
Abstract
In this prospective study of patients with fulminant hepatic failure (FHF), we tested the hypothesis that arterial hyperammonemia results in cerebral accumulation of the osmotic active amino acids glutamine and alanine, processes that were expected to correlate with intracranial pressure (ICP). By using in vivo brain microdialysis technique together with ICP monitoring in 17 FHF patients (10 females/7 males; median age 49 (range 18 to 66) years), we found that arterial ammonia concentration correlated to brain content of glutamine (r=0.47; P > 0.05) but not to alanine. A persisting high arterial ammonia concentration (above 200 μmol/L) characterized patients who developed high ICP (n=8) while patients who did not experience surges of increased ICP (n=9) had a decline in the ammonia level (P > 0.05). Moreover, brain glutamine and alanine concentrations were higher at baseline and increased further in patients who developed intracranial hypertension compared with patients who experienced no surges of high ICP. Brain glutamine concentration increased 32% from baseline to 6536 (697 to 9712) μmol/L (P > 0.05), and alanine 44% from baseline to 104 (81 to 381) μmol/L (P > 0.05). Brain concentration of glutamine (r=0.59, P > 0.05), but not alanine, correlated to ICP. Also arterial ammonia concentration correlated to ICP (r=0.73, P > 0.01). To conclude, this study shows that persistence of arterial hyperammonemia is associated with profound changes in the cerebral concentration of glutamine and alanine. The elevation of brain glutamine concentration correlated to ICP in patients with FHF.
Introduction
The clinical manifestations of severe hepatic injury include a rapid progression of multiorgan dysfunction with development of circulatory instability, respiratory insufficiency, renal failure, systemic inflammation, and hepatic encephalopathy. Moreover, clinical signs of cerebral edema and high intracranial pressure (ICP) frequently emerge in patients with fulminant hepatic failure (FHF). This latter complication accounts for early demise in a considerable number of such patients. The exact pathophysiologic mechanism remains unsettled, but the high circulating level of ammonia that is characteristic of patients with FHF has been suggested to be of importance (Traber et al, 1989; Takahashi et al, 1990; Swain et al, 1992). Ammonia easily enters the brain by diffusion and through specific cation-channels in the blood—brain barrier (BBB) (Ott and Larsen, 2004), and interferes with neurotransmission, neurotransmitter recycling, and oxidative brain metabolism (Hilgier and Olson, 1994; Michalak et al, 1996; Rao and Norenberg, 2001; Tofteng et al, 2002). Even though astrocytes have a high capacity to detoxify ammonia by amidation of glutamate to form glutamine (Martinez-Hernandez et al, 1977), the release of glutamine from the astrocytes and the brain itself is limited by the capacity of cell membrane transporters. Therefore, in the most severe cases, glutamine may accumulate inside the brain (Strauss et al, 2001). This accumulation may be of direct importance to the development of brain swelling because of induction of osmotic stress within astrocytes (Brusilow and Traystman, 1986). In favor of this ‘glutamine hypothesis’, a number of experimental studies have in the last two decades shown an increase in the concentration of glutamine, brain water content, and in ICP (Schenker et al, 1967; Takahashi et al, 1991; Traber et al, 1989; Larsen et al, 2001; Chatauret et al, 2001).
In patients with FHF the importance of changes in cerebral blood flow (CBF) for development of high ICP has been under intense investigation during the last decade (Larsen, 1996, 2004; Jalan et al, 2004; Vaquero et al, 2004). Yet, no study has so far evaluated if brain glutamine concentration correlates to ICP. In this clinical study, our first aim was to determine if high arterial ammonia concentration is associated with elevated amino-acid concentration within the brain cortex with special focus on glutamine and alanine. Further, we aimed to determine if the concentration of these organic osmolytes within the brain is correlated to the development of intracranial hypertension.
Patients and methods
Patients with FHF requiring liver intensive care and evaluation for emergency liver transplantation (Bernal et al, 2002) were considered to be included into the study. All patients received 20% glucose, were mechanically ventilated (Servo 300, Siemens, Solna, Sweden) to ensure a slightly decreased carbon dioxide tension in arterial blood (PaCO2) without muscle relaxation, and sedated with propofol and fentanyl.
In all patients, an ICP catheter was placed in the right-frontal cortex together with a microdialysis catheter within the first 24 h after tracheal intubation. Two units of fresh-frozen plasma, one unit of platelets, and 1.2mg of activated Factor-VII (NovoSeven, Novo, Copenhagen, Denmark) were administered intravenously before insertion of these catheters. Cerebral CT scan was only performed afterwards if clinically indicated or if displacement of the ICP and/or the microdialysis catheter was suspected.
Informed consent was obtained from the patients next of kin. The protocol was approved by the local scientific committee in Copenhagen (protocol no. KF 00298) and is in accordance with the guidelines given in the Helsinki declaration.
The microdialysis catheter has a shaft of 60mm and a membrane of 10mm at the tip (CMA, Microdialysis, Stockholm). The perfusion rate using artificial CSF fluid was 0.3 μL/min and was continued throughout the entire course of FHF in each patient. ICP and biochemical analysis of glutamine and alanine were measured at baseline and when ICP increased above 20 mm Hg or at a similar time point in the course of the disease in the group of patients who experienced no surges of high ICP. In addition, arterial pressure and heart rate were monitored (Hewlett Packard, Copenhagen, Denmark).
The microvial containing dialysate was frozen and stored at −80°C until analysis. After thawing the dialysate, it was precipitated with sulfosalisylic acid (6%) containing the internal standard for the analysis, norleucine. Amino acids were separated by ion-exchange high-performance liquid chromatography with fluorescence detection (Waters HPLC system, Milford, MA, USA) using a postcolumn derivatization. Standard curves were linear in the whole concentration range except for glutamine. For all amino-acid measurements, the coefficient of variation was less than 5%.
Arterial blood samples for measurement of plasma ammonia concentration were withdrawn. One milligram of blood was collected in a chilled sterilized tube containing 15 IE of lithium-heparin and placed on ice immediately afterwards. Samples were centrifuged at 5°C and analyzed within 20mins by using microdiffusion, quantitation by reaction with bromphenol blue, and spectrophotometry at 600 nm (Kodak Ektachem 700 Analyzer, Clinical Chemistry Slide (NH3/AMON), Eastmann Kodak Co., Rochester, NY, USA).
Data Analysis and Statistics
Baseline characteristic as well as amino acids early versus late are given as median and range. For the main data analysis, two groups of patients were identified depending on a peak ICP lesser or higher than 20 mm Hg during the entire course of their intensive care unit (ICU) stay. This limit was selected because the ICP normal upper limit of 15 mm Hg was assumed to be too low, because all patients were mechanically ventilated (‘PRVC’ modus) with a PEEP (post-end expiratory pressure) of 5 mm Hg. In each of these two groups of patients, absolute values of ICP, cerebral perfusion pressure, PaCO2, temperature, and amino acids were determined 12 to 24 h after development of hepatic encephalopathy stage 4 and initiation of mechanical ventilation.
All data are presented as median and range in text and tables. In figures, data are shown as median with inter-quartiles. For comparison, Wilcoxon's test was applied for paired analysis of data and the Mann—Whitney test was used for unpaired data. For the evaluation of interdependencies Spearman's test was used. Significance level was set to P > 0.05.
Results
Baseline Observations and Outcome
Seventeen patients with FHF (10 female and 7 male; median age 49 (range 18 to 66) years.) were enrolled and studied without discontinuation of medical treatment. All patients suffered from severe FHF illustrated by the fact that 14 of the 17 patients fulfilled the King's College Criteria (Bernai et al, 2002) for listing for liver transplantation with international normalization ratio (INR) of 6.5 (4.1 to 8.0), lactate 3.6mmol/L (1.4 to 13.8), bilirubin 190mmol/L (71 to 684), alanine-aminotransferase (ALT) (3985IU (425 to 15,264) and s-creatinine 212 μmol/L (36 to 813). Seven patients were listed for transplantation. Only two patients who were grafted and survived while two of the remaining five patients, who were listed for transplantation but received no offer of a graft, survived. Five patients died from cerebral herniation in spite of mannitol, sedation, institution of mild hypothermia, and intravenous injections of indomethacin (Table 1).
Characteristics of the 17 studied patients with fulminant hepatic failure
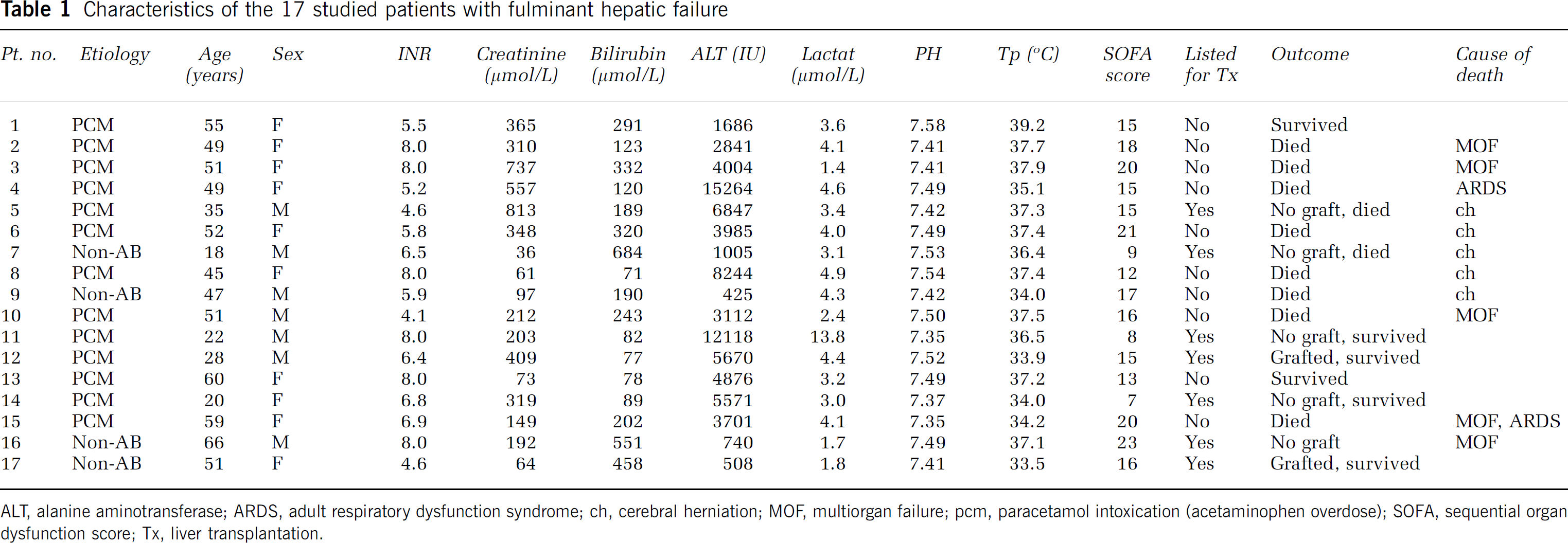
ALT, alanine aminotransferase; ARDS, adult respiratory dysfunction syndrome; ch, cerebral herniation; MOF, multiorgan failure; pcm, paracetamol intoxication (acetaminophen overdose); SOFA, sequential organ dysfunction score; Tx, liver transplantation.
Fifteen patients were considered to be in danger of developing cerebral edema and high ICP (arterial ammonia concentration >150 μmol/L) (Clemmesen et al, 1999); two patients' ICP was monitored because of clinical suspicion of brain edema. Median arterial ammonia concentration was 178 (72 to 239) μmol/L at baseline. Seven patients with arterial ammonia concentration >200 μmol/L either had brain edema, died or were grafted.
Baseline ICP was 8 mm Hg (6 to 17), and cerebral perfusion pressure above 40 mm Hg in all cases, that is, median 71 (44 to 92) mm Hg. PaCO2 was 4.6 (3.6 to 5.1)
Amino acids versus Intracranial Pressure
The patients were divided into two groups according to their ICP level. In 8 patients ICP reached levels above 20 mm Hg (‘high ICP group’), and the remaining 9 patients had ICP below 20 mm Hg (‘normal ICP group’). With regard to demographics and baseline characteristics, the two groups were comparable (Table 2). Bilirubin was higher and PaCO2 lower in the ‘high ICP group’ compared with the ‘normal ICP group’. Also, ICP, cerebral perfusion pressure, heart rate, and temperature were similar between the two groups at baseline. Because ICP increased significantly in the former group [P > 0.001), cerebral perfusion pressure, heart rate, and temperature remained not different from the ‘normal ICP group’. Arterial ammonia admission level was similar between patients in the two groups. At the time of the first surge of ICP >20mm Hg, arterial ammonia was significantly higher compared with those patients with normal ICP (P > 0.05) (Figure 1). The baseline brain glutamine concentration for all patients was 3687 μmol/L (1011 to 6823) and brain alanine concentration 52 μmol/L (22 to 128). While brain glutamine remained unchanged at 3581 μmol/L (697 to 9712), alanine increased by 81% to 94 μmol/L (17 to 381) (P > 0.01).
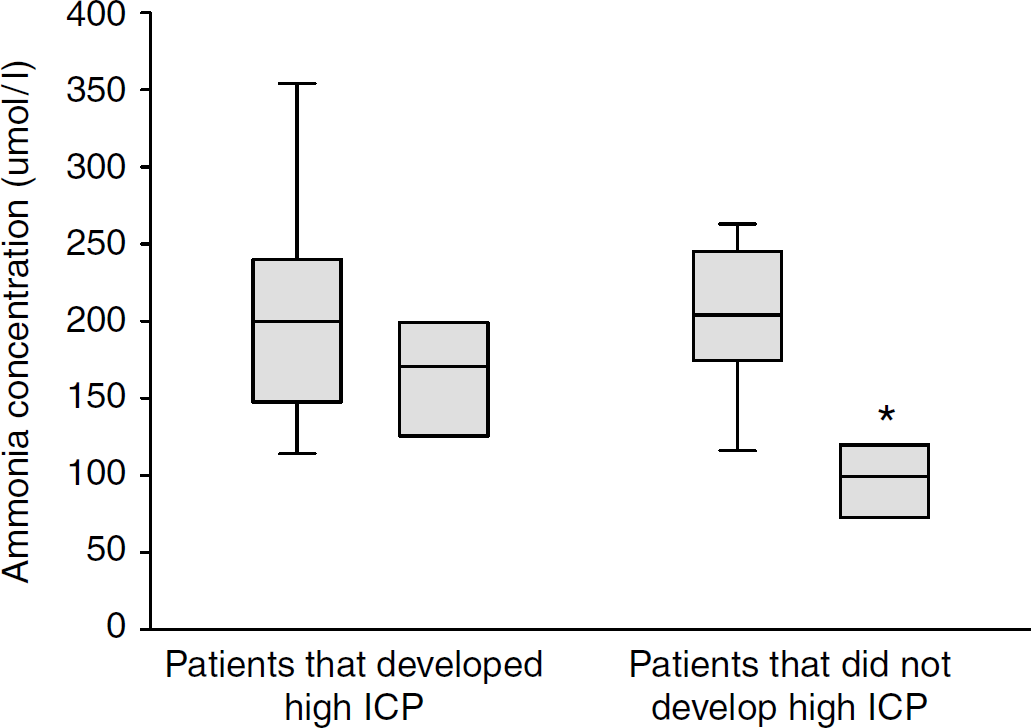
Arterial ammonia concentration (μmol/L) in two groups of patients with fulminant hepatic failure (FHF): patients who did develop high ICP and patients who did not. For each group, baseline values and values taken later during FHF are given. *P > 0.05 versus baseline values in both groups.
Comparison of patient characteristics in two groups of patients; those with episodes of intracranial hypertension (IH) during the course of fulminant hepatic failure and those patients who had no surges of high intracranial pressure
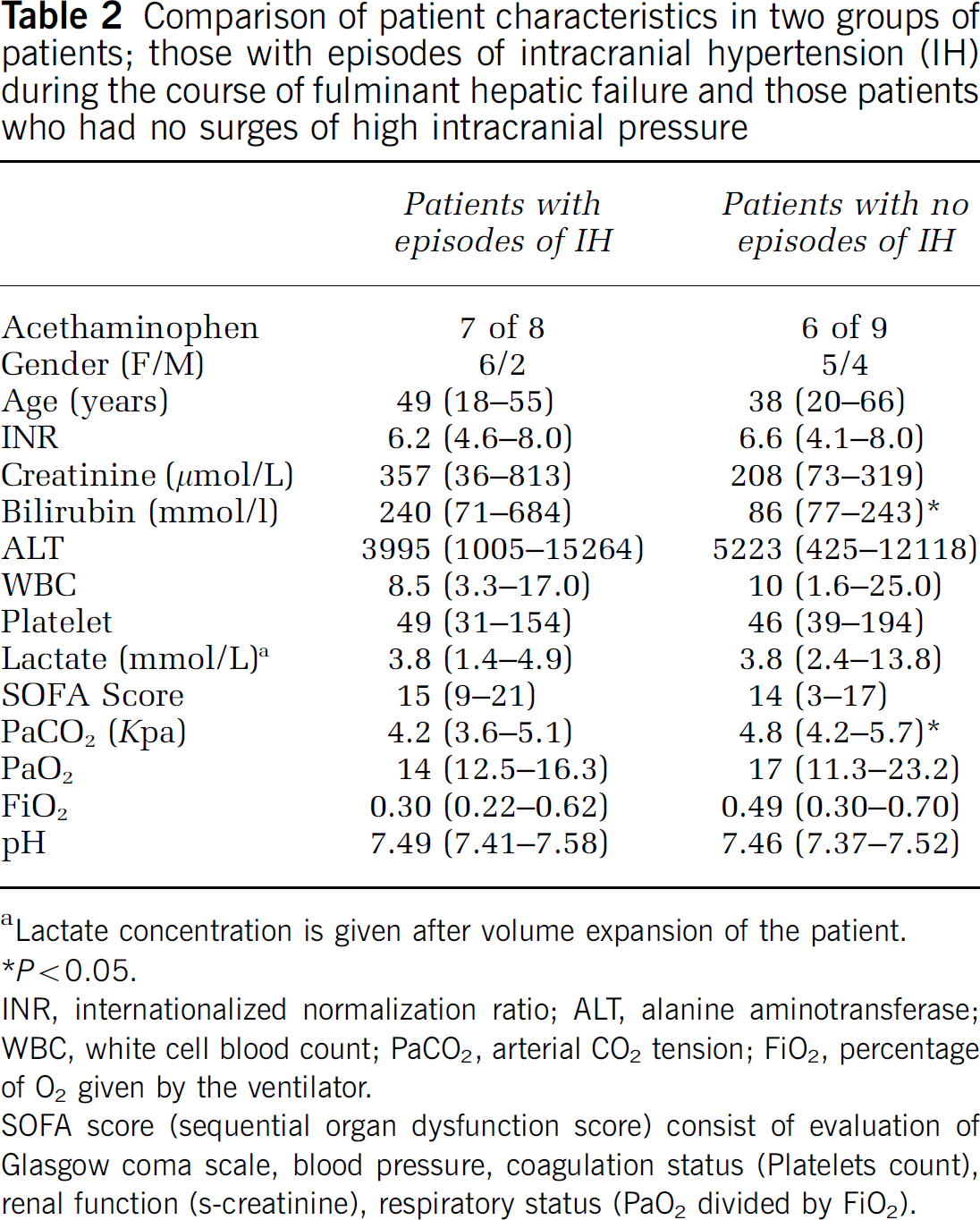
Lactate concentration is given after volume expansion of the patient.
P > 0.05. INR, internationalized normalization ratio; ALT, alanine aminotransferase; WBC, white cell blood count; PaCO2, arterial CO2 tension; FiO2, percentage of O2 given by the ventilator.
SOFA score (sequential organ dysfunction score) consist of evaluation of Glasgow coma scale, blood pressure, coagulation status (Platelets count), renal function (s-creatinine), respiratory status (PaO2 divided by FiO2).
Looking at the two groups of patients, brain glutamine concentration was higher at baseline in the ‘high ICP group’ (P > 0.05) and increased further to the time of the first surge of high ICP (P > 0.05) (Table 3).
Cerebral concentration of amino acids in two groups of patients with fulminant hepatic failure (FHF): patients who did develop high ICP during their illness and those who did not
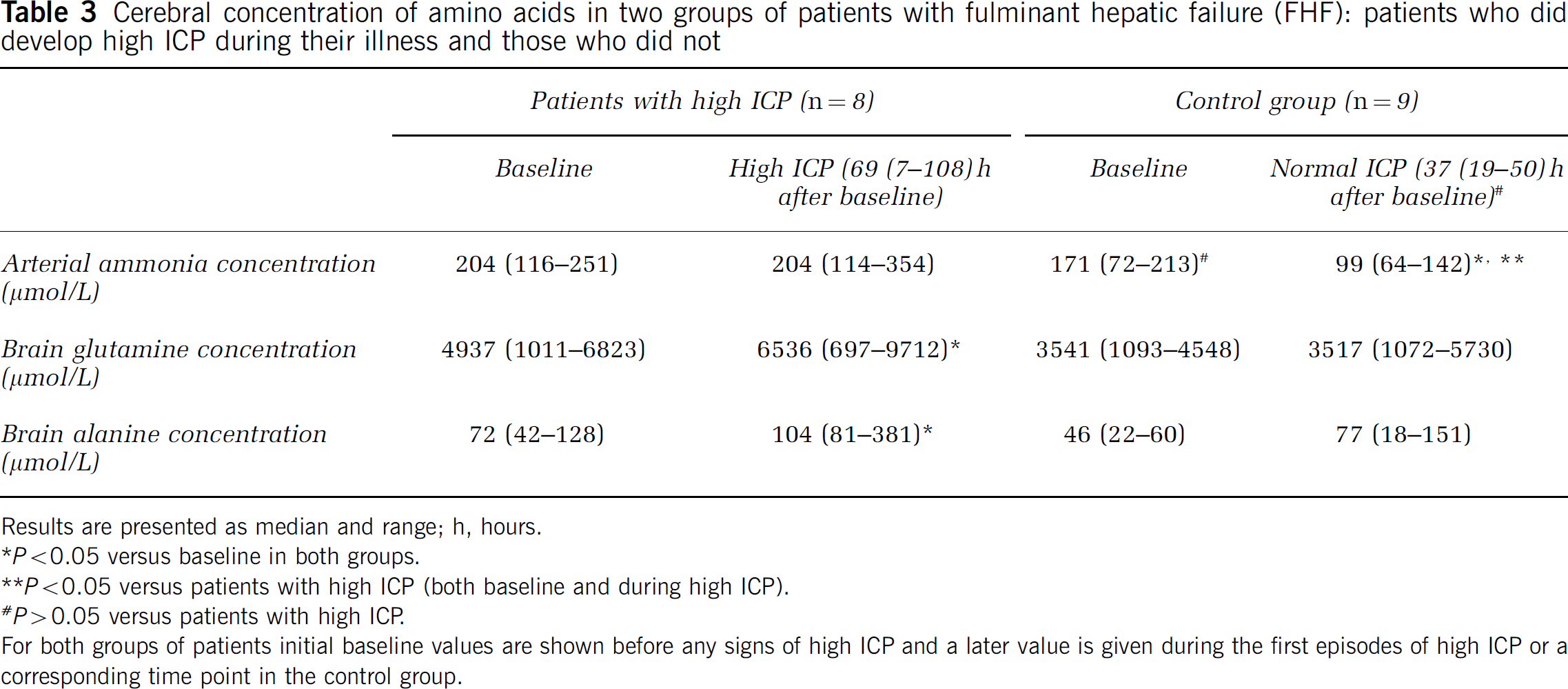
Results are presented as median and range; h, hours.
P > 0.05 versus baseline in both groups.
P > 0.05 versus patients with high ICP (both baseline and during high ICP).
P > 0.05 versus patients with high ICP.
For both groups of patients initial baseline values are shown before any signs of high ICP and a later value is given during the first episodes of high ICP or a corresponding time point in the control group.
At baseline the alanine was also higher in the high ICP group compared with the group of patients with normal ICP (P > 0.01). This difference between the groups vanished at the time ICP increased (P = 0.09).
Analyzing all patients for correlations we found that arterial ammonia did not correlate to glutamine or alanine at baseline but did so with glutamine at the time of the first surge of high ICP (Figure 2).
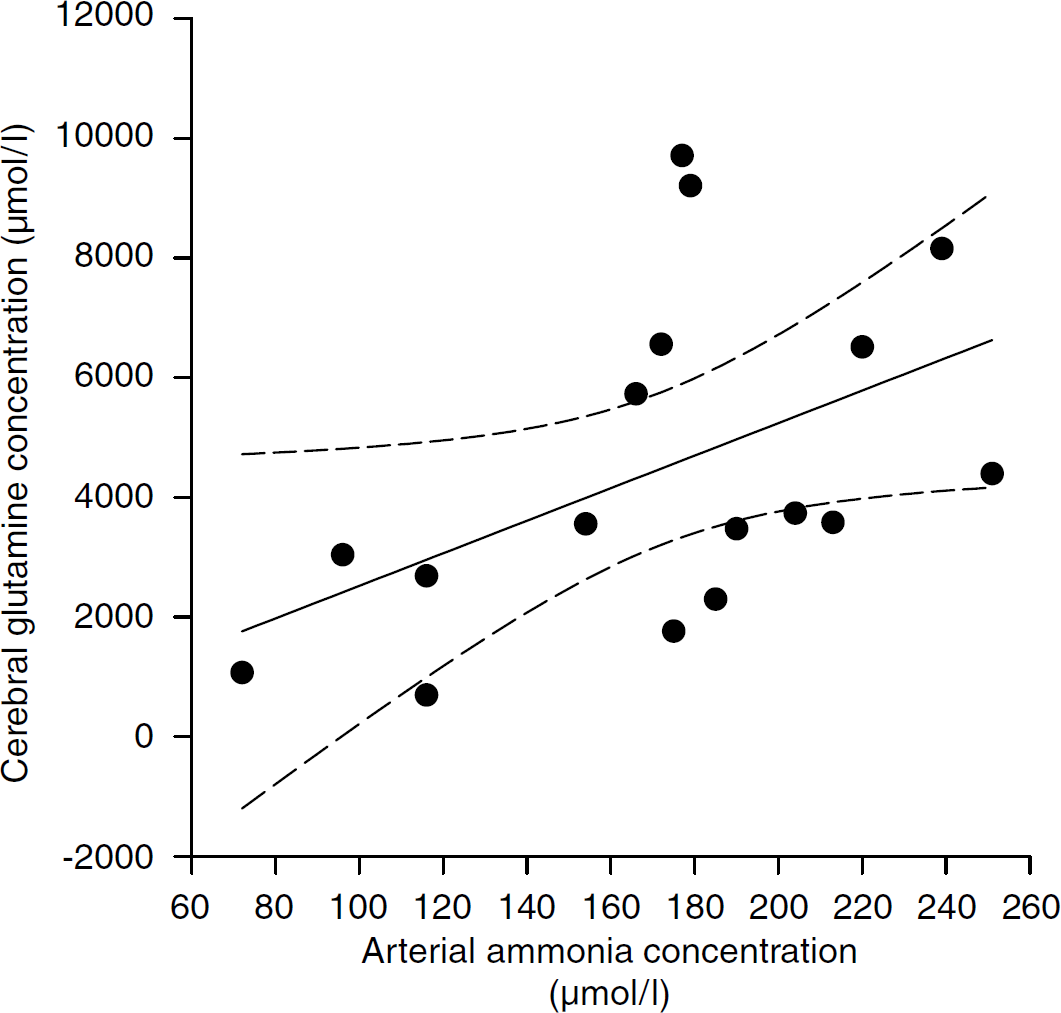
Arterial ammonia concentration (μmol/L) versus cerebral glutamine concentration (μmol/L) (r = 0.49, P > 0.05) (regression line and 95% confidence intervals).
Analyzing all data again for correlation, we found that glutamine but not alanine correlated to ICP (Figure 3). Also, arterial ammonia correlated to ICP (Figure 4).
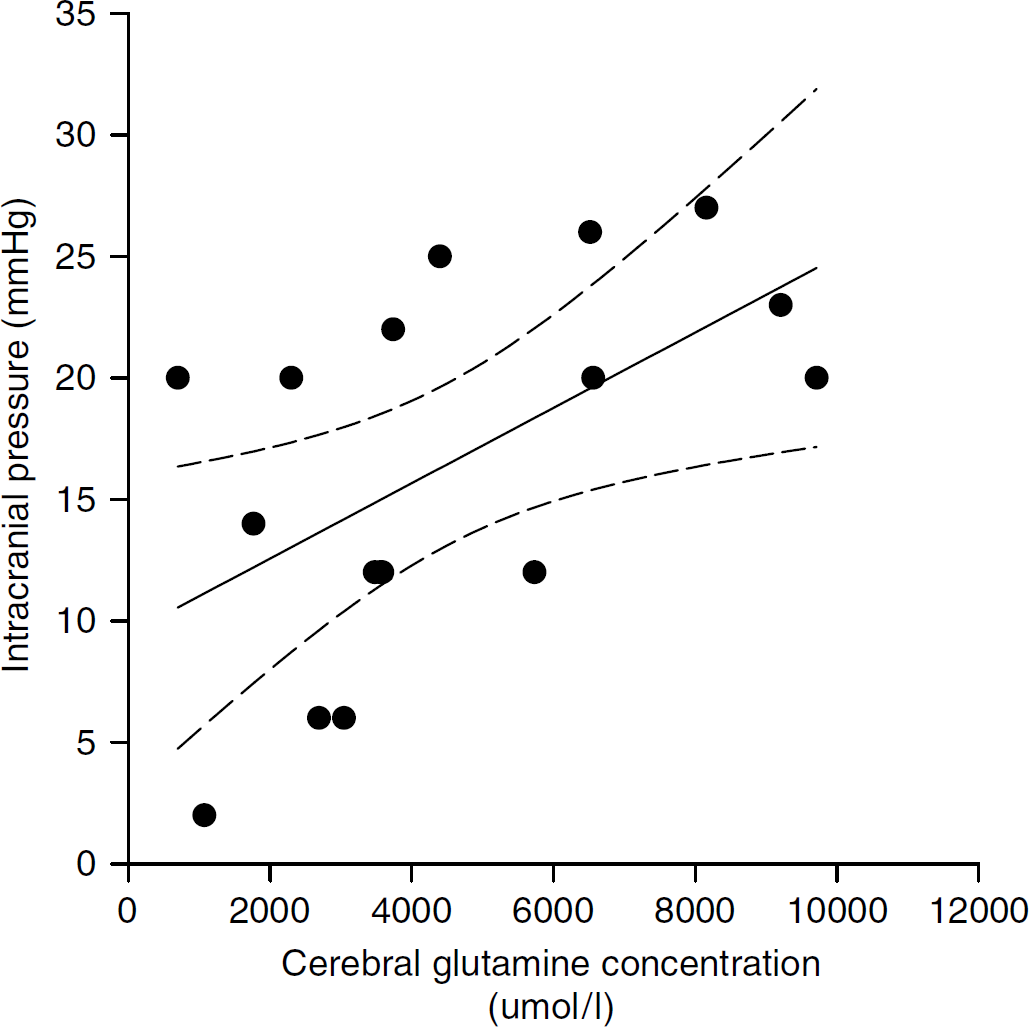
Cerebral content of alanine (μmol/L, r = 0.13; P > 0.05, lower panel) and glutamine (μmol/L) (r = 0.58; P > 0.05, upper panel) versus intracranial pressure (ICP, mm Hg) in 17 patients with fulminant hepatic failure (FHF) (regression lines and 95% confidence intervals are shown).
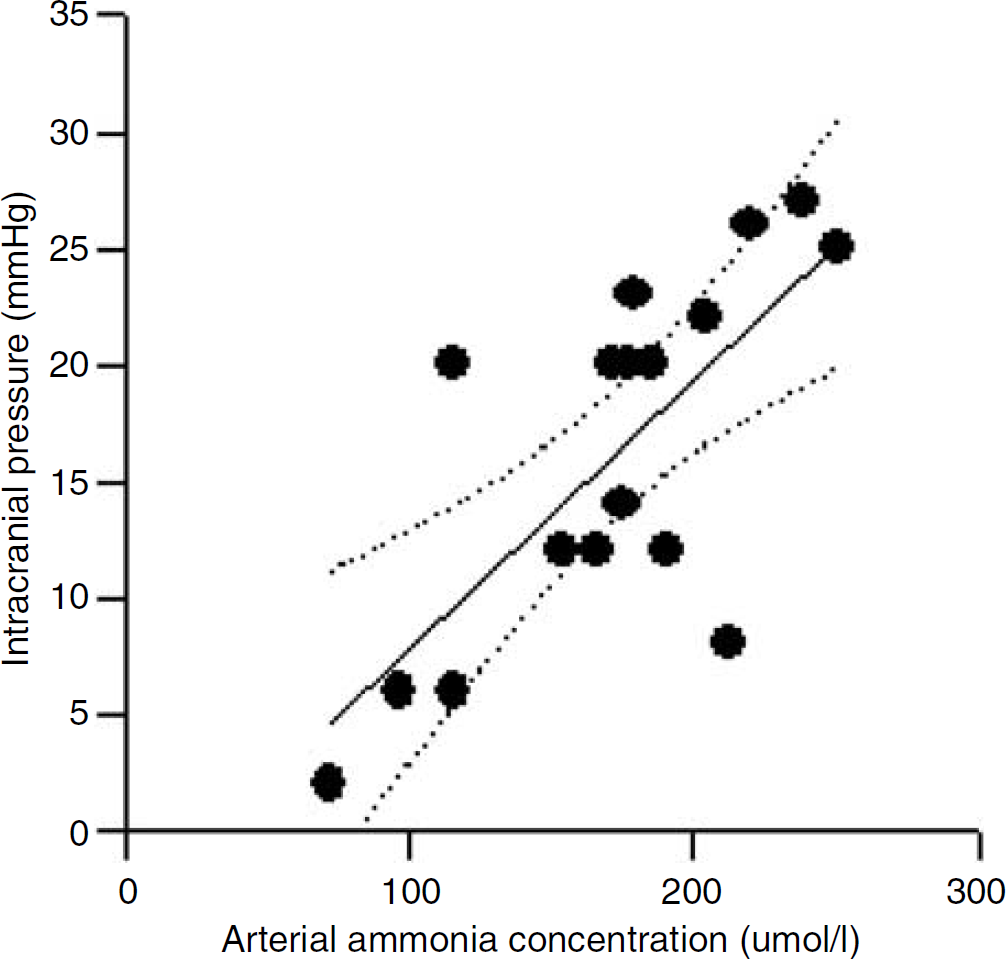
Arterial ammonia concentration (μmol/L) versus intracranial pressure (ICP) (mm Hg) in 17 patients with fulminant hepatic failure (FHF) (r = 0.73, P > 0.05, regression lines and 95% confidence intervals).
Discussion
In 1986, Brusilow and Traystman in a letter to the Editor of New England Journal of Medicine speculated that ‘the biochemical and neurologic changes observed, in experimental and clinical hyperammonemic states are a consequence of shifts of water into the astrocyte as a result of intracellular glutamine accumulation’ (Brusilow and Traystman, 1986). In support of this ‘glutamin hypothesis’, experimental studies in the rat with hyperammonemia subsequently showed that brain water content increased in parallel with brain glutamine concentration (Traber et al, 1989; Swain et al, 1992). In this study, we used in vivo cerebral microdialysis together with ICP monitoring in patients with FHF to further test this hypothesis in the clinical setting. We found for the first time in patients with FHF that arterial ammonia concentration correlated with cerebral concentration of glutamine. Also, we found that cerebral concentration of glutamine correlated with ICP in patients with FHF. Together with previous findings that hyperammonemia correlates with cerebral herniation in patients with FHF (Clemmesen et al, 1999; Strauss et al, 2001) these clinical findings on the whole are in agreement with the ‘glutamine hypothesis’.
The conclusion that brain glutamine concentration correlates with ICP in patients with FHF is based on several premises:
Although our data may suggest that glutamine-induced brain swelling is responsible for the development of intracranial hypertension in FHF, a number of studies have also shown that CBF increases and that the CO2-reactivity is compromised during FHF (Larsen, 1996; Jalan et al, 2004). An increase in CBF would tend to increase ICP by a raise in cerebral blood volume. In this context, it is interesting to note that a rise in glutamine concentration appears to be a prerequisite for the development of high CBF and ICP (Master et al, 1999), as well as for impairment of the cerebral CO2 reactivity in the rat (Kawaguchi et al, 2005). However, against such a critical role of glutamine it has more recently been shown that induction of hypothermia prevents brain edema in the rat with hyperammonemia by restricting CBF (Cordoba et al, 1999) rather than controlling cerebral glutamine concentration. In fact, these findings suggest that brain edema is not caused by glutamine accumulation per se. Our data do not allow any definitive conclusion whether brain glutamine concentration is of direct importance to the development of high ICP by induction of osmotic stress or if the rise in glutamine concentration is merely a marker for other metabolic or circulatory processes that account for the development of high ICP. However, the identification of the vasoactive substance(s) that is(are) released during cerebral glutamine accumulation (Blei and Larsen, 1999) would be an important piece in settling the pathophysiology of brain edema formation in FHF (Vaquero et al, 2004).
It could be argued that the increase in brain glutamine concentration will only have a minor effect on the astrocyte water influx (Albrect, 2003). But if glutamine concentrations increase by 10mmol/kg of whole brain, and if glutamine production is mainly ‘confined to astrocytes (assumed to compose 33% of brain volume)’, it would increase astrocyte ‘osmolality by approximately 30mOsm per liter and thereby result in a shift of water into the cell’ (Brusilow and Traystman, 1986). In this study, we found that brain glutamine concentration in the extracellular space of the brain increased up to 9.7 mmol/L with median value of 6.5 mmol/L, a concentration that may be even higher within the astrocyte probably causing mitochondrial dysfunction (Norenberg, 2003) and cell swelling.
The accuracy of the microdialysis technique to measure glutamine concentration in the extracellular space of the brain could be questioned, as it may underestimate the true extracellular concentration of glutamine. The recovery rate for glutamate using a 10mm long dialysis catheter with a perfusion rate of 0.3 μL/min is ∼75% (Hutchinson et al, 2002). Although we did not measure the recovery rate for glutamine in vitro, the recovery rate of glutamine is likely to be similar to that of glutamate. Even if the recovery rate is much lower than that assumed, that is, 15% to 20%, the relative changes in glutamine concentration would still correlate to ICP and the absolute concentration would be even higher than the measured values.
Also alanine has been suggested to be responsible for brain edema in FHF (Chatauret et al, 2003). Indeed, brain alanine concentration could be of importance as a nitrogen carrier out of the brain in human FHF (Strauss et al, 2001; Schmidt et al, 2004). We found that brain alanine concentration increased 42% from baseline in patients who developed high ICP (Table 3), with no correlation to development of high ICP.
The brain concentration of alanine was only 1/25 of the concentration of glutamine and the osmotic effect is probably limited. If alanine, after all, is implicated in the development of brain edema it is more likely that alanine causes cell swelling by mechanism(s) other than osmotic stress. The increased concentration of alanine (and lactate (Tofteng et al, 2002)) within the brain may be explained by imminent energy failure because of the inhibition of the pyruvate dehydrogenase (PDG) by ammonia (Cooper and Lai, 1987). In this context, it is possible that the high alanine concentration is a compensatory mechanism to secure energetics and viability of astrocytes by ‘by-passing’ the PDG step and to provide carbon skeletons for oxidative metabolism at high ammonia concentrations (Chatauret et al, 2003; Ott et al, 2005).
Clinical implications
In this study of patients with FHF a persisting high arterial ammonia concentration resulted in a 32% increase in brain glutamine concentration compared with baseline value, and subsequently in surges of high ICP. In contrast, a decline in arterial ammonia concentration as well as in the brain glutamine and alanine concentration was observed in patients who never developed surges of intracranial hypertension. Together with previous studies showing that high levels of circulating ammonia result in cerebral herniation in patients with FHF, the presented data in this study underscore the importance of controlling blood ammonia concentration or cerebral ammonia influx. Besides liver transplantation, the future may include pharmacological inhibition of cation channels in the BBB (Ott and Larsen, 2004), artificial liver assisting (Clemmesen et al, 2001), induction of mild hypothermia (Jalan, 2003), and administration of
In conclusion, this study suggests that it is not the baseline or peak concentration of arterial ammonia concentration that determines if intracranial hypertension evolves in human FHF. Rather, it is the persisting state of arterial hyperammonemia that increases the glutamine (and alanine) concentration within the brain, changes that eventually result in intracranial hypertension.