increases in preconditioned cortex, with no corresponding change in MeCP2 mRNA expression, consistent with speculation that MeCP2 is posttranscriptionally regulated (Shahbazian et al, 2002). Accordingly, MeCP2 protein is directly regulated by miR-132 in neurons: decreased miR-132 increases MeCP2 expression (Klein et al, 2007). Although neither miRNAs nor MeCP2 have previously been linked to ischemic preconditioning, these studies support the concept that both could serve as novel effectors of the molecular mechanisms underlying ischemic preconditioning-induced tolerance.
Materials and methods
Transient Focal Ischemia
Adult male C57BL/6J mice (25 to 30 g; Charles River Laboratories, Wilmington, MA, USA) were maintained under diurnal conditions (12 h light/dark cycle) on an
MicroRNA Microarray
RNA was isolated from each cortex using the
MicroRNA Microarray Data Analysis
The miRNA microarray data were analyzed using a Web-based miRNA microarray analysis program created by Rob Lusardi (Slowdog Software, Portland, OR, USA). The ratio of the median intensities for each signal was calculated: 1 indicated equal quantities of target miRNA in the ipsilateral and contralateral cortices, < 1 reduced quantities of target miRNA in the ipsilateral cortex, and > 1 increased quantities of target miRNA in the ipsilateral cortex, relative to contralateral cortex. Each ratio was log2-transformed to produce a normally distributed data set amenable to standard statistical analysis: average log ratio (ALR) of miRNA expression = log2(Cy5/Cy3), where Cy5 and Cy3 were the probe intensities of a single miRNA in the ipsilateral and contralateral cortices, respectively, from the same mouse. A log ratio of 0 indicated no change between ipsilateral and contralateral cortices, positive values indicated increased miRNA expression in ipsilateral cortex, and negative values decreased expression in ipsilateral cortex, relative to contralateral cortex. The ALR of miRNA expression in preconditioned, ischemic, or tolerant cortex was compared with that of miRNAs in sham cortex, and Student's
Target prediction was limited to high-confidence miRNAs, defined as those present in all experimental replicates. Analysis was performed using miRanda (version 2005; www.microrna.org) (John et al, 2004) that allows queries of several miRNAs and reports the total number of gene targets (ENSG), mRNA transcripts (ENST), and the number of ‘hits’ that are predicted sites for miRNA binding based on mRNA complementarity (miRNA/ENST pair). The mRNA binding sites are located in the 3′ UTR, numbered 5′ to 3′ from nucleotide 1 beginning just after the stop codon.
Quantitative Real-Time Polymerase Chain Reaction
We analyzed miRNA expression in mouse cortex by qRT-PCR using the miRCURY RNA miRNA PCR System with miR-132 and control U6 primer sets labeled with SYBR Green (Exiqon Inc., Woburn, MA, USA). The mRNA qRT-PCRs were performed using primer sets for MeCP2, SLC2A3, and 18S RNA with
MeCP2 Knockout Mice
MeCP2 knockout (KO) mice (strain B6.129P2(C)-
Immunoblots
Nuclear fractionation of mouse brain cortices was performed using the CelLytic NuCLEAR Extraction Kit (Sigma, St Louis, MO, USA). Proteins separated by one-dimensional gel electrophoresis were transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). Protein blots were incubated in 1:1,000 dilution of mouse antihuman MeCP2 antibody (ab50005; Abcam, Cambridge, MA, USA) then incubated in 1:10,000 dilution of goat antimouse horseradish-peroxidase-conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA, USA). Protein bands were detected using enhanced chemilumines-cence (Amersham Biosciences) and Kodak BioMax film (Eastman Kodak Co., Rochester, NY, USA). Blots were scanned and quantified using the Kodak Image Station 2000rt and Kodak 1D version 3.6 software. Protein bands were background-subtracted, normalized to α-tubulin III (46 kDa; Sigma T8660), and the normalized intensity was expressed as relative to the average control tissue. Statistical analysis was performed by analysis of variance (ANOVA), with a
Immunohistochemistry
Mouse brains were flash frozen and sectioned on a Cryostat CM3050S (Leica Microsystems Inc., Bannockburn, IL, USA) into 12-μm-thick sections. Whole-brain mounts were fixed, permeabilized, blocked, and incubated with rabbit antimouse MeCP2 antibody (07-013; Millipore/Upstate), then incubated with goat antirabbit Cy3-conjugated antibody (111-165-003; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Slides were mounted with Vectasheild (Vector Labs, Burlingame, CA, USA) containing DAPI (4′,6-diamidino-2-phenylindole) and images were captured on a Leica TCS SP2 microscope (Leica Microsystems Inc.). For Nissl stain, whole-brain mounts were incubated in cresyl violet, covered in permount, and coverslipped. Each slice was scanned, and stained and unstained areas of the treated hemisphere were quantified using ImageJ 1.32j (NIH) to calculate the infarct volume as a percentage of the unlesioned hemisphere.
Results
Ischemic Preconditioning Alters miRNA Expression in Adult Mouse Cortex
We generated preconditioned, ischemic, and tolerant mice using varying durations of MCAO (Figure 1A). Representative images of TTC-stained sections reveal the extent of injury in mouse brains (Figure 1B). Infarct volume was quantified using TTC-stained sections (
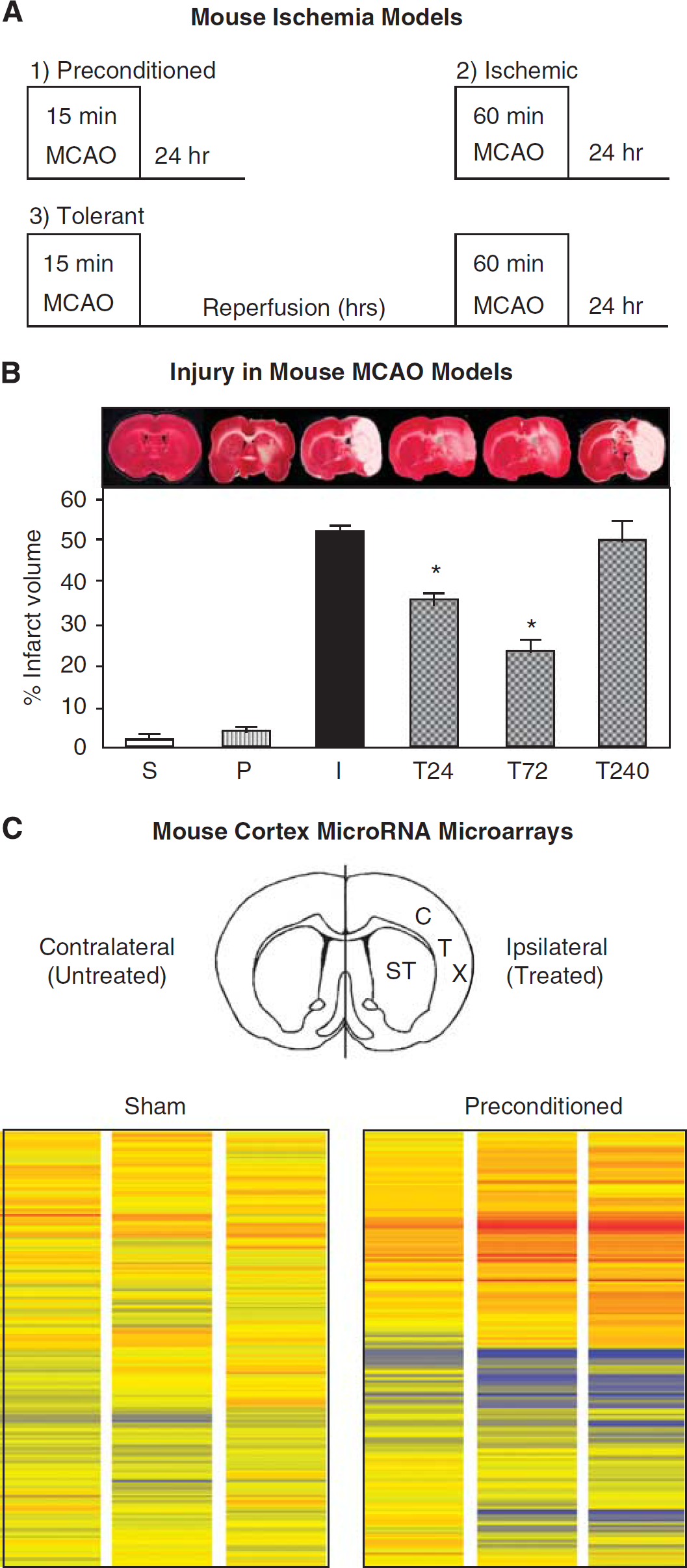
Ischemic preconditioning and microRNA microarrays in adult mouse cortex. (
Statistical Analysis Reveals Distinct Changes in miRNA Expression in Preconditioned, Ischemic, and Tolerant Mouse Cortices
We first evaluated the consistency of miRNA expression within each group to identify animal-to-animal and/or diurnal variations in miRNA expression. For each mouse, the ALR of miRNA expression from one animal (Replicate) was compared with the ALR of miRNA expression for all replicates in the same group (Total); each point in Figure 2A represents one unique miRNA. Representative graphs show a positive correlation between one individual replicate relative to the average of the total replicates sampled within each group for sham (Figure 2Ai), preconditioned (Figure 2Aii), ischemic (Figure 2Aiii), and tolerant (Figure 2Aiv) animals. These studies show that miRNA expression is consistent within a treatment group and that regulation of miRNAs is not random in individual mice.
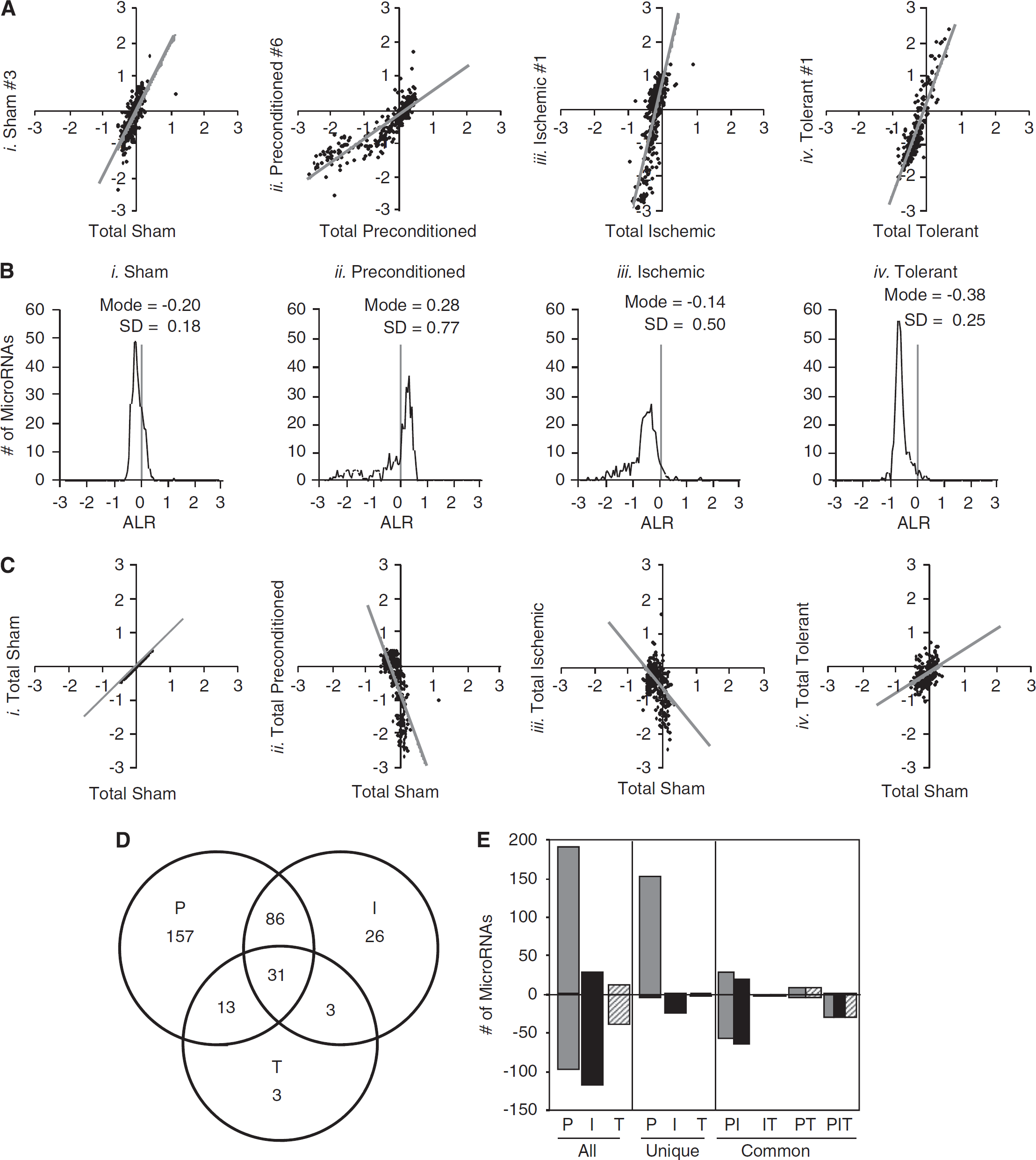
Distinct changes in microRNA expression in preconditioned, ischemic, and tolerant mouse cortices. (
We then analyzed miRNA expression in the ipsilateral cortex of mice in each group, relative to contralateral cortex (Figure 2B). A histogram of the ALR for sham miRNAs (
We then compared miRNA expression in the preconditioned, ischemic, and tolerant mice to those of the sham mice (Figure 2C). Graphs show the total ALR for each miRNA in a group plotted against the total ALR of miRNAs in sham. Sham versus sham is provided to illustrate the expected outcome of a positive correlation in unchanged miRNA expression (Figure 2Ci). The graphs show that the ALRs of miRNAs from preconditioned cortices are negatively correlated to sham values (Figure 2Cii), and a similar negative correlation is seen in the ALR of miRNAs from ischemic cortices (Figure 2Ciii). In contrast, there is a positive correlation between the ALR of miRNAs from tolerant cortices relative to sham (Figure 2Civ). These results show distinct changes in miRNAs in the ipsilateral cortex of preconditioned, ischemic, and tolerant cortices, relative to sham.
Individual miRNAs from preconditioned, ischemic, and tolerant cortices were then examined by
Prominent messenger RNA targets of high-confidence preconditioning-regulated microRNAs (total number of unique miRNAs targeting the 3′ UTR/number of potential miRNA/mRNA binding sites)
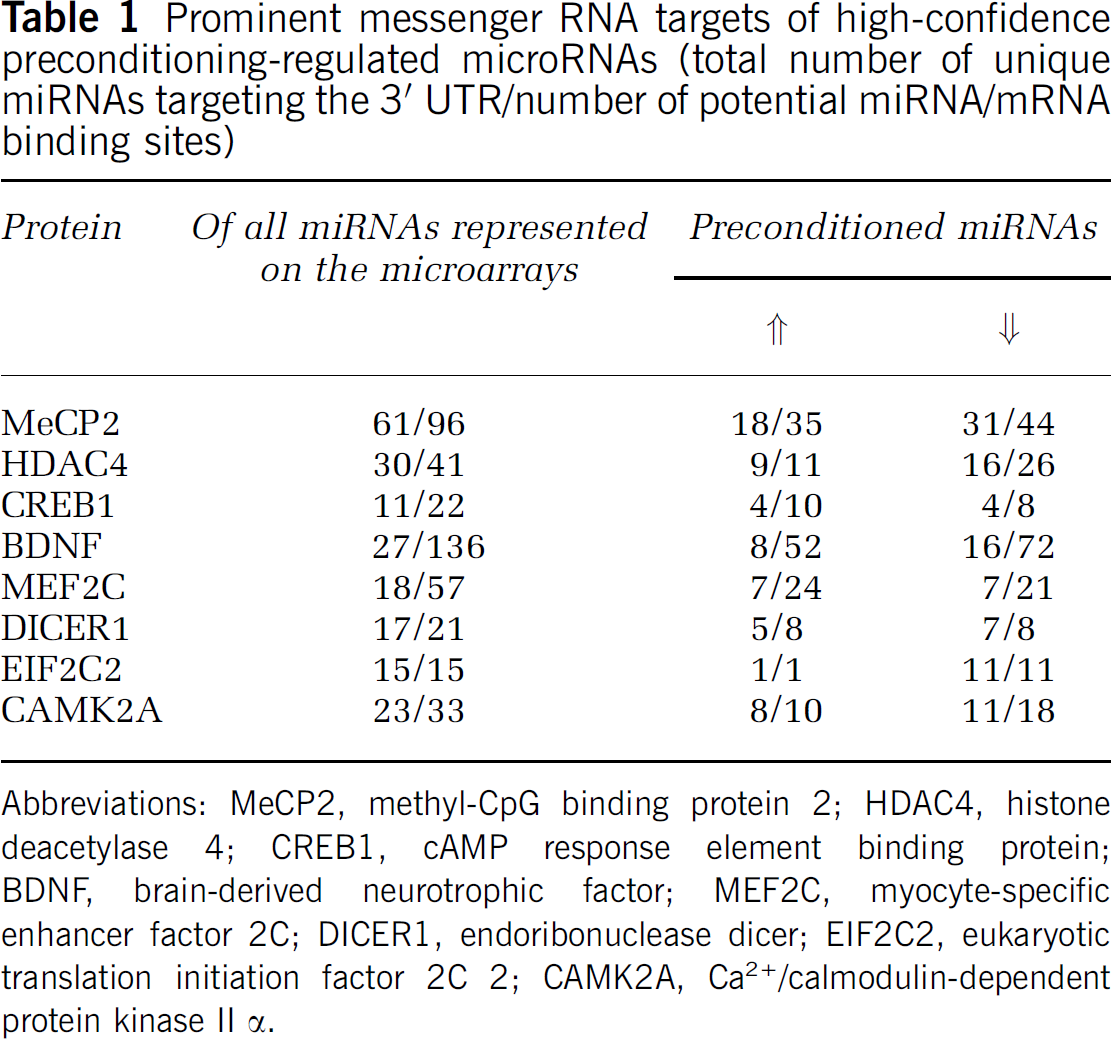
Abbreviations: MeCP2, methyl-CpG binding protein 2; HDAC4, histone deacetylase 4; CREB1, cAMP response element binding protein; BDNF, brain-derived neurotrophic factor; MEF2C, myocyte-specific enhancer factor 2C; DICER1, endoribonuclease dicer; EIF2C2, eukaryotic translation initiation factor 2C 2; CAMK2A, Ca2+/calmodulin-dependent protein kinase II α.
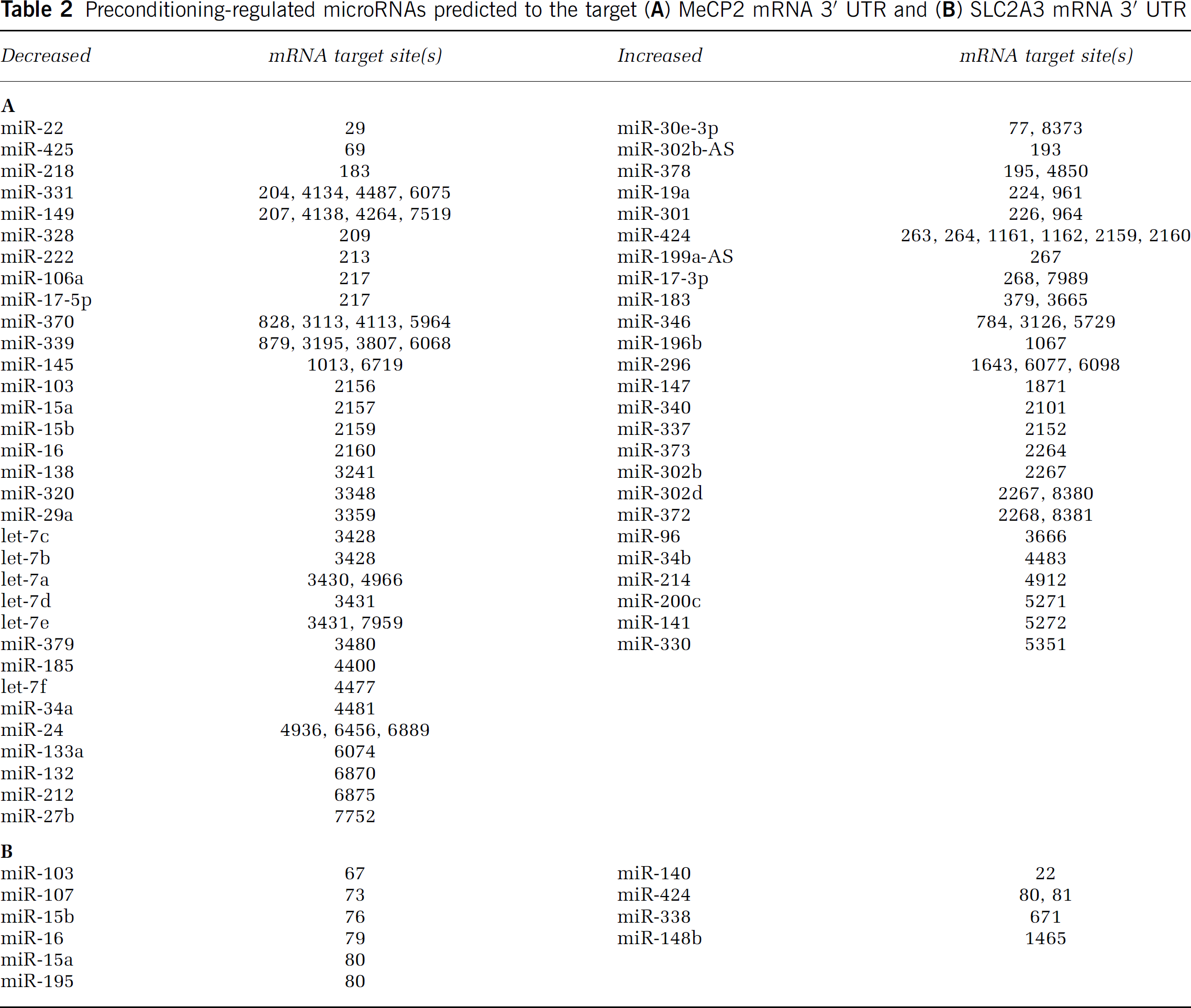
Preconditioning-regulated microRNAs predicted to the target (
Target Prediction of the miRNAs Regulated in Preconditioned Mouse Cortex
We used bioinformatic software tools to identify potential mRNA targets of the miRNAs significantly regulated in preconditioned mouse cortex (John et al, 2004), and restricted our analysis to high-confidence miRNAs, defined as those miRNAs detected in all microarray replicates (nine sham, six preconditioned, six ischemic, three tolerant). Given the cooperative nature of miRNAs and that target regulation is more potent when several miRNAs bind to a given mRNA (Doench and Sharp, 2004), the number of ‘hits,’ or miRNA binding sites on an mRNA 3′ UTR is an important consideration in target prediction. Thus, we used miRanda (version 2005) for target prediction as it can query several miRNAs at one time to identify predicted mRNA targets of a cohort of miRNAs. Significantly regulated (
The mRNA predicted to be most heavily targeted by the preconditioning-regulated miRNAs encodes for MeCP2. DNA microarray studies show that MeCP2 mRNA
Ischemic Preconditioning Decreases miR-132 and Increases MeCP2 Protein, but has no Effect on MeCP2 mRNA Levels
Those miRNAs predicted by miRanda to target the MeCP2 3′ UTR are depicted in Figure 3A; among the decreased miRNAs is miR-132. Klein et al (2007) have shown that MeCP2 expression is controlled by miR-132: decreased miR-132 leads to increased MeCP2 expression in neurons, whereas increased miR-132 leads to decreased MeCP2 expression. Results of our miRNA microarray data show that miR-132 expression is significantly decreased in preconditioned (P) cortex (Figure 3B, −2.26 ± 0.485 ALR,
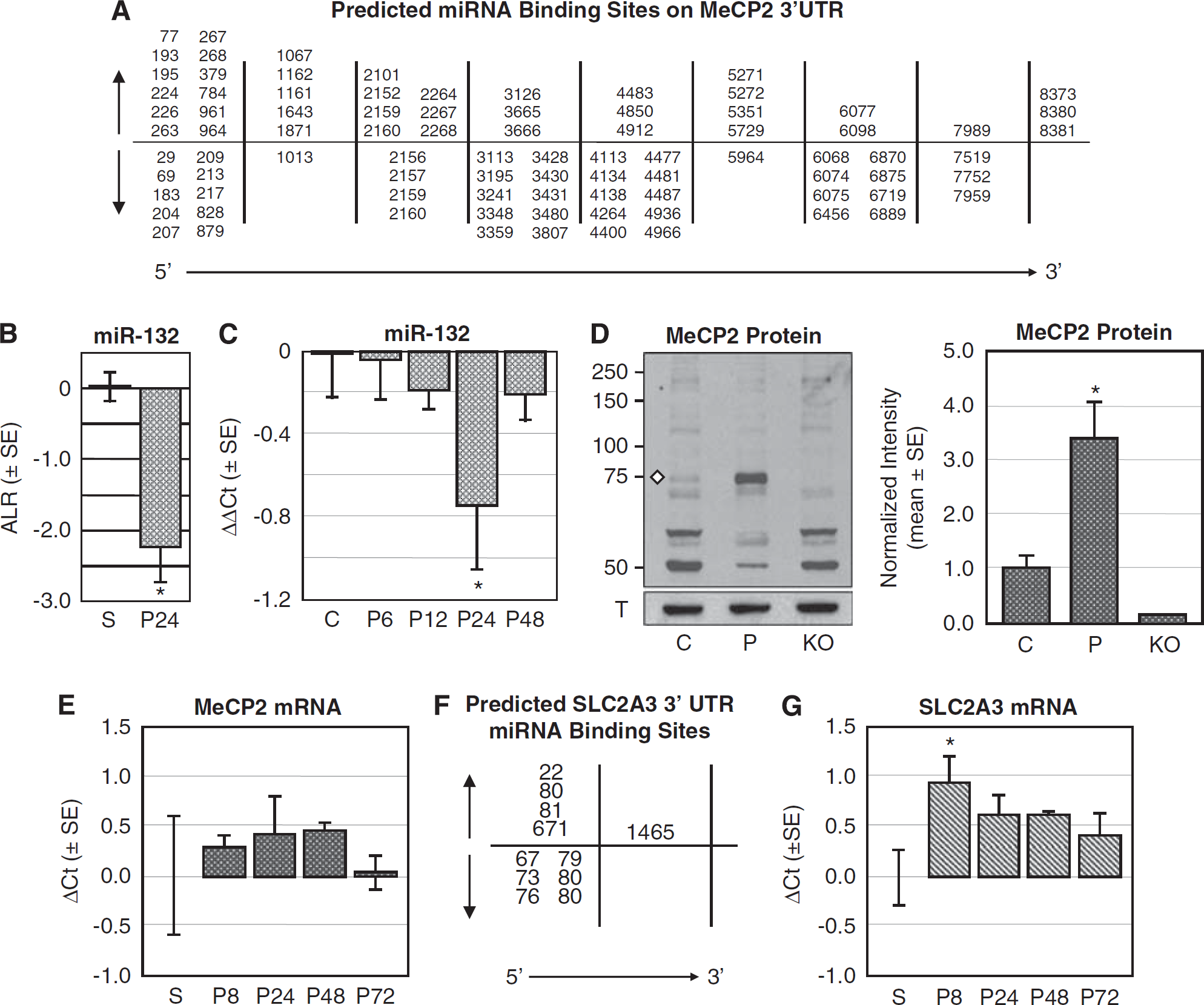
Decreased miR-132 expression correlates with increased MeCP2 protein, but no change in MeCP2 mRNA, in preconditioned mouse cortex. (
We then used immunoblot analysis to quantify MeCP2 protein expression in preconditioned mouse cortex. A representative immunoblot shows MeCP2 expression in control (C) and preconditioned (P) mouse cortical nuclear lysates (Figure 3C). The MeCP2 antibody (ab50005; Abcam) detects more than one protein band on the immunoblots, thus we also examined nuclear lysates from MeCP2 KO mice to confirm that the 72kDa protein band (white diamond) is indeed MeCP2. α-Tubulin III (T), which served as a loading control, is shown in the bottom panel. Quantitative analysis of MeCP2 protein expression for control (C,
DNA microarray studies do not find MeCP2 mRNA regulated in preconditioned cortex (Stenzel-Poore et al, 2003). Thus, we quantified expression of MeCP2 mRNA by qRT-PCR at 8, 24, 48, and 72 h after preconditioning (
MeCP2 Knockout Mice Show Increased Susceptibility to Preconditioning Ischemia
We next examined the cellular distribution and temporal expression of MeCP2 protein in control, sham, and at 8, 24, 48, and 72 h after preconditioning by immunohistochemistry (
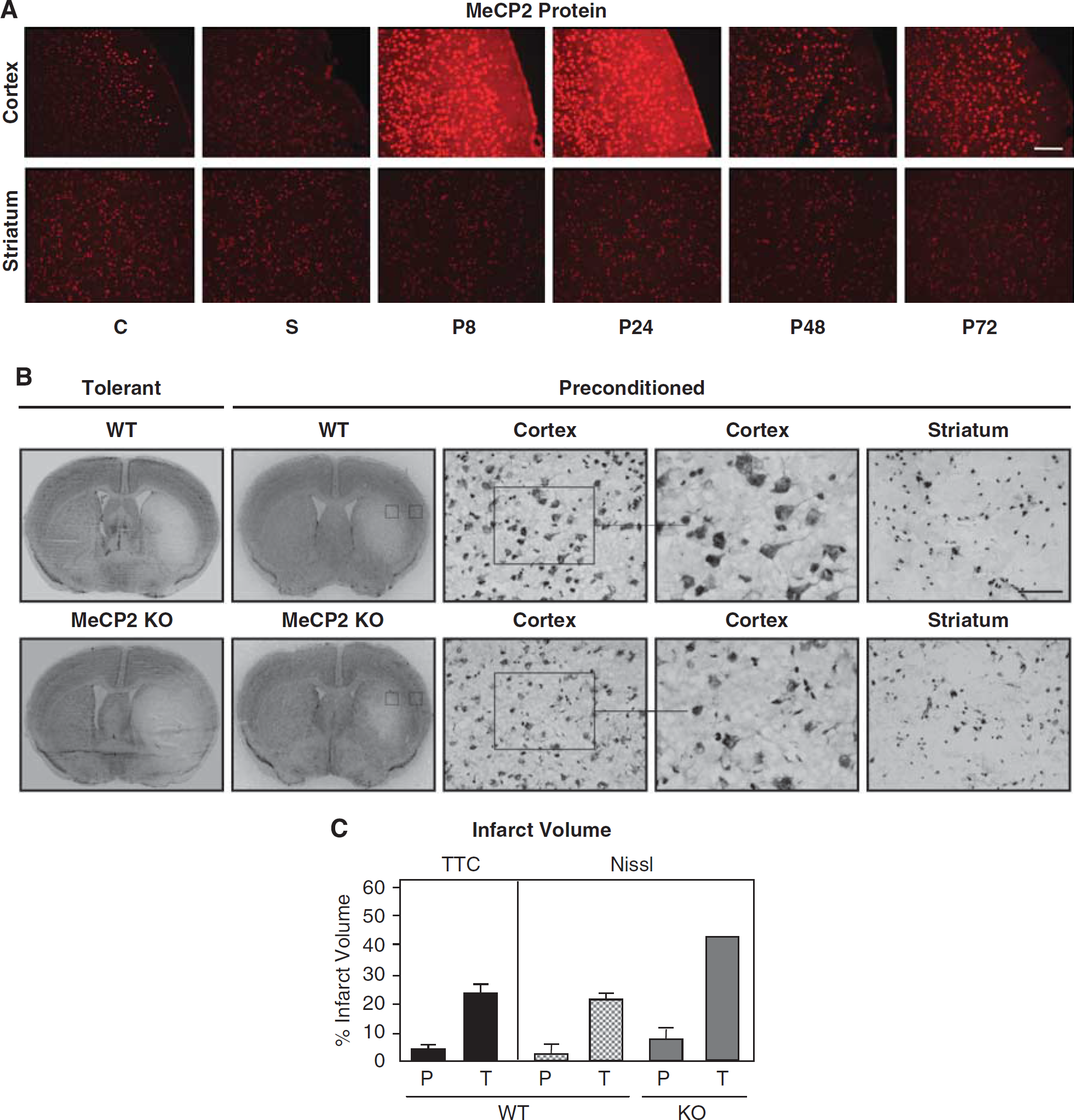
Increased susceptibility to ischemia in MeCP2 KO mouse cortex. (
Discussion
Cerebral miRNA expression is regulated by transient ischemia (Dharap et al, 2009; Jeyaseelan et al, 2008), traumatic brain injury (Redell et al, 2009), and several other neurologic disorders (Kuss and Chen, 2008). Ischemic preconditioning-induced tolerance requires
We examined MeCP2 protein because as a transcriptional repressor (Fuks et al, 2003), MeCP2 could be an effector of genomic reprogramming, and had not been examined in the context of preconditioning. Thus our finding that MeCP2 expression is rapidly increased in preconditioned cortex provided a potential link between two key, yet seemingly paradoxical, features of ischemic preconditioning-induced tolerance: the requirement for new protein synthesis (Barone et al, 1998) and genomic reprogramming of the response to ischemia that leads to a transient repression of gene expression (Stenzel-Poore et al, 2003). MeCP2 is a potent transcriptional repressor, yet recent studies show that MeCP2 is a complex regulator of transcription (Chahrour et al, 2008): MeCP2 as a transcriptional activator requires CREB1, and as a transcriptional repressor requires histone deacetylase. Since MeCP2 had not previously been examined in the context of preconditioning or tolerance, it also served as a novel protein to test the power of miRNA target prediction. These studies support the concept that, in addition to preconditioning-induced changes in gene transcription and increased mRNA levels, mechanisms of preconditioning-induced protein expression could include posttranscriptional regulation of mRNAs by miRNAs, consistent with studies showing miRNA regulation of protein expression independent of changes in mRNA levels (Baek et al, 2008; Selbach et al, 2008).
Functionally, miRNAs expressed within dendrites regulate translation of proteins mediating dendritic growth (Schratt et al, 2006). Further, miRNAs are important for regulating synaptic plasticity, and miRNAs target (and are targeted by) plasticity mediators such as CREB, fragile X mental retardation protein, and MeCP2 (Smalheiser and Lugli, 2009). A recent study has shown that miR-132 directly regulates MeCP2 protein expression in rat cortical neurons: increased miR-132 leads to decreased MeCP2 protein, whereas decreased miR-132 leads to increased MeCP2 protein (Klein et al, 2007). This finding is consistent with our data showing decreased miR-132 and increased MeCP2 protein in ischemic preconditioned mouse cortex. Our microarrays and qRT-PCR both showed miR-132 significantly decreased in preconditioned cortex. On the basis of our target analysis showing that many miRNAs, including miR-132, are predicted to bind to the 3′ UTR of MeCP2 mRNA, we used immunoblot and immunohistochemistry studies to evaluate MeCP2 protein expression and show, for the first time, that MeCP2 is rapidly increased in preconditioned cortex. As miR-132 expression is decreased and MeCP2 protein increased in preconditioned cortex, we suggest that MeCP2 mRNA is translationally repressed by miRNAs in control brain and that preconditioning leads to derepression of MeCP2 mRNA by miRNAs with resultant synthesis of MeCP2 protein. When we examined the effect of preconditioning and tolerance in MeCP2 KO mice, we found increased susceptibility to ischemia, consistent with studies showing increased cell death in cerebellar granule neurons in MeCP2 KO mice exposed to excitotoxicity and hypoxic-ischemia (Russell et al, 2007). Further, MeCP2 KO mice show increased susceptibility to hypoxia in telencephalic neuronal networks that involve disturbed potassium channel function, suggesting that hypoxia might contribute to the vulnerability of male Rett patients who are either not viable or severely disabled (Fischer et al, 2009). Although these initial studies in MeCP2 KO mice are not conclusive, they do provide further evidence that MeCP2 is necessary for the induction of ischemic preconditioning.
Given that the MCAO treatments are identical except for duration of occlusion, we expected miRNAs regulated by preconditioning also to be regulated by ischemia. We predict that these common miRNAs mount a response to subsequent injury, but that cell death pathways induced by the longer duration of ischemia overcome this response. Although we focused on miRNAs decreased in preconditioned cortex, many uniquely regulated miRNAs were increased in preconditioned cortex. These miRNAs could serve to repress translation of mRNAs not essential for neuroprotection as a mechanism of energy conservation; studies focused on the role of these miRNAs in tolerance are currently in progress. The studies presented herein set the stage to address additional questions such as which miRNAs specifically target MeCP2 to regulate protein expression. Our use of a low-stringency prediction program (miRanda, version 2005) allowed us to assess the cooperative potential across miRNA changes. Current studies examining specific miRNA/MeCP2 interactions and their therapeutic potential are focused on five preconditioning-decreased miRNAs predicted by three increasingly stringent bioinformatic prediction programs (miRanda, TargetScan, and PicTar) to target MeCP2 mRNA. In addition, recent studies show that MeCP2 is not restricted to neuronal cells as previously thought, but is also expressed in glia (Ballas et al, 2009; Maezawa et al, 2009). Given that miRNAs can activate translation in quiescent cells but repress translation in proliferating cells (Vasudevan et al, 2007), differential regulation of target proteins could occur in neurons and glia. We trust that these studies will contribute to our understanding of the mechanisms underlying ischemic preconditioning-induced tolerance, and have potential to translate into novel strategies for the treatment of ischemic brain injury: the induction of tolerance.
Footnotes
Acknowledgements
We acknowledge the support of NIH (R21NS054220, JAS), the NIH Neuroscience Microarray Consortium, and Dr Holly Dressman, Director of the Duke University Microarray Facility. We thank Mr Rob Lusardi (Slowdog Software, Portland, OR, USA) for creating the miRNA microarray analysis program used for the data analysis. We thank Jaclyn Shingara and Dr David Brown of Ambion/Applied Biosystems for early contributions to miRNA microarrays in rat brain. RPS envisioned a role for miRNAs in ischemic tolerance, JAS designed and supervised all experimental aspects of this project, GP and TY performed mouse neurosurgeries, JAS isolated all RNAs for microarray studies and qRT-PCR studies, TAL and JAS analyzed miRNA microarray data, JQL sectioned mouse brains, CDF performed immunoblot and immunohistochemistry, CLF performed immunoblot, and maintained the MeCP2 KO mice. JAS, TAL, and RPS wrote the paper.
The authors declare no conflict of interest.