
Abstract
It has been proposed that proinflammatory mechanisms are involved in the pathogenesis of brain edema in acute liver failure (ALF). The aim of this study was to assess the contribution of cerebral inflammation to the neurologic complications of ALF and to assess the antiinflammatory effect of mild hypothermia. Upregulation of CD11b/c immunoreactivity, consistent with microglial activation, was observed in the brains of ALF rats at coma stages of encephalopathy. Interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) mRNAs were increased two to threefold in the brains of ALF rats compared with that in sham-operated controls. The magnitude of increased expression of proinflammatory cytokines in the brain was correlated with the progression of encephalopathy and the onset of brain edema. Significant increases in IL-1β, IL-6, and TNF-α levels were also found in the sera and cerebrospinal fluid (CSF) of these animals. Mild hypothermia delayed the onset of encephalopathy, prevented brain edema, and concomitantly attenuated plasma, brain, and CSF proinflammatory cytokines. These results show that experimental ALF leads to increases in brain production of proinflammatory cytokines, and afford the first direct evidence that central inflammatory mechanisms play a role in the pathogenesis of the cerebral complications of ALF. Antiinflammatory agents could be beneficial in the management of these complications.
Keywords
Introduction
Acute liver failure (ALF) that results from viral infection or toxic liver injury is a life-threatening condition. Hepatic encephalopathy and brain edema are serious neurologic complications of ALF, and their appearance heralds the need for emergency liver transplantation (Cordoba and Blei, 1996). Several theories have been proposed to explain the pathogenesis of hepatic encephalopathy and brain edema, among which ammonia toxicity has received the most attention (Butterworth, 2003; Vaquero et al, 2003). However, ammonia is not the only pathophysiologic entity with the potential to adversely affect cerebral function in ALF. In particular, there is evidence that inflammation may also play a significant role. For example, large clinical studies have convincingly shown a higher prevalence of the “systemic inflammatory response syndrome” in ALF patients (Rolando et al, 2000). Systemic inflammatory response syndrome is a response to the presence of proinflammatory cytokines, such as the interleukins, IL-1β and IL-6, as well as to tumor necrosis factor-α (TNF-α), and increased circulating levels of cytokines have been reported consistently in ALF patients (Muto et al, 1988; Sekiyama et al, 1994; Nagaki et al, 2000; Jalan et al, 2003). Cytokines may also be formed and released by the necrotic liver. Studies on arteriovenous difference by Jalan et al (2003) suggest a net production of proinflammatory cytokines in the brains of patients with ALF. However, direct evidence for a role of increased brain cytokines in the pathogenesis of the cerebral complications of ALF is still lacking. Therefore, the aim of this study was to measure TNF-α, IL-1β, and IL-6 mRNA, and protein expression as a function of neurologic status in the brains of rats with ALF because of hepatic devascularization. These cytokines were chosen in view of earlier reports of their selective increase in both experimental and human ALF (above). In addition, given the recent upsurge of interest in hypothermia in the management of patients with ALF (Jalan et al, 1999, 2003; Vaquero et al, 2007; Vaquero and Butterworth, 2007) and the earlier report that hypothermia led to a delay in the onset of coma and brain edema in hepatic devascularized rats (Rose et al, 2000), the effect of hypothermia on cytokine expression in the serum and brains of these animals was also examined.
Materials and methods
Surgical Procedures
Adult male Sprague-Dawley rats (200 to 250 g) purchased from Charles River (Saint-Constant, Quebec, Canada) were tested routinely for common pathogens and were free from infection at the time of surgery. Rats were anesthetized using isoflurane, and an end-to-side portacaval anastomosis was performed according to the guidelines of Lee and Fisher (1961). Briefly, rats underwent a laparotomy, the inferior vena cava and portal vein were isolated and clamped using an anastomosis clamp (Roboz Instruments Inc, Washington DC, USA), and an elliptical portion 1.5 times the diameter of the portal vein was removed. The portal vein was ligated and cut, and an end-to-side anastomosis was performed under a dissecting microscope. Total surgery time was < 30 mins. Sham-operated control rats, matched for weight, were anesthetized similarly and the inferior vena cava was clamped for 20 mins. After surgery, all animals were individually housed with free access to food and water under constant conditions of temperature, humidity, and light cycles. Twenty-four hours after portacaval anastomosis, rats were reanesthetized and subjected to hepatic artery ligation (HAL). After HAL, arterial blood glucose levels were monitored and glucose was administered subcutaneously as needed to maintain normoglycemia. Body temperature was monitored every 15 min and maintained at 37°C ± 0.5°C using heating pads. Hypothermia occurred spontaneously in the absence of external heating, and body temperature in the hypothermic ALF animals was again maintained at 33°C ± 0.5°C using heating pads when necessary. A group of animals was killed at precoma (before the appearance of encephalopathy and brain edema) 6h after HAL (ALF-6h). A second group (ALF-coma) was killed at the coma stage of encephalopathy (defined as the loss of righting and corneal reflexes), at which time the animals had significant brain edema. Hypothermic animals (ALF-33) were killed in parallel with time-matched comatose normothermic ALF animals and sham-operated controls. Their brains were rapidly removed, dissected on ice, and were immediately frozen in isopentane. All tissues were stored at −70°C until use. All the above surgical methods were performed in accordance with the Guidelines of Canadian Council of Animal care and were approved by the Animal Research Committee at Saint-Luc Hospital (C.H.U.M.).
Brain Water Measurement
Brain water was quantitated using the wet-weight/dry-weight method. Half of the brain sample was weighed before and after 48 h of incubation in a 120°C oven. Water content of the brain samples are expressed as percentage of water content according to the following equation: %water = (wet weight-dry weight)/wet weight x 100.
Ammonia Concentration
Plasma and cerebrospinal fluid (CSF) ammonia concentrations were determined using a commercial ammonia test kit (Sigma Aldrich, St-Louis, MO, USA) on the basis of the enzymatic method using the glutamate dehydrogenase reaction (Bergmeyer and Beutler, 1985).
Immunohistochemistry
Animals were deeply anesthetized using pentobarbital (60 mg/kg). After being transcardially perfused with 240-mL ice-cold saline followed by 240 mL of neutral-buffered formalin (containing 4% formaldehyde, 0.5% sodium phosphate buffer, 1.5% methanol, pH 7.0), their brains were removed, postfixed in 10% formalin at 4°C for 12h, and transferred into an ice-cold phosphate-buffered saline (PBS) solution for storage. Coronal sections that were 50-μm thick were obtained using a vibratome from −4.0 to −5.5 mm relative to the bregma according to the rat brain atlas of Paxinos and Watson (1986). Sections were incubated with 0.3% hydrogen peroxide in PBS for 10 min to block the endogenous peroxidase activity. After washing with PBS, the sections were blocked with 2% horse serum and 0.5% Triton X100 in PBS, washed, and incubated at 4°C overnight with mouse anti-CD11b/c (OX-42) (1/1000) (Cedarlane Laboratories, Burlington, NC, USA). After washing with PBS, the sections were incubated for 1 h with horse antimouse biotinylated secondary antibody (1:100) (Vector Laboratories, Burlingame, CA, USA). Furthermore, after washing with PBS, the sections were incubated with the Vectastain ABC reagent (Vector Laboratories) and immunoreactivity was detected by incubation with 3–3'-diaminobenzidine containing urea-hydrogen peroxide (Sigma Aldrich). The sections were mounted on Superfrost Plus slides (Fisher Scientific, Pittsburgh, CA, USA), dehydrated stepwise in ethanol and xylene, and coverslipped with Permount (Fisher Scientific). Sections without primary antibodies were used as negative controls and showed no immunoreactivity. Quantitative analysis was performed by counting the immunopositive cells in 10 selected representative areas of 0.5 mm2. Cell counts were performed by an investigator who was unaware of the animal treatment group.
Measurement of Cytokine Gene Expression by Quantitative Real-Time Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
In view of the established diurnal variations of IL-1β and TNF-α mRNAs, all experiments were performed at the same time of day (pm). Animals were transcardially perfused with 240-mL ice-cold saline to remove the residual blood from the brain. Total RNA was isolated from rat brain cortex using the Trizol reagent (Invitrogen Ltd, Carlsbad, CA, USA), according to the manufacturer's instructions. cDNA was synthesized using thermoscript RT-PCR system (Invitrogen). Expression levels were assessed using real-time PCR in a RotorGene 3000™ Real time DNA detection system (Corbett Life Science, Sydney, Australia) with the Quantitect SYBRGreen I PCR kit (Qiagen, Valencia, CA, USA). Oligonucleotide primers were designed using the Primer 3 software (Rozen and Skaletsky, 2000) on the basis of the following GeneBank accession numbers: V01217 (β-actin), M98820 (IL-1β), M26744 (IL-6), and X66539 (TNF-α), and included at least one intron. The specificity of the oligonucleotide primers was verified using the program BLASTN from the National Center for Biotechnology Information (NCBI, Bethesda, MD, USA). For each primer pair, the amplified cDNA fragments were verified by agarose gel to confirm the absence of the intron and of any nonspecific products. The forward and reverse primers used were 5'-CCACAGCTGA GAGGGAAATC-3' and 5'-TCTCCAGGGAGGAAGAGGAT-3' for β-actin; 5'-TCTTCTCATTCCTGCTCGTG-3' and 5'-GATG AGAGGGAGCCCATTT-3' for TNF-α; 5'-CTCAACTGTGAAA TAGCAGCTTTC-3' and 5'-GGACAGCCCAAGTCAAGG-3' for IL-10; and 5'-CTTCACAAGTCGGAGGCTTAAT-3' and 5'-ACAGTGCATCATCGCTGTTC-3' for IL-6. A relative quantification was performed by comparing the threshold cycle values of samples with serially diluted standards. Expression levels were normalized to the housekeeping gene, β-actin.
Plasma, Cerebrospinal Fluid Collection, and ELISA (Enzyme-Linked Immunosorbent Assay)
Plasma and CSF samples were collected 6h after HAL (precoma) and at the coma stage of encephalopathy. Cisterna magna catheters were installed in groups of animals to collect CSF as described earlier (Swain et al, 1992a). In brief, the animal's head was mounted with the skull in a horizontal position on a stereotaxic apparatus. A 3-cm incision was made on the skin from the back of its head, and the overlying connective tissue was removed to expose the skull. A small hole was drilled in the skull using a dental burr (009) on the midline immediately rostral to the interparietal-occipital bone suture. The hole was drilled in such a way that the occipital bone could be used as a guideline while inserting the cannula (PE-10 tubing, Clay Adams, Parsipanny, NJ, USA). The catheter was inserted into the cisterna magna. Correctness of placement was accompanied by a spontaneous flow of clear CSF. Protein levels of IL-1β, IL-6 IL-10, and TNF-α were measured in plasma and in CSF samples using commercial ELISA kits (R&D Systems, Minneapolis, MN, USA; Biosource, Camarillo, CA, USA) according to the manufacturer's instructions. The plates were read at 450 nm and the absorbances were converted to pg/mL using standard curves prepared with recombinant cytokines.
Statistical Analysis
All data are expressed as the mean ± s.e.m., and statistical analysis was performed using ANOVA (one-way analysis of variance) followed by Tukey's post hoc analysis. A probability of P < 0.05 was chosen to establish significance between the groups. Data were analyzed using the Prism 4.0 software (Prism 4.0, San Diego, CA, USA).
Results
Neurologic Status and Brain Edema
After hepatic devascularization, normothermic ALF rats (maintained at 37°C) developed progressive encephalopathy, which progressed to loss of corneal reflex (coma). At 6h after surgery (ALF-6h), ALF rats showed no overt neurologic symptoms. Hypothermia (ALF rats maintained at 33°C) significantly delayed the onset of encephalopathy. Sham-operated controls and ALF-6h rats showed normal behavior and unchanged reflexes throughout the entire period of the experiments.
Brain water content was not significantly different between sham-operated control rats and normothermic rats 6h after HAL (79.4 ± 0.07% versus 79.5 ± 0.14%), but was elevated significantly in normothermic rats that were at the coma stage of encephalopathy (80.8 ± 0.06%, P < 0.001). Hypothermia (ALF-33) significantly attenuated brain water content (ALF-33 versus ALF-coma: 79.5 ± 0.15% versus 80.8 ± 0.06%, P < 0.001) in ALF rats compared with normothermic rats killed at equivalent time points (Figure 1).
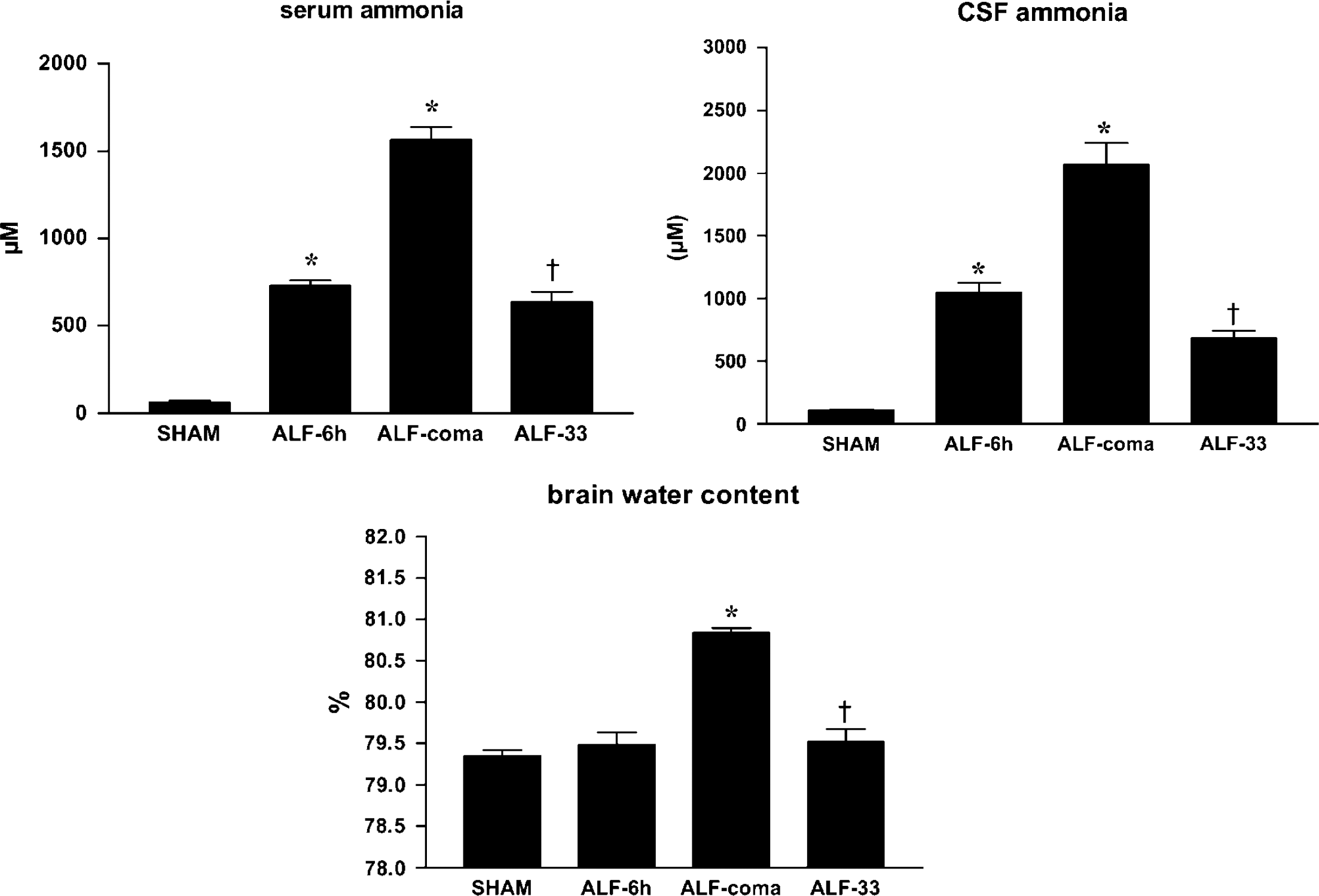
Cerebrospinal fluid and plasma ammonia and brain water content in ALF rats maintained normothermic (ALF-6h, ALF-coma) or hypothermic (ALF-33) compared with sham-operated controls (sham). Data represent mean ± s.e.m. of 10 animals in each group. *P < 0.001 versus sham; †P < 0.001 versus ALF-coma.
Plasma and Cerebrospinal Fluid Ammonia
Serum ammonia levels were elevated in normothermic ALF rats starting 6 h after HAL (ALF-6 h versus sham-operated controls: 729 ± 32 versus 62 ± 9 μmol/L, P < 0.001) with further increases at the coma stage of encephalopathy (1566 ± 72 μmol/L, P < 0.001). Cerebrospinal fluid ammonia levels were also increased 6 h after HAL (ALF-6 h versus sham-operated controls: 1049 ± 78 versus 109 ± 7 μmiol/L, P < 0.001) with further increases again apparent at the coma stage of encephalopathy (2067 ± 176 μmol/L, P < 0.001). Hypothermia significantly attenuated the rise of both serum (ALF-33 versus ALF-coma: 637 ± 57 versus 1566 ± 72 μmol/L, P < 0.001) and CSF ammonia concentrations (ALF-33 versus ALF-coma: 682 ± 62 versus 2067 ± 176 μmol/L, P < 0.001) (Figure 1).
Effects of Acute Liver Failure on Microglial Activation
Formaldehyde-fixed floating cerebral cortical sections from ALF rats stained with CD11b/c (OX-42), the major histocompatibility complex class II antigen marker, reveal microglial activation, which was correlated with the onset of brain edema and coma stage of encephalopathy (Figure 2). Comparable increases in OX-42-positive cells were also observed in the thalamus and hippocampus (Table 1).
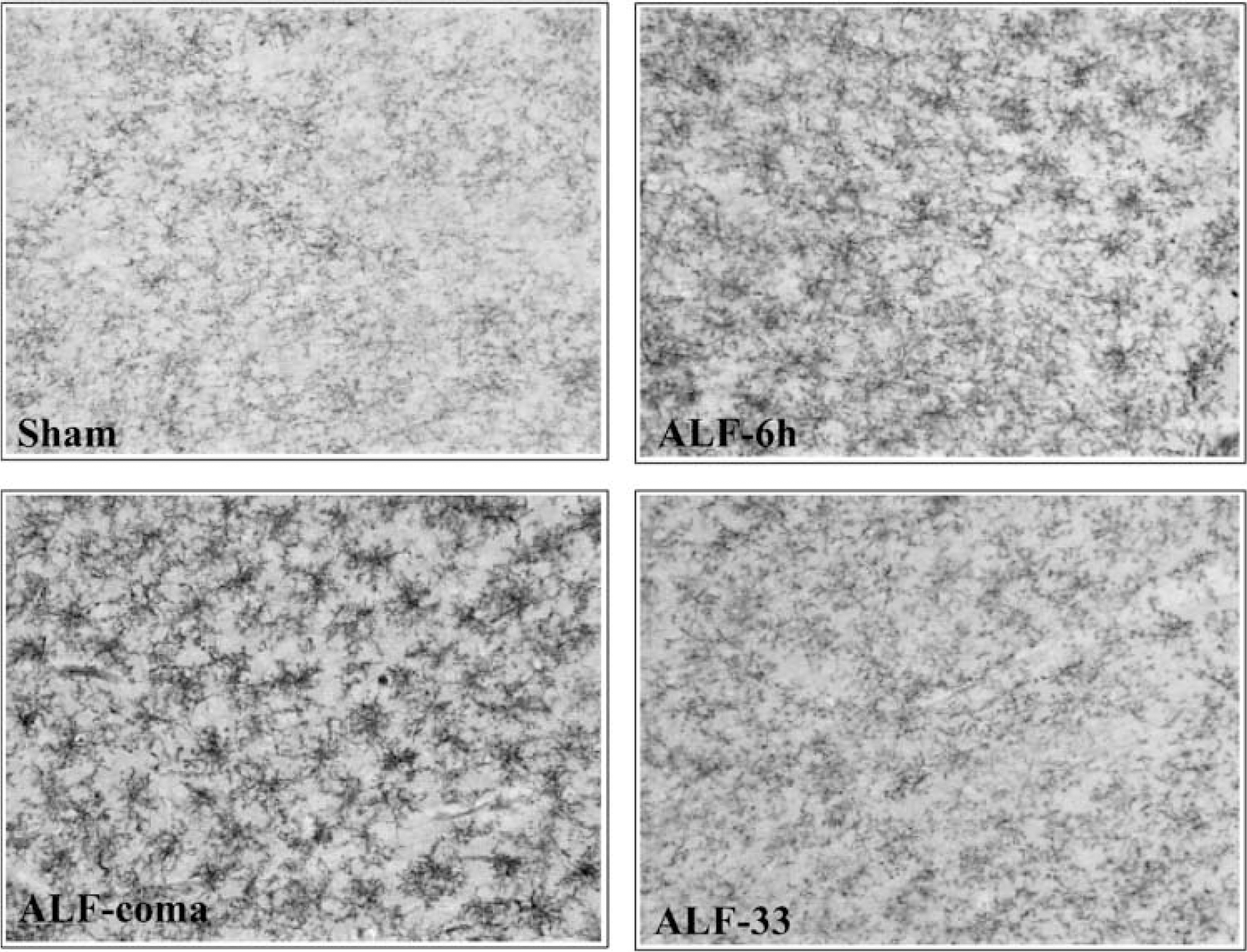
Microglial activation in the brains of rats with ALF at the coma stage of encephalopathy. Representative micrographs showing the effect of ALF on OX-42 staining in cerebral cortex from sham-operated controls (sham) and comatose ALF rats (ALF-coma) (original magnification 200 x).
Effect of hypothermia on microglial activation in various brain regions of ALF rats

ALF, acute liver failure.
OX-42-positive cells (per x100 optical fields) in brain regions of sham-operated controls (sham), ALF-rats 6 h after HAL (ALF-6 h), ALF rats at coma stage of encephalopathy (ALF-coma), and in hypothermic ALF rats (ALF-33). Data represent mean ± s.e.m. of 10 measurements per animal (n = 6)
P < 0.001 versus sham-operated controls
P < 0.001 versus ALF-coma).
Effects of ALF and Hypothermia on Circulating IL-1β, IL-6, and TNF-α
In normothermic ALF rats, serum IL-1β protein levels were increased 3.8-fold at 6h after HAL (sham-operated controls versus ALF-6h: 36.7 ± 10.6 versus 140.2 ± 10.4 pg/mL, P <0.05) and IL-6 levels were increased 40-fold (sham-operated controls versus ALF-6h: 72.5 ± 1.8 versus 2933 ± 196 pg/mL, P < 0.001), whereas TNF-α concentrations remained at the level of sham-operated controls (sham versus ALF-6h: 3.7 ± 0.3 versus 3.4 ± 0.3pg/mL) (Figure 3). At coma stage, IL-1β concentrations were increased 6.5-fold (239.3 ± 49.3 pg/mL, P < 0.001), IL-6 levels were increased 47-fold (3419 ± 218.3 pg/mL, P < 0.001), and TNF-α concentrations were increased 2.8-fold (4.2 ± 0.7 pg/mL) (P < 0.001). In hypothermic ALF rats, increases in serum levels of IL-1β, IL-6, and TNF-α were attenuated by 55% (P <0.01), 81% (P < 0.001), and 60% (P < 0.001), respectively, compared with normothermic ALF rats (Figure 3). Concentrations of the antiinflammatory cytokine IL-10 were increased 16-fold (P < 0.001) in the plasma of normothermic ALF animals, but this increase was only marginally influenced by hypothermia (data not shown).
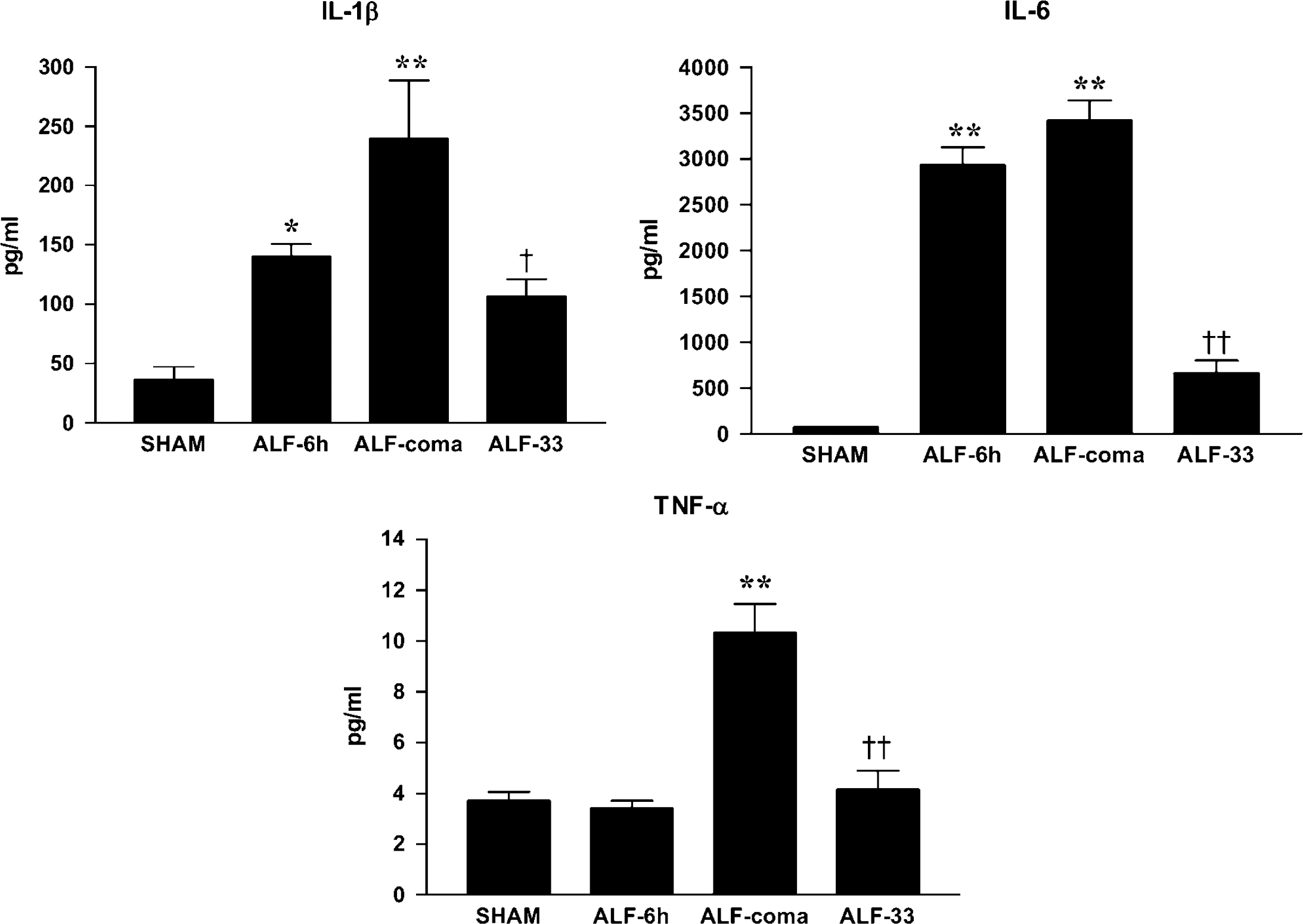
Plasma IL-1β, IL-6, and TNF-± levels in ALF rats maintained normothermic (ALF-6 h, ALF-coma) or hypothermic (ALF-33) compared with sham-operated controls (sham). Data represent mean ± s.e.m. of 10 animals in each group. *P < 0.05 versus sham; **P < 0.001 versus sham; †P < 0.01 versus ALF-coma; ‡P < 0.001 versus ALF-coma.
Effects of ALF and Hypothermia on Cerebral (CSF) IL-1β, IL-6, and TNF-α
The IL-1β protein levels in CSF were increased 2.3-fold in normothermic ALF rats at the coma stage of encephalopathy (ALF-coma versus sham-operated controls: 5.4 ± 0.6 versus 2.4 ± 0.3 pg/mL, P < 0.001), but were not changed significantly in the 6h (precoma) after-HAL group of animals (Figure 4). IL-6 protein concentrations were increased in normothermic ALF rats 6h after HAL (sham-operated controls versus ALF-6h: 35.8 ± 4.0 versus 60.7 ± 4.8 pg/mL, P <0.05) with further increases at the coma stage of encephalopathy (108.2 ± 9.5 pg/mL, P < 0.001). The TNF-α levels in CSF were not changed significantly in the ALF-6 h group, but were increased significantly at the coma stage of encephalopathy (sham-operated controls versus ALF-coma: 1.1 ± 0.2 versus 2.3 ± 0.2pg/mL, P < 0.001). In hypothermic ALF rats, increases in CSF levels of IL-1β, IL-6, and TNF-α were attenuated by 65% (P < 0.001), 67% (P < 0.001), and 34% (P <0.05), respectively, compared with normothermic ALF rats (Figure 4). The antiinflammatory cytokine, IL-10, was increased fivefold (P < 0.001) in the CSF of normothermic ALF animals, but again this increase was marginally influenced (P >0.05) by hypothermia (data not shown).
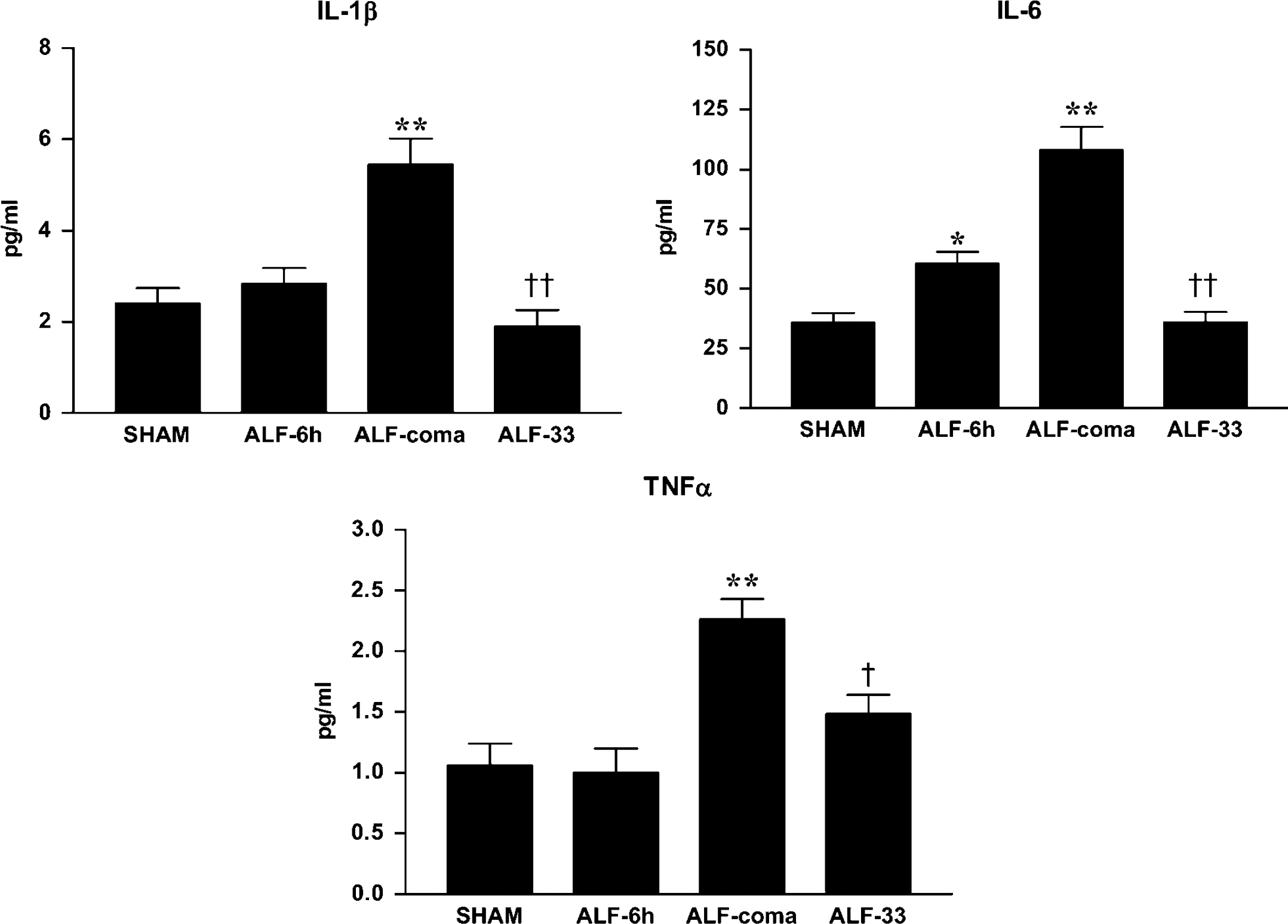
Cerebrospinal fluid IL-1β IL-6, and TNF-± levels in ALF rats maintained normothermic (ALF-6 h, ALF-coma) or hypothermic (ALF-33) compared with sham-operated controls (sham). Data represent mean ± s.e.m. of 10 animals in each group. *P <0.05 versus sham; **P < 0.001 versus sham; †P < 0.01 versus ALF-coma; ‡P < 0.001 versus ALF-coma.
Effects of Hypothermia on IL-1β, IL-6, and TNF-± Gene Expression in ALF Rats
Brain samples from ALF rats were thoroughly perfused transcardially with saline to prevent the influence of circulating cytokines on measurements of brain cytokines. Using quantitative real-time PCR, cytokine mRNA levels were normalized to β-actin mRNA levels. The IL-1β gene expression in the brain was elevated 3.2-fold in normothermic comatose ALF rats (ALF-coma versus sham-operated controls: 324.5 ± 41.3 versus 100 ± 10.2, P < 0.001), but was not changed significantly 6h after HAL (Figure 5). Brain IL-6 mRNA levels were increased 2.1-fold in normothermic ALF rats 6h after HAL (sham-operated controls versus ALF-6 h: 100 ± 13.6 versus 208.6 ± 14.0, P <0.05) with further increases at the coma stage of encephalopathy (388.0 ± 42.0, P < 0.001). TNF-± gene expression in the brain was increased 2.8-fold in normothermic ALF rats at the coma stage (sham-operated controls versus ALF-coma: 100.1 ± 8.8 versus 282.1 ± 26.5, P < 0.001) but was not changed significantly 6h after HAL. In hypothermic ALF rats, increases in brain IL-1β, IL-6, and TNF-± mRNA levels were attenuated by 89%, 47%, and 37%, respectively (P <0.001), compared with that in normothermic ALF rats (Figure 5).
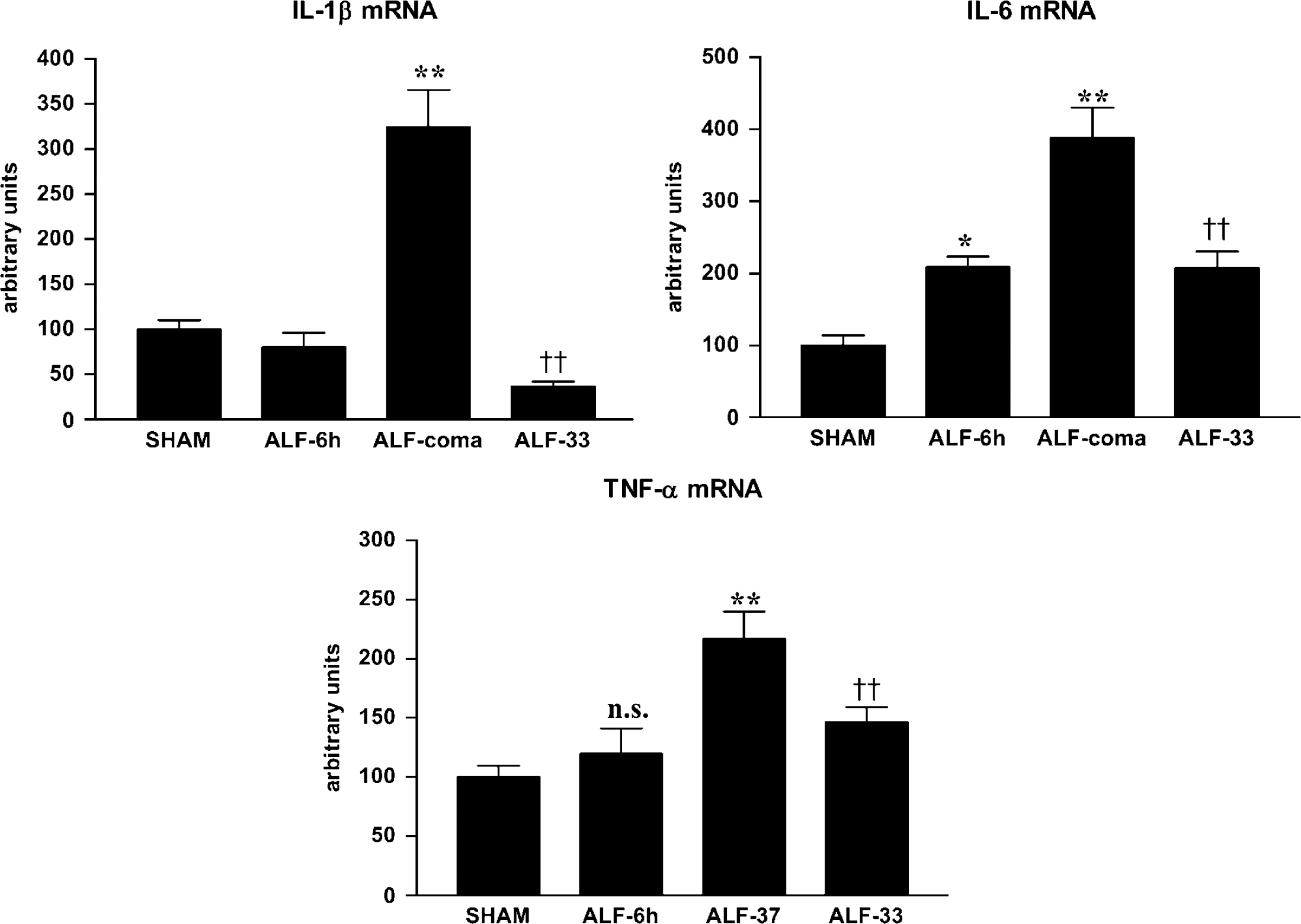
Brain IL-1β IL-6, and TNF-± mRNA levels in ALF rats maintained normothermic (ALF-6 h, ALF-coma) or hypothermic (ALF-33) compared with sham-operated controls (sham). Data represent mean ± s.e.m. of 10 animals in each group. *P <0.05 versus sham; **P < 0.001 versus sham; †P <0.05 versus ALF-coma; ‡P < 0.001 versus ALF-coma; NS: not significantly different from sham.
Discussion
Results of this study provide direct evidence for brain-derived proinflammatory mechanisms in the pathogenesis of the neurologic complications of ALF. This evidence includes activation of microglia, as shown by increased immunoreactivity of the major histocompatibility complex marker, OX-42, together with the increased production of brain proinflammatory cytokines. Increased cytokine transcripts in the hepatic devascularized rat model of ALF were found to be selective in terms of their identity, the magnitude of the increase in their expression, and the timing of the increased expression in relation to the progression of ALF. Early increases in IL-6 mRNA levels were followed by more substantial increases in the expression of IL-1β and TNF-± and occurrence of severe encephalopathy and brain edema was associated with a generalized increase in the expression of all three cytokines. Although astrocytes when activated, are theoretically an alternative source of cytokines, there is no evidence for reactive astrocytosis in this model of ALF (Bélanger et al, 2002). The finding of increased brain proinflammatory cytokines in association with encephalopathy in ALF adds to a growing body of evidence implicating these cytokines in neuropsychiatric disorders. Earlier studies show that expression of TNF-± in the brain results in poor performance in cognitive tasks (Fiore et al, 1996), and that altered expression of TNF-± and IL-1 in the brain leads to impaired sleep quality (Fang et al, 1998); in this regard, it is interesting to note that hepatic encephalopathy is characterized in its early stages by sleep disorders and cognitive deficits.
It may be tempting to speculate that the increased brain cytokines in this study originated from infection. Alternatively, apparent increases in brain cytokines could have originated from blood contamination of the brain tissue. However, neither possibility is likely, because the presence of infection was precluded by rigorous screening for bacteria and other pathogens in all animals before entry into the study. Blood contamination of the brain tissue was prevented by transcardial perfusion of all animals to remove residual blood from the brain before cytokine assays. Serum concentrations of TNF-±, IL-1β and IL-6 were increased significantly in hepatic devascularized rats (this study), and this cytokine profile resembles that reported earlier in rodent models of ALF resulting from acetaminophen (Cover et al, 2006) or azoxymethane (Bémeur et al, 2007) hepatotoxicity, as well as in sera from patients with ALF because of acetaminophen toxicity or viral hepatitis (Sekiyama et al, 1994). In this study, increases in IL-1β IL-6, and TNF-± levels were measured as a function of severity of liver failure and were sensitive to mild hypothermia. Given the similar identity of the increased serum and brain cytokines in this study, it may be tempting to conclude that increased brain cytokines originated from failing liver. There are known routes whereby peripheral cytokines can directly cross the blood-brain barrier (Licinio and Wong, 1997). However, a uniquely peripheral source of cytokines is unlikely, given the present findings of increased expression of genes coding for TNF-±, IL-1β and IL-6 together with the increased expression of their respective proteins in the brain.
The precise mechanism(s) that are responsible for microglial activation in ALF are unknown. However, one potential mechanism involves ammonia toxicity. ALF results in sustained hyperammonemia, and brain ammonia concentrations in the 1 to 5 mmol/L range have been reported (Swain et al, 1992b). Ammonia inhibits ±-ketoglutarate dehydrogenase in rat brain mitochondria (Cooper and Lai, 1987), resulting in decreased glucose oxidation and in lactate accumulation. Increased brain lactate production has been shown unequivocally in experimental ALF (Zwingmann et al, 2003), and CSF lactate levels are positively correlated with the severity of hepatic encephalopathy in both experimental (Therrien et al, 1991) and human (Yao et al, 1987) ALF. Results of this study show that increases in TNF-±, IL-6, and IL-1β levels in the brain follow a time course that is comparable with that of brain ammonia (Swain et al, 1992b), and CSF (Chatauret et al, 2001) and brain (Zwingmann et al, 2003) lactate suggestive of a pathophysiologic link. Moreover, cultured astroglia exposed to lactate in concentrations equivalent to those reported in brain in experimental ALF cause increased production and release of these cytokines (Andersson et al, 2005). Conversely, there is evidence to suggest that TNF-± IL-6, and IL-1β have the capacity to increase permeability of cerebrovascular endothelial cells to ammonia (Duchini et al, 1996; de Vries et al, 1996), suggesting that their presence could increase the blood-brain barrier permeability to ammonia providing an explanation for the disproportionately increased brain ammonia reported in experimental ALF (Swain et al, 1992b).
Mild hypothermia is increasingly being considered as a useful tool to prevent and treat intracranial hypertension in ALF (Nagaki et al, 2000; Vaquero and Butterworth, 2007; Jalan et al, 2004). Several mechanisms by which hypothermia exerts its beneficial effects have been proposed. Such mechanisms include improvement in hepatic function (Vaquero et al, 2007), improvement of brain energy metabolism (Chatauret et al, 2001, 2003; Zwingmann et al, 2004), and effects on the expression of genes coding for oxidative stress proteins (Sawara et al, 2005). Results of this study suggest an additional mechanism, namely reduction of brain proinflammatory cytokines. Further studies are needed to determine whether mild hypothermia is beneficial when instigated after the onset of ALF. Approaches aimed at the reduction of brain levels of proinflammatory cytokines have the potential to limit the cerebral consequences of ALF.
Footnotes
The authors state no conflict of interest.