
Abstract
Neurovascular remodeling has been recently recognized as a promising target for neurologic therapies. Hopes have emerged that, by stimulating vessel growth, it may be possible to stabilize brain perfusion, and at the same time promote neuronal survival, brain plasticity, and neurologic recovery. In this review, we outline the role of vascular endothelial growth factor (VEGF) in the ischemic brain, analyzing how this growth factor contributes to brain remodeling. Studies with therapeutic VEGF administration resulted in quite variable results depending on the route and time point of delivery. Local VEGF administration consistently enhanced neurologic recovery, whereas acute intravenous delivery exacerbated brain infarcts due to enhanced brain edema. Future studies should answer the following questions: (1) whether increased vessel density translates into improvements in blood flow in the hemodynamically compromised brain; (2) how VEGF influences brain plasticity and contributes to motor and nonmotor recovery; (3) what are the actions of VEGF not only in young animals with preserved vasculature, on which previous studies have been conducted, but also in aged animals and in animals with preexisting atherosclerosis; and (4) whether the effects of VEGF can be mimicked by pharmacological compounds or by cell-based therapies. Only on the basis of such information can more definite conclusions be made with regard to whether the translation of therapeutic angiogenesis into clinics is promising.
Introduction
The promotion of vascular remodeling has been recently recognized as a particularly promising therapeutic strategy in brain illnesses (Greenberg and Jin, 2005; Chen and Chopp, 2006; del Zoppo and Mabuchi, 2003; Hansen et al, 2008). Hopes have emerged that by stimulating the growth of new vessels, it may be possible to stabilize brain perfusion, and at the same time promote neuronal survival, plasticity, and functional recovery, once a stroke has occurred (Chen and Chopp, 2006; del Zoppo and Mabuchi, 2003).
Indeed, experimental studies in the past few years have pointed out that tissue remodeling after stroke is a highly dynamic process, which involves close and finely tuned interactions between neuronal, glial, and vascular cells. On injury, these cells release various trophic factors through which they mutually influence each other, thus creating a permissive environment in which successful neurologic recovery may take place (Chen and Chopp, 2006; Chopp et al, 2008).
Several angiogenic factors have been investigated in experimental studies in recent years, analyzing whether functional recovery may be therapeutically enhanced by these molecules. Among these, at least four molecules, namely vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), granulocyte-colony stimulating factor (G-CSF), and erythropoietin (Epo), have entered preclinical studies in humans (Hendel et al, 2000; Ehrenreich et al, 2002; Ren and Finklestein, 2005; Schäbitz and Schneider, 2007).
Vascular endothelial growth factor promotes angiogenesis in a particularly potent manner (Ferrara et al, 2003; Rosenstein and Krum, 2004; Yasuhara et al, 2004). As such, VEGF is an interesting model molecule, from which observations on brain–vessel interactions might be generalized to other growth factors. This review summarizes the existing literature on VEGF in the ischemic brain, analyzing how this growth factor stimulates neurologic recovery, at the same time evaluating whether and perhaps how VEGF, similar to other angiogenic factors, may be used therapeutically. This review claims that, in view of their potential side effects, particularly careful animal studies are required when pleiotropic growth factors are considered for neurologic therapies.
Neurovascular unit as a therapeutic target in stroke
Brain neurons and astrocytes critically depend on the integrity of a functional capillary system that supplies oxygen and energy-rich substrates to the tissue (Han and Suk, 2005). To safeguard this continuous supply, neurons and astrocytes release various angiogenic factors, such as VEGF, angiopoietins, bFGF, G-CSF, Epo, transforming growth factor-β, and insulin-like growth factor-1 (Jain and Munn, 2000; Zhang and Chopp, 2002; Park et al, 2003), which together with guidance molecules, namely semaphorins, ephrins, netrins, and their receptors (Adams and Klein, 2000; Carmeliet, 2003), induce endothelial proliferation, directed vessel growth, and formation of a functional vascular network.
Similar to neurons and astrocytes, endothelial cells also release growth factors, such as brain-derived neurotrophic factor, nerve growth factor, and neurotrophins, which promote the viability of neurons, providing a sustained protection under conditions of oxygen deprivation (Carmeliet, 2003; Hennigan et al, 2007; Lok et al, 2007). At the same time, through an interaction with the receptors of these growth factors, namely Tropomyosin receptor kinase A (TrkA), Tropomyosin receptor kinase B (TrkB), and p75 neurotrophin receptor (p75NTR) (Hennigan et al, 2007), these growth factors promote reorganization processes in the brain, facilitating long-term potentiation and synaptic plasticity (Bliss and Collingridge, 1993) and promoting axon outgrowth through an interaction with guidance cues, such as semaphorins, ephrins, slits, and netrins (Bagri and Tessier-Lavigne, 2002; Dickson, 2002; Carmeliet, 2003).
The mutual interactions between endothelial cells, neurons, and astrocytes have led to the concept of the neurovascular unit (Hawkins and Davis, 2005; del Zoppo and Mabuchi, 2003; Lok et al, 2007), upon which these cells should not be regarded as separate entities but as integrated networks that need to respond to injuries in a coordinated, synergistic manner so that recovery becomes possible (Chopp et al, 2008). This idea has consequences for therapeutic concepts, which should consider that treatments targeting this integrated unit may achieve more successful results than treatments targeting single cell types (Lok et al, 2007). Indeed, the physiologic interactions between neurons and vessels provide insights into endogenous protective responses that the brain itself activates once a stroke occurs. This raises hopes that by strengthening these mechanisms, it may be possible to promote recovery for therapeutic purposes.
The vascular endothelial growth factor family
Vascular endothelial growth factor is a pivotal regulator in angiogenesis that is markedly increased in ischemic brain tissues. As such, the growth factor well exemplifies how recovery processes are initiated and regulated in the stroke brain. In humans, the VEGF family consists of five homodimeric members (VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor), among which VEGF-A stimulates angiogenesis in a particularly powerful manner (Olsson et al, 2006; Hansen et al, 2008). Vascular endothelial growth factor-A (in the following referred to as VEGF) exists in at least six isoforms (VEGF121, VEGF145, VEGF165, VEGF183, VEGF189, and VEGF206), which differ in their binding affinity to VEGF receptors.
Vascular endothelial growth factor is ubiquitously expressed in the brain under physiologic conditions, mainly by choroid plexus epithelial cells, but also by astrocytes and microglia (Monacci et al, 1993; Marti and Risau, 1998). In brain hypoxia and ischemia, VEGF expression is rapidly increased within hours in a hypoxia-inducible factor (HIF)-1 and HIF-2-dependent manner (Forsythe et al, 1996; Marti and Risau, 1998; Abumiya et al, 1999; Lennmyr et al, 2005), and VEGF remains elevated for as long as 1 month after stroke (Hai et al, 2003; Wang et al, 2004). As such, VEGF is not only upregulated on glial cells (Marti and Risau, 1998; Abumiya et al, 1999) but is also induced on neurons (Marti and Risau, 1998), microvascular smooth muscle cells (Brogi et al, 1994; Abumiya et al, 1999), pericytes (Yonekura et al, 1999), and leukocytes (Abumiya et al, 1999). After stroke, VEGF expression is not only restricted to ischemic areas but is also found in remote cortex regions (Stowe et al, 2007). Hence, additional mechanisms besides hypoxia might be involved in the regulation of this growth factor.
Role of vascular endothelial growth factor in postischemic angiogenesis
In the process of angiogenesis, loss of vascular integrity and degradation of cell matrix are crucial initiating steps. The dilation of ischemic vessels that is evoked by the release of nitric oxide (NO) (Distler et al, 2003) is rapidly followed by the loss of tight junction integrity, caused by the phosphorylation of junctional proteins (Takenaga et al, 2009), which further goes along with the degradation of the basal lamina (del Zoppo and Mabuchi, 2003). These events are rapidly activated within only 1 to 2 h after stroke, resulting in an increase in vascular permeability that facilitates extravasation of plasma proteins into the neighboring tissue (Figure 1). Extravasated plasma proteins provide mechanical support for migrating endothelial cells.
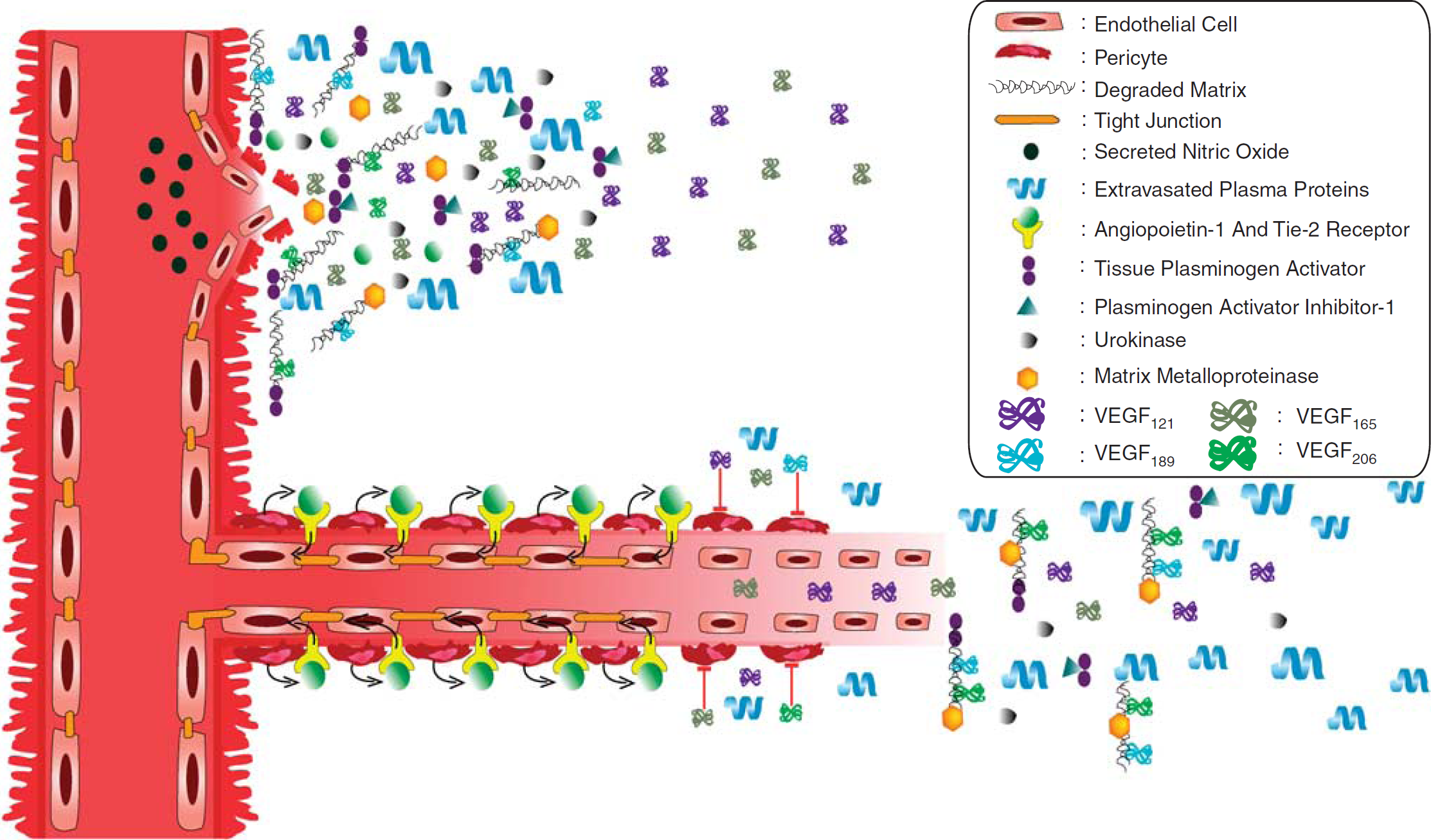
Upper. Role of VEGF in postischemic angiogenesis. In the ischemic brain tissue, loss of vascular integrity is a crucial step, which results in the extravasation of plasma proteins into the neighboring tissue. With the help of matrix metalloproteinases (MMPs), tissue-plasminogen activator, plasminogen activator inhibitor (PAI)-1, urokinase, and angiopoietins, namely angiopoietin-1, the basal membrane is degraded, which prepares the stage for VEGF that induces the proliferation of endothelial cells. Nitric oxide (NO) has an important role in the initiation of the blood–brain barrier breakdown. The highly basic isoforms, VEGF189 and VEGF206 (shown in blue and green), are involved in angiogenesis, mainly as matrix-bound short distance guidance cues, whereas VEGF121 and VEGF165 (shown in purple and gray) are diffusible growth signals acting over larger distances. Lower. While migrating through brain matrix, endothelial cells arrange themselves in cell monolayers and form tube-like structures. Pericytes from the surrounding area proliferate and arrange around the abluminal surface of newly formed vessels. These pericytes release angiopoietin-1, which induces the formation of tight junctions by binding to Tie-2 receptor, which is localized on endothelial cells. VEGF counteracts the maturation of newborn blood vessels by disrupting the pericyte coverage of the vessels.
With the help of supporting cells located in the perivascular space, extracellular matrix constituents, namely laminins, collagen type IV, fibronectin, heparin sulfates, and heparin sulfate proteoglycans (del Zoppo and Mabuchi, 2003), are degraded. The following are the major effectors of this process: (1) matrix metalloproteinases, which already exist in proforms and are enzymatically activated upon stroke; (2) plasminogen activator inhibitor, and urokinase, which are de novo expressed along the infarct border on endothelial cells, astrocytes, and neurons; (3) tissue-plasminogen activator, which forms complexes with plasminogen activator inhibitor-1; and (4) angiopoietins, namely angiopoietin-1 and its natural antagonist angiopoietin-2 (see Figure 1; for review see Distler et al, 2003; del Zoppo and Mabuchi, 2003; Lo, 2008; Yepes et al, 2009).
Degradation of extracellular matrix prepares the stage for growth factors and guidance molecules. Together with other angiogenic factors, VEGF induces the proliferation of endothelial cells that migrate along a gradient of chemotactic cues (Yancopoulos et al, 2000; Ruhrberg et al, 2002; Carmeliet, 2003). The highly basic isoforms, VEGF189 and VEGF206, are involved in this process mainly as matrix-bound short distance guidance cues, whereas VEGF121 and VEGF165 are more diffusible growth signals acting over larger distances (Figure 1; Houck et al, 1992; Park et al, 1993). Endothelial migration is supported by pericytes that release several angiogenic factors, including VEGF, which migrate away from vessels into the perivascular space, where they release proteases including matrix metalloproteinases and urokinase, guiding endothelial cells toward their targets (Dore-Duffy and LaManna, 2007).
While migrating through the brain matrix, endothelial cells arrange themselves into cell monolayers and form tube-like structures (Figure 1). Mesenchymal cells in the surrounding area proliferate and move to the abluminal surface of the newly formed vessels. Endothelial cells and pericytes start to interact with each other (Hanahan, 1997; Carmeliet, 2000; Dore-Duffy and LaManna, 2007), pericytes releasing angiopoietin-1 that stabilizes newborn vessels through its receptor Tie-2 (Figure 1; Croll and Wiegand, 2001; Zacharek et al, 2007). Newborn vessels still need to build a functional blood–brain barrier by formation of tight junctions between endothelial cells. Pericytes again control this blood–brain barrier formation in an angiopoietin-1/Tie-2-dependent manner (Figure 1; Thurston et al, 1999; Wang et al, 2007b).
It has recently been suggested that VEGF itself counteracts the maturation of newborn blood vessels by disrupting the pericyte coverage of vessel sprouts (Figure 1; Greenberg et al, 2008). In this study, the authors evaluated the effects of platelet-derived growth factor (PDGF), showing that PDGF's receptor, PDGFRβ, forms a complex with one of VEGF's receptors, VEGFR-2, once PDGF is in place (Greenberg et al, 2008). Inhibition of VEGFR-2 prevented the assembly of this receptor complex and restored the maturation of vessels (Greenberg et al, 2008).
After hypoxia–ischemia, VEGF and angiopoietins are expressed in a temporally and spatially orchestrated manner. As such, angiopoietin-1 is induced slightly later than VEGF in the peri-infarct border zone, starting at 1 to 2 days after stroke (Croll and Wiegand, 2001). At the same time, the expression of angiopoietin-1's counterplayer, angiopoietin-2, which may have angiogenic or antiangiogenic effects depending on its microenvironment (Jones et al, 2001; Zhu et al, 2005), decreases (Croll and Wiegand, 2001). The temporospatially coordinated release of VEGF and angiopoietins well illustrates that the right factors need to be in place at the right time point so that neurologic recovery becomes possible. Interestingly, the upregulation of VEGF takes place at a time point at which the extracellular matrix is degraded, whereas upregulation of angiopoietin-1 and downregulation of angiopoietin-2 coincide with the resolution of brain edema. Hence, molecular processes and pathophysiological changes go closely along with each other.
The vascular endothelial growth factor receptor family
Vascular endothelial growth factor binds two related receptor tyrosine kinases (RTKs), namely VEGFR-1 (flt-1) and VEGFR-2 (flk-1, KDR), consisting of seven extracellular immunoglobulin-like domains, a transcellular domain, and a consensus tyrosine kinase sequence (Shibuya et al, 1990; Terman et al, 1991). Besides VEGFR-1 and VEGFR-2, a third RTK, i.e., VEGFR-3, exists, which binds to VEGFC and VEGFD but not to VEGF (Karkkainen et al, 2002). In addition to RTKs, a coreceptor, neuropilin-1 (NRP-1), has been identified (Soker et al, 1996), which is otherwise involved in axon-repulsive semaphorin-3A signaling (Neufeld et al, 2002), presenting VEGF165 to VEGFR-2, thus enhancing the signal response of VEGFR-2 (Soker et al, 1998).
Vascular endothelial growth factor receptor-2 mediates most of the mitogenic, angiogenic, and permeability-enhancing effects of VEGF (Ferrara et al, 2003; see Table 1). VEGFR-2−/− mice exhibit a severe lack of vasculogenesis and failure to develop blood islands, resulting in early death in utero (Shalaby et al, 1995). Vascular endothelial growth factor receptor-2 exhibits several tyrosine residues that are phosphorylated in response to VEGF (Roskoski, 2008), inducing a broad intracellular signal response involving phosphatidylinositol-3 kinase (PI3K)/Akt, Ras GTPase-activating protein, tyrosine kinase Src, and Raf-mitogen-activating protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK)-1/2 pathways (Ferrara et al, 2003).
Expression of VEGF and angiopoietin receptors in the healthy and ischemic brain
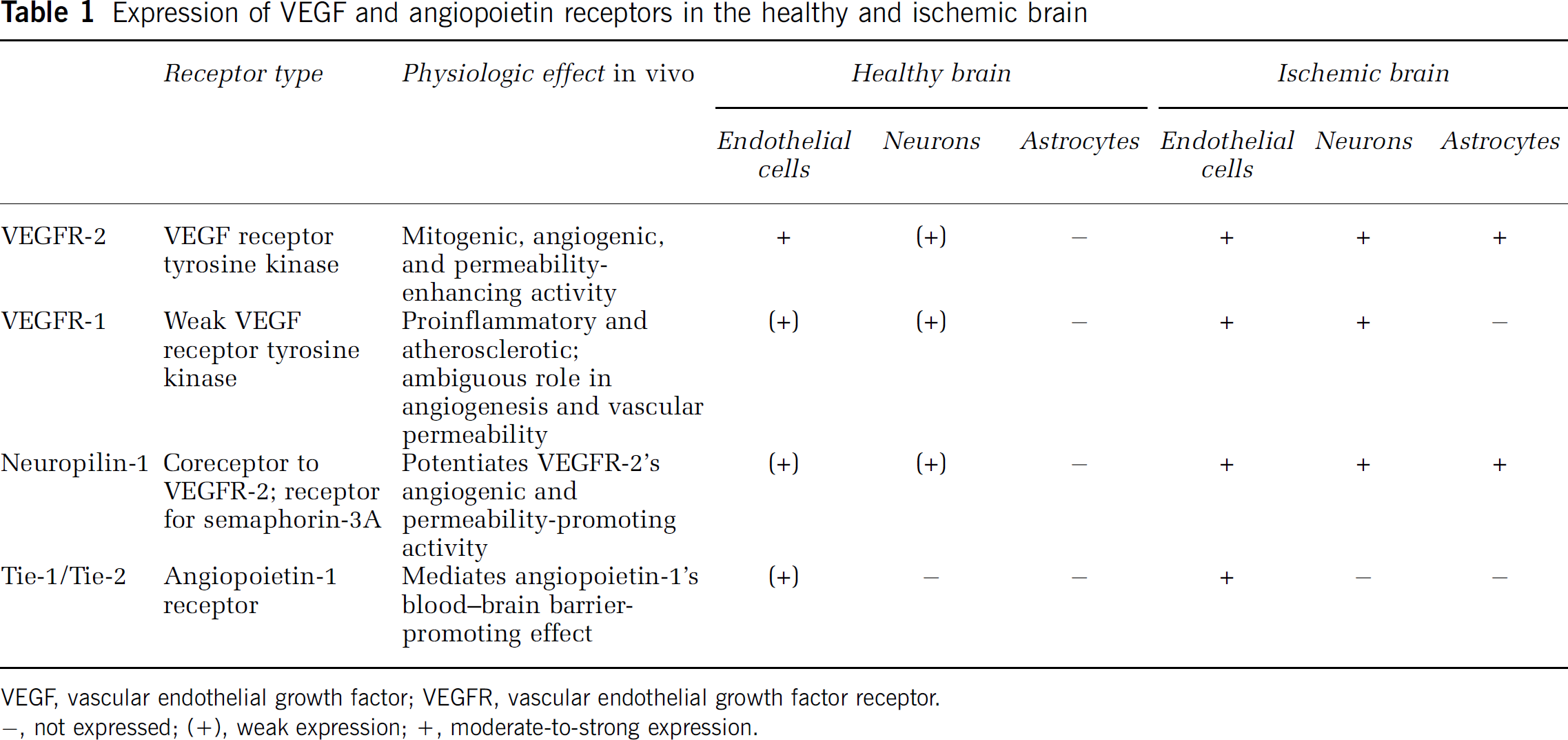
VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
−, not expressed; (+), weak expression; +, moderate-to-strong expression.
Compared with VEGFR-2, VEGFR-1 less potently influences angiogenesis and vascular permeability. Vascular endothelial growth factor receptor-1 was proposed to act as a decoy receptor (Park et al, 1994), representing a scavenger that prevents VEGF binding to VEGFR-2 (Roskoski, 2008). Indeed, in embryonic tissues, VEGFR-1 is a negative regulator of new vessel growth (Fong et al, 1995). The binding affinity of VEGFR-1 to VEGF is about an order of magnitude higher than that of VEGFR-2, whereas its tyrosine kinase activity is an order of magnitude lower than that of VEGFR-2 (Shibuya, 2006; Roskoski, 2008), which explains its partly antagonistic role. Being strongly expressed in macrophages, VEGFR-1 promotes inflammatory responses in peripheral tissues and perhaps atherosclerosis (Shibuya, 2006). As such, the role of VEGFR-1 might vary between the pathophysiological contexts in which it is active.
Vascular endothelial growth factor receptors in brain hypoxia and ischemia
The functions of VEGF receptors that were so far discussed are relevant not only exclusively for brain microvessels, but they are features of vessels in other tissues as well. In the adult brain, VEGFR-2 is found in newborn microvascular endothelium (Marti et al, 2000; Beck et al, 2002) that exhibits the capacity of proliferation and growth, and it is also weakly expressed in healthy neurons (Beck et al, 2002) (see Table 1). The density of VEGFR-2+ capillaries increases within 48 h after stroke (Marti et al, 2000) and it is further elevated in the presence of VEGF (Wang et al, 2005) (Table 1). Both the level of VEGF expression and its cellular distribution change in response to stroke. As such, VEGFR-2 more strongly appears on ischemic neurons (Jin et al, 2000; Matsuzaki et al, 2001; Beck et al, 2002; Wick et al, 2002; Kilic et al, 2006a) and astrocytes (Lennmyr et al, 1998; Kilic et al, 2006a; Choi et al, 2007) (Table 1).
Observations of an altered expression have also been made for VEGFR-1, which exhibits a weak expression on endothelial cells (Beck et al, 2002) and neurons (Beck et al, 2002; Choi et al, 2007) under physiologic conditions and is upregulated on hypoxic–ischemic endothelial cells (Lennmyr et al, 1998; Plate et al, 1999; Beck et al, 2002), neurons (Lennmyr et al, 1998; Matsuzaki et al, 2001; Wick et al, 2002), and pericytes (Levine et al, 2004; Dikov et al, 2005) during the first 3 days after the insult (Table 1); for the VEGFR-2 coreceptor NRP-1, which is expressed at a low level on healthy neurons (Beck et al, 2002) and astrocytes (Beck et al, 2002; Choi et al, 2007) and is elevated on ischemic endothelial cells (Beck et al, 2002), neurons (Zhang et al, 2001; Beck et al, 2002; Hou et al, 2008), and astrocytes (Zhang et al, 2001; Beck et al, 2002) over as long as 30 days (Table 1); and for angiopoietin receptors Tie-1 and Tie-2, which are hardly found in the healthy brain (Lin et al, 2001) but are more robustly expressed in ischemic cerebral vessels (Lin et al, 2000, 2001; Croll and Wiegand, 2001; Zhang and Chopp, 2002) over 28 days (Table 1).
Vascular endothelial growth factor is not the only growth factor that, together with its receptors, is regulated upon ischemia in the brain parenchyma. De novo expression on neurons and astrocytes has previously been shown also for bFGF and its receptor (Liu et al, 2006), for G-CSF and its receptor (Schneider et al, 2005), and for Epo receptor (Li et al, 2007). Parenchymal expression might represent a more general feature of growth factors involved in neurovascular remodeling. Whether and how these different molecules interact with each other remains to be shown.
Functionality of vascular endothelial growth factor receptors in the hypoxic–ischemic brain
The fact that VEGF and its receptors are upregulated upon stroke should alter the responsiveness of the brain tissue. This might explain why VEGF acts not only as an angiogenic but also as a neuroprotective and plasticity-promoting growth factor. To exert its function and initiate cytosolic signal responses, several requirements have to be fulfilled, which relate to (1) the phosphorylation of receptors; (2) the interaction of VEGF receptors, namely of RTK with non-RTK; and (3) the interaction of VEGF receptors with integrins.
Autophosphorylation of VEGF Receptors
Activation of RTK occurs upon VEGF binding through autophosphorylation (Roskoski, 2008). Two forms of autophosphorylation exist, i.e., cis-autophosphorylation, which implies that an RTK monomer catalyzes its own phosphorylation, and trans-autophosphorylation, in which two RTK monomers forming a homodimer mutually phosphorylate each other when exposed to VEGF (see Figure 2; Roskoski, 2008). Previous studies have shown that RTK VEGFR-2 is indeed phosphorylated in the presence of VEGF after stroke and that VEGFR-2 phosphorylation goes along with downstream signal responses mediating neuronal survival and blood–brain barrier permeability (Kilic et al, 2006a).
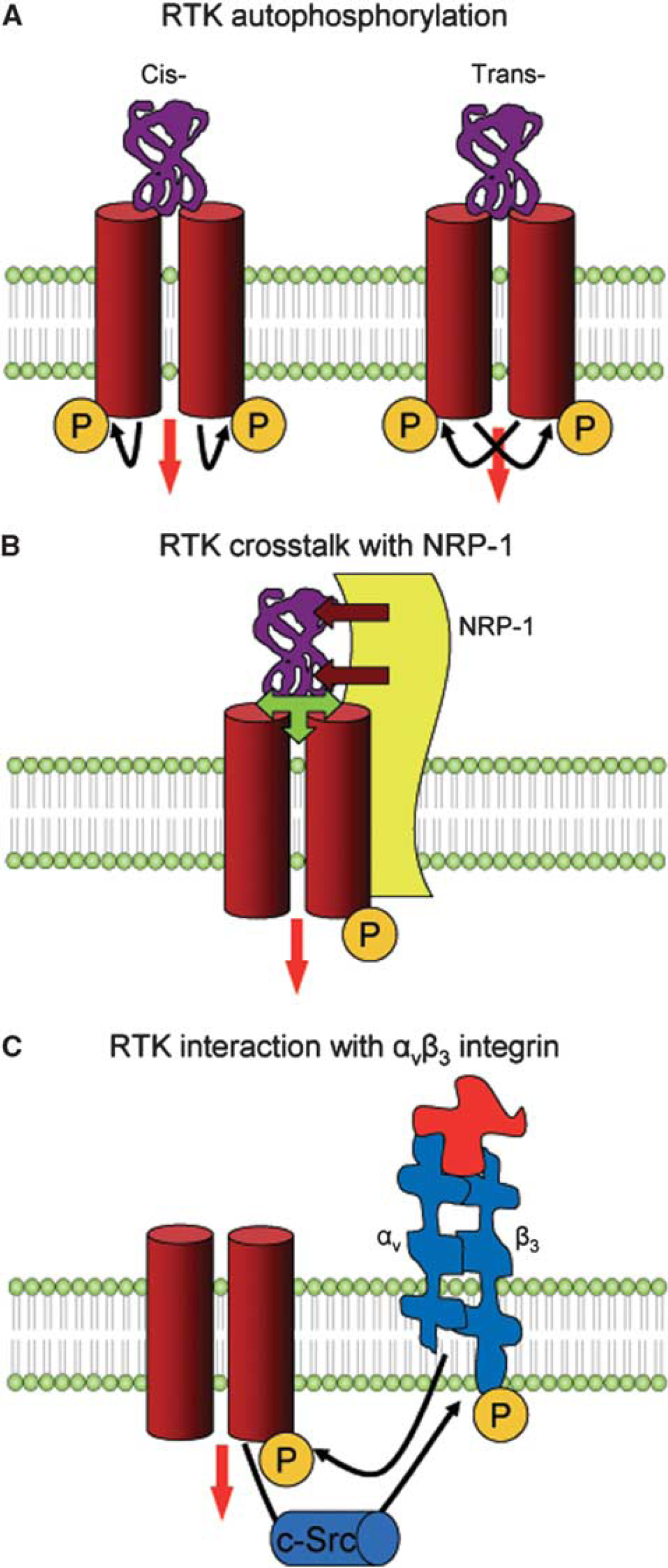
Mechanisms of VEGF receptor activation. Three major mechanisms have been identified with regard to the activation of receptor tyrosine kinase (RTK). (
Besides forming homodimers, RTKs probably form heterodimers that, similar to homodimers, are believed to transactivate each other by reciprocal phosphorylation. Evidence for heterodimeric RTK transphosphorylation has previously been shown in pharmacological and immunoprecipitation studies for VEGFR-1 and VEGFR-2 (Autiero et al, 2003), as well as for VEGFR-2 and VEGFR-3 (Dixelius et al, 2003). Whether transphosphorylation is relevant for VEGF receptor signaling in the brain remains unclear.
VEGF Receptor Cross Talk
Tyrosine kinase activity is influenced by the cross talk of RTK with non-RTK coreceptors, namely with NRP-1, as previously shown for VEGFR-2 (Figure 2). When interacting with VEGFR-2, NRP-1 increases the binding affinity of VEGFR-2 to VEGF165 (Figure 2; Soker et al, 1996, 1998). As a result, a ternary receptor complex is formed (RTK–NRP-1–VEGF), which exhibits enhanced signal transduction properties (Kawamura et al, 2008). The fact that RTK and NRP-1 are coexpressed upon ischemia argues in favor of a joint function of the two receptor subtypes. Observations that NRP-1 indeed promotes the activation of VEGFR-2 upon VEGF165 treatment were recently made in vitro using porcine aortic endothelial cells (Kawamura et al, 2008). Evidence that this mechanism is also relevant in vivo after brain ischemia is still lacking.
Interactions of VEGF Receptors and Integrins
Integrins are receptors for cell matrix molecules, such as collagen, laminin, fibronectin, and vitronectin, which control the arrangement of vascular cells and the formation of the blood–brain barrier (del Zoppo and Mabuchi, 2003). Integrins exhibit rapid expression changes after stroke. The collagen receptor α1β1 strongly decreases within 2 h after middle cerebral artery (MCA) occlusion in areas that develop neuronal injury (Tagaya et al, 2001), whereas the vitronectin receptor αvβ3, which mediates endothelial and smooth muscle cell migration (Leavesley et al, 1993), was found to increase on noncapillary (>7.5 μm) microvessels (Okada et al, 1996; Abumiya et al, 1999).
Observations that αvβ3 and VEGF were colocalized after ischemia (Abumiya et al, 1999) led to protein interaction studies showing that VEGFR-2 and β3 integrin are capable of mutually phosphorylating each other (Figure 2; Mahabeleshwar et al, 2007). The phosphorylation of β3 integrin was shown to be mediated by tyrosine kinase c-Src (Mahabeleshwar et al, 2007), whereas VEGFR-2 phosphorylation was facilitated by αvβ3's ligand, the matrix-bound sixth immunoglobulin-like domain of cell adhesion molecule L1 (Figure 2; Hall and Hubbell, 2004). Deactivation of αvβ3 by a pharmacological inhibitor reduced VEGFR-2 phosphorylation after MCA occlusion, pointing toward the notion that αvβ3–VEGFR-2 interaction is relevant in vivo (Shimamura et al, 2006).
Owing to their regionally select expression that differs between capillary and noncapillary microvessels (Abumiya et al, 1999), integrins may adjust vascular migration to environmental tissue requirements (del Zoppo and Mabuchi, 2003). As such, the upregulation of αvβ3 on noncapillary, but not on capillary, microvessels may provide an explanation for why precapillary arterioles are so important for vascular sprouting.
Effects of other growth factors on VEGF and VEGF receptors
Besides VEGF, several other growth factors also enhance the formation of new vessels. Some of them, such as angiopoietin-1 and Epo, mobilize endothelial precursor cells or recruit hematopoietic precursor cells from blood (Losordo and Dimmeler, 2004). This process of vasculogenesis, i.e., the formation of new vessels from immature cells, has long been considered to be a feature of embryonic tissues, but is meanwhile accepted to take place also in the adult brain in parallel to angiogenesis, i.e., the growth of new vessels out of existing vessels (Asahara and Kawamoto, 2004).
However, not all growth factors directly promote new vessel growth. In fact, some factors, including bFGF and Epo, have been suggested to exert their angiogenic function by stimulating the expression of VEGF and VEGF receptors on endothelial cells (Pepper and Mandriota, 1998; Wang et al, 2004). In case of bFGF and Epo, pharmacological inhibition of VEGFR-2 abolished the effects of growth factors on capillary tube formation and new vessel growth (Seghezzi et al, 1998; Tille et al, 2001; Wang et al, 2004), indicating that VEGF indeed mediates their angiogenic effects. In case of Epo, de novo expression and secretion of VEGF on neural progenitor cells is regulated through an activation of PI3K/Akt and ERK-1/2 pathways (Wang et al, 2008). Besides VEGF, Epo was shown to elevate bFGF, angiopoietin-2, and Tie-1 levels in the ischemic brain (Keogh et al, 2007).
Vascular endothelial growth factor-mediated vasculogenesis has previously also been shown for G-CSF, which promotes the recruitment of VEGFR-1+, VEGF-positive neutrophils, as well as of circulating VEGFR-2+ endothelial progenitor cells in blood (Ohki et al, 2005). Blockade of VEGFR-1 and, to a lesser extent, that of VEGFR-2 abrogated the effect of G-CSF on new vessel growth, indicating that myelomonocytic cells have a role in the modeling of vessels (Ohki et al, 2005). Not only growth factors but also statins increase VEGF, VEGFR-2, and brain-derived neurotrophic factor levels in the brain, putatively by increasing subventricular zone precursor cell migration (Chen et al, 2005). Hence, VEGF seems to reflect a downstream signal in neurovascular remodeling that acts as a common target of various angiogenic molecules. The combined evidence of various growth factor studies suggests that angiogenesis and vasculogenesis are closely linked in the adult ischemic brain and that VEGF has a crucial role in both processes.
Implications of angiogenesis for the ischemic brain
This review so far described the responses of the VEGF system and how it contributes to the angiogenesis that is observed in the surrounding area of an evolving brain infarct. The question arises with regard to the benefit that the brain has from increasing its vessel density. There are four major concepts with regard to this question, which are subsequently discussed.
According to the hypoxia hypothesis, ischemia-induced expression of HIF-1 and HIF-2 drives neovascularization in the infarct border zone through the expression of VEGF, aiming at stabilizing the perfusion of tissue (Marti et al, 2000). Unfortunately the formation of new capillaries is slow. The density of new vessels starts to increase not before 48 h after stroke (Seylaz et al, 1999; Marti et al, 2000; Pinard et al, 2000). Vascular endothelial growth factor delivery increases angiogenesis, but does not significantly change the time line in which new capillaries are formed (Sun et al, 2003; Wang et al, 2005). Thus, there are few reasons to assume that angiogenesis has relevant effects on brain hemodynamics during an acute ischemic event. If at all, angiogenesis can protect the brain against subsequent ischemic episodes.
The capillary recruitment hypothesis postulates that VEGF has a second effect besides promoting vessel growth, i.e., the improvement in vascular perfusion by a relaxation of vessels. In fact, VEGF delivery acutely improved the cerebral blood flow (CBF) of the ischemic tissue during the first 3 h after cerebral thromboembolism, when applied at high doses through a systemic delivery (Zhang et al, 2000). Whether this effect reflects a restoration of vascular reactivity, e.g., by boosting endothelial NO levels, as suggested by in vitro studies (Wu et al, 1996), remains to be shown. It is still unknown to what extent this hemodynamic improvement contributes to the survival effect of VEGF. In the experiments described above (Zhang et al, 2000), no neuroprotective effect was observed after acute VEGF treatment.
According to the neurotrophic hypothesis, newly formed vessels are rich sources of trophic molecules that promote survival of ischemic neurons at risk. Thus, vascular density is elevated not to enhance hemodynamics but to provide a microenvironment in which injured neurons are capable of surviving (Chen and Chopp, 2006; Chopp et al, 2008). In contrast to the two preceding concepts, this is the first and only hypothesis that specifically relates to the brain tissue. The fact that neurotrophic responses are important for tissue survival is supported by the dynamic expression changes that growth factors and their receptors reveal in ischemic microvessels, neurons, and glial cells. From a system biologic point of view, the formation of new vessels is an energy intensive process. As stroke is a metabolically deprived condition, in which any kind of energy supply is highly valuable, it would be surprising if neurotrophic interactions would be the only reason for the formation of a new vessel.
An alternative interpretation of the enhanced angiogenesis after stroke is the removal of dead tissue. According to the clean-up hypothesis, newly formed vessels allow macrophages to enter the tissue and remove cell debris (Manoonkitiwongsa et al, 2001). In fact, the density of newly formed vessels closely correlated with that of macrophages in the ischemic brain, which supports that view (Manoonkitiwongsa et al, 2001). Vascular endothelial growth factor increased new vessel formation and macrophage infiltration in the brain (Manoonkitiwongsa et al, 2004, 2006). The clean-up hypothesis brings together two features of VEGF, its angiogenic and permeability-promoting function. The main goal of the newly formed vessels is not to provide oxygen and nutrients, but to guide inflammatory cells into tissue areas of damage. Yet, does it need vascular networks that are uniquely formed with the aim of cell removal? How about this concept in view of the high blood flow levels of the mammalian brain under normal, physiologic conditions? Blood flow is much higher in the brain than in other tissues. Although the clean-up hypothesis may not only be relevant for brain tissues but also for peripheral tissues, there are no convincing answers to this question.
Taken together, although angiogenesis is a key feature in the remodeling of ischemic brain tissue, the process still remains to some extent a conundrum. There is no commonly accepted explanation of why new vessel formation takes place. Perhaps it is inadequate to search for a single explanation for such a multifaceted process. Perhaps we have developed wrong concepts in the past by focussing our interests on single cells or molecules. Despite this, it is tempting that we may have to understand the basis of neurovascular interactions better to develop more adequate treatment concepts for stroke. To date, we have not completely reached this goal.
Therapeutic effects of vascular endothelial growth factor
The effects of therapeutic VEGF delivery in the ischemic brain were analyzed in a total of 22 animal studies, which investigated the actions of this growth factor on ischemic injury, blood–brain barrier disturbances, CBF, and neurologic recovery (summarized in Tables 2 and 3). In all, 14 of these studies were performed in rats, 7 in mice, and 1 in gerbils. A total of 17 studies used reversible proximal (i.e., intraluminal) MCA occlusions. Three studies examined transcortical MCA occlusions (two permanent and one reversible), one study cerebral thromboembolism, and one venous thrombosis.
Animal studies with local VEGF delivery
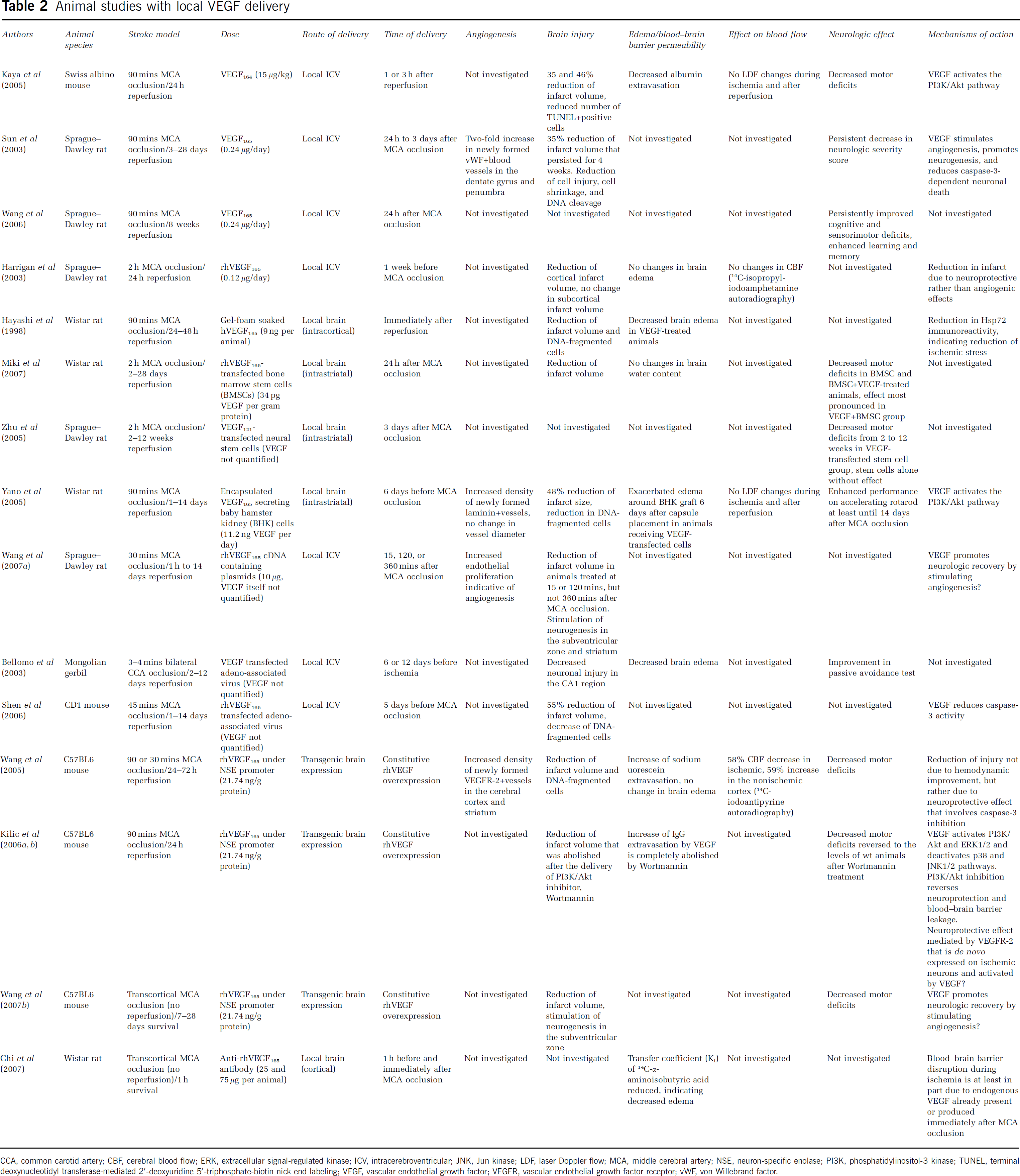
CCA, common carotid artery; CBF, cerebral blood flow; ERK, extracellular signal-regulated kinase; ICV, intracerebroventricular; JNK, Jun kinase; LDF, laser Doppler flow; MCA, middle cerebral artery; NSE, neuron-specific enolase; PI3K, phosphatidylinositol-3 kinase; TUNEL, terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; vWF, von Willebrand factor.
Animal studies with systemic VEGF delivery
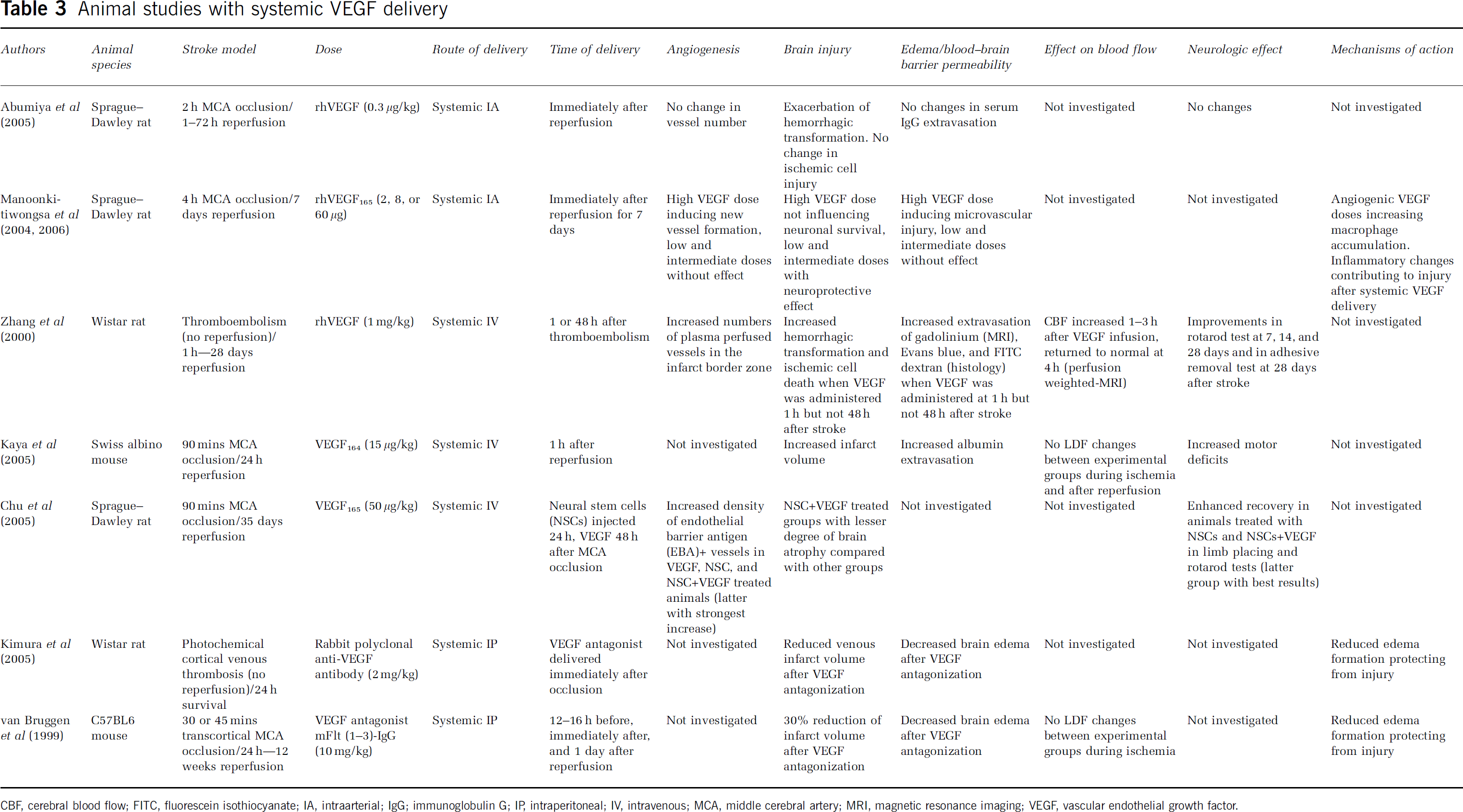
CBF, cerebral blood flow; FITC, fluorescein isothiocyanate; IA, intraarterial; IgG; immunoglobulin G; IP, intraperitoneal; IV, intravenous; MCA, middle cerebral artery; MRI, magnetic resonance imaging; VEGF, vascular endothelial growth factor.
Recombinant VEGF, mostly of human origin, was delivered in 10 studies. Vascular endothelial growth factor-secreting cells were administered in three studies (neural precursor/stem cells in one, bone marrow-derived stem cells in one, and baby hamster kidney cells in one study). Adeno-associated virus vectors were used in two studies, plasmids containing VEGF cDNA in one study, and transgenic mouse models expressing human VEGF under a neuronal promoter in three studies. Three studies evaluated VEGF antagonists. To understand the mechanisms of action of VEGF, these studies were also included in the analysis.
In 15 studies, VEGF or VEGF antagonist was locally applied either by intracerebroventricular or by local brain delivery, or was induced by brain-specific expression. Seven studies used systemic delivery strategies (intravenous in three, intraarterial in two, and intraperitoneal in two studies) for VEGF/VEGF antagonist. A total of 15 studies focussed on the acute injury phase, whereas 7 studies involved postacute assessments of functional neurologic recovery up to 12 weeks after stroke. A total of 962 ischemic animals were examined, 427 of which served as control animals (402 vehicle treated, 25 no injection), 448 of which received VEGF treatment (either in the form of delivery of the VEGF protein, of VEGF-secreting cells or viral vectors), 42 of which were constitutively VEGF overexpressing animals, and 45 received VEGF antagonists.
Effects on neuronal survival and blood–brain barrier integrity
Studies with a therapeutic VEGF application showed quite different effects depending on the route of growth factor delivery. All 12 studies examining the effects of a local (i.e., intracerebroventricular or brain) application of VEGF, of VEGF-secreting cells, or of vectors on structural histologic injury consistently showed a reduction in ischemic brain damage, which was evaluated by infarct volumetry in 11 and by analysis of disseminated neuronal injury in 7 studies (see Table 2). All the 10 studies examining the impact of VEGF on functional neurologic recovery showed sensorimotor, coordination, and cognitive improvements that persisted over as long as 12 weeks after stroke (Table 2). Along with the reduction in ischemic injury, brain edema was reduced in two studies (Hayashi et al, 1998; Bellomo et al, 2003) and remained unchanged in four studies (Harrigan et al, 2003; Wang et al, 2005; Kilic et al, 2006a; Miki et al, 2007), whereas a single study using encapsulated VEGF-secreting cells reported an exacerbation of edema around the grafting site (Yano et al, 2005). The extravasation of serum proteins was reduced in one study (Kaya et al, 2005) and was increased in two studies (Wang et al, 2005; Kilic et al, 2006a, 2006b). Vascular endothelial growth factor antagonization by neutralizing antibodies reduced edema formation in another study (Chi et al, 2007).
A completely different picture resulted from the seven studies using systemic, i.e., intraarterial, intravenous, or intraperitoneal application strategies. Four out of five studies with systemic (intraarterial, intravenous) VEGF delivery showed detrimental effects (Zhang et al, 2000; Abumiya et al, 2005; Kaya et al, 2005; Manoonkitiwongsa et al, 2004, 2006), reflected by an increase in brain injury in two studies, an increased hemorrhagic transformation in two studies, and an increased microvascular injury and inflammation in one study (Table 3). One of these studies showed increased permeability of the blood–brain barrier to gadolinium, Evans blue, and FITC (fluorescein isothiocyanate) dextran (Zhang et al, 2000), one study showed an increase in albumin extravasation (Kaya et al, 2005), and one study showed no changes in IgG (immunoglobulin G) extravasation (Table 3). Both studies using VEGF antagonists showed a reduction in infarct volume and brain edema (van Bruggen et al, 1999; Kimura et al, 2005), further showing that VEGF has a detrimental role when applied systemically (in this case intraperitoneally) (Table 3).
The only study showing an unequivocally beneficial effect of VEGF after systemic (intraarterial) administration used a postacute delivery strategy, in which the growth factor was applied together with stem cells starting 2 days after stroke (Chu et al, 2005). This suggests that time point, in addition to route of delivery, might have a role in the actions of VEGF in the ischemic brain. It is noteworthy that all studies showing unfavorable effects of VEGF used acute application strategies, in which the growth factor was delivered the day before, immediately after, or 1 h after stroke (Table 3).
Role of hemodynamic changes
One study with systemic (i.e., intravenous) VEGF delivery reported that VEGF acutely influences cerebral hemodynamics after stroke (Zhang et al, 2000). In that study, the authors used a permanent focal brain ischemia model, i.e., cerebral thromboembolism in rats, in which an exacerbation of brain edema and histologic injury were observed. Using arterial spin-labeling techniques, the authors showed a short-lasting increase of CBF that persisted over as long as 3 h after VEGF delivery (Zhang et al, 2000). No CBF changes were observed in nonischemic animals. The authors concluded that the enhanced blood flow may be attributed to an enhanced vasorelaxation that was interpreted as the response of ischemic vasculature to this growth factor (Zhang et al, 2000).
To analyze whether hemodynamic changes are involved in the survival-promoting actions of VEGF in paradigms going along with structural neuroprotection, Wang et al (2005) studied the effects of VEGF on CBF by 14C-iodoantipyrine autoradiography in a model of constitutive central nervous system-specific VEGF overexpression. Using intraluminal MCA occlusions, the authors observed that VEGF increased regional CBF in areas outside the MCA territory, but reduced regional CBF in the ischemic tissue (Wang et al, 2005). These data were interpreted as hemodynamic steal phenomena, indicating that VEGF may redirect blood from hemodynamically compromised to nonischemic regions. In view of the observation that VEGF reduced infarct size in a stroke model and that neuroprotection went along in the reduction of caspase-3 levels in ischemic brain areas (Wang et al, 2005), the authors concluded that direct neuroprotective rather than hemodynamic effects are involved in the survival-promoting effects of VEGF.
Mechanisms underlying the neuroprotection of vascular endothelial growth factor
Several in vitro and in vivo studies addressed the question with regard to how VEGF protects against ischemia. Expression studies for VEGFR-2 and VEGFR-1 (Svensson et al, 2002; Brockington et al, 2006; Kilic et al, 2006b; Tolosa et al, 2008), similar to pharmacological studies with selective VEGFR agonists or antagonists (targeting either VEGFR-2 or VEGFR-1) (Wick et al, 2002; Brusselmans et al, 2005; Ding et al, 2005), showed that VEGFR-2 is required, whereas VEGFR-1 is dispensable for the neuronal survival-promoting effects of VEGF (Table 4). On the basis of in vitro studies, NRP-1 seems to act as a coreceptor for VEGFR-2, which enhances the binding of VEGF to this RTK (Shraga-Heled et al, 2007), thereby increasing the neuroprotective signal effect of VEGFR-2 in models of neuronal hypoxia–ischemia (Brusselmans et al, 2005) (Table 4). In vivo studies confirmed that VEGFR-2 is phosphorylated and activated in response to VEGF after stroke (Kilic et al, 2006a). There is little doubt that this RTK mediates the neuroprotective functions of VEGF. To which extent NRP-1 is involved in the neuroprotective effect of VEGF in vivo after stroke still remains to be shown.
Effects of VEGF in neuronal culture and simple in vivo models
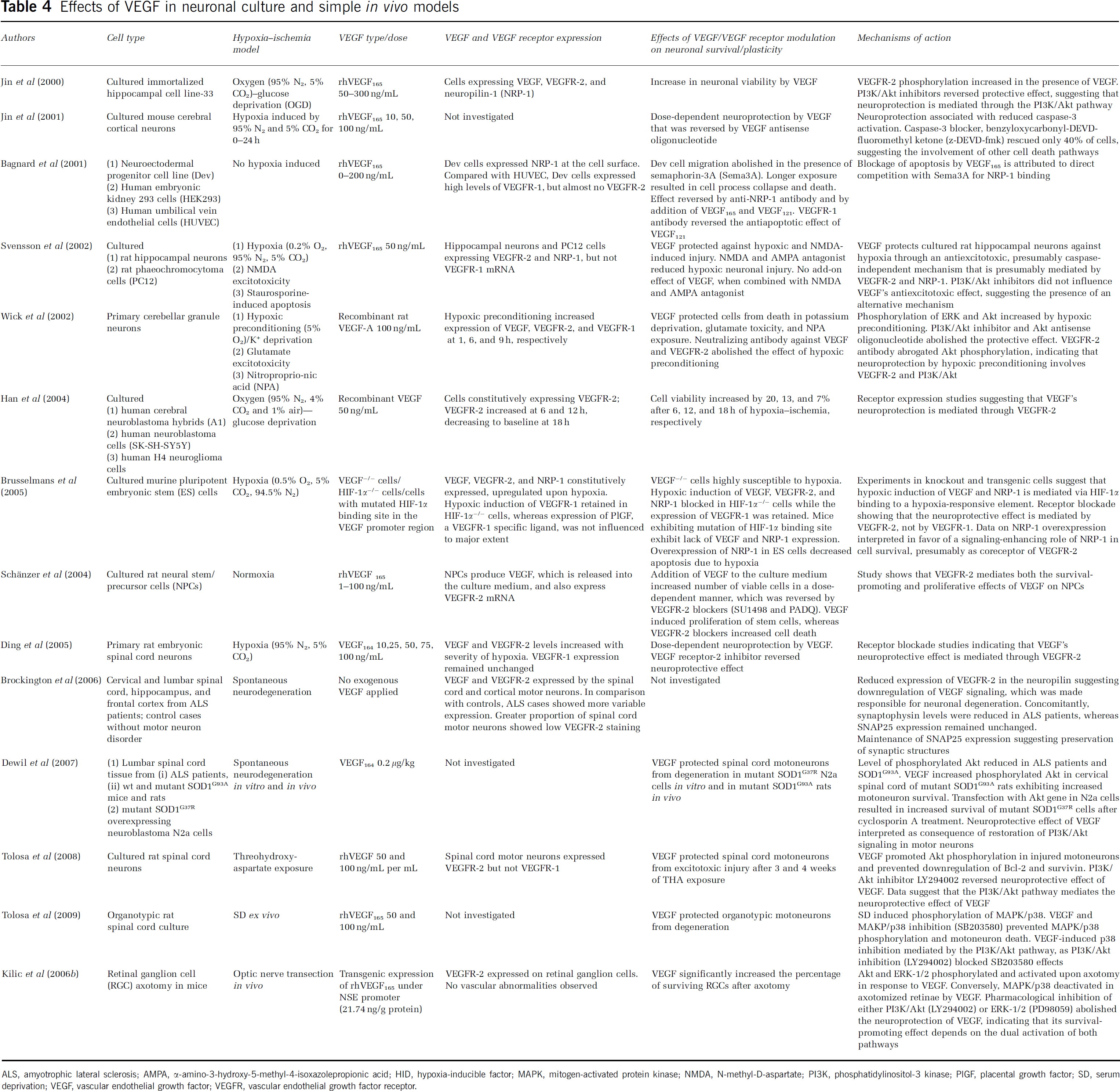
ALS, amyotrophic lateral sclerosis; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; HID, hypoxia-inducible factor; MAPK, mitogen-activated protein kinase; NMDA, N-methyl-D-aspartate; PI3K, phosphatidylinositol-3 kinase; PlGF, placental growth factor; SD, serum deprivation; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Vascular endothelial growth factor receptor-2 is a tyrosine kinase that phosphorylates several cytosolic kinases, including phospholipase-Cγ, PI3K/Akt, Ras-GTPase-activating protein, proteins of the Src family, and Raf, which in turn activates the MEK pathway (Matsumoto and Claesson-Welsh, 2001). Protein expression and phosphorylation studies after stroke have shown that VEGF stimulates the phosphorylation of Akt (Kaya et al, 2005; Yano et al, 2005; Kilic et al, 2006a; Dewil et al, 2007; Tolosa et al, 2008) and of MEK's downstream kinase ERK-1/2 (Kilic et al, 2006a, 2006b) (Table 4). At the same time, VEGF deactivates stress kinases p38 and Jun kinase-1/2 by desphosphorylation, presumably through indirect effects, which are mediated in a PI3K/Akt-dependent manner (Tolosa et al, 2009) (Table 4).
Pharmacological studies in in vitro models of neuronal hypoxia–ischemia or serum deprivation (Gerber et al, 1998; Jin et al, 2000; Wick et al, 2002; Tolosa et al, 2008, 2009), in in vivo models of delayed neuronal degeneration (Kilic et al, 2006b), and in in vivo models of intraluminal MCA occlusion (Kilic et al, 2006a) showed that deactivation of the PI3K/Akt pathway by delivery of pharmacological inhibitors or antisense oligonucleotides reverses the neuroprotective effects of VEGF, indicating a causative role of this signaling pathway in the survival effects of VEGF (Table 4). However, inhibition of MAPK (mitogen-activated protein kinase)/p38 mimicked at least some of the effects of VEGF (Tolosa et al, 2009) (Table 4). Importantly, the delivery of caspase-3 blockers only partly reversed the survival-promoting actions of VEGF (Jin et al, 2001) (Table 4), indicating that, besides caspase-3, other executive death pathways are involved in the survival-promoting actions of VEGF.
Strategies for counteracting the permeability changes induced by vascular endothelial growth factor
The observation that VEGF promotes neuronal survival and stimulates new vessel growth has prompted the question as to whether it may be possible to stimulate neurovascular remodeling without inducing blood–brain barrier disturbances and, consecutively, brain edema. The lack of an intact barrier is a physiologic feature of newborn blood vessels (Hanahan, 1997; Carmeliet, 2000; Wang et al, 2005), and according to recent studies, VEGF actively counteracts the maturation of the blood–brain barrier (Greenberg et al, 2008) (Table 5). The question arises whether it may at all be possible to stimulate new vessel growth in stroke without risking edema formation. Indeed, the induction of permeability by VEGF seems to be an active process, which is controlled by several signaling pathways (see Tables 2 and 3). This raises the question with regard to possibilities to dissect the angiogenic and blood–brain barrier permeability actions of VEGF.
Effects of VEGF on endothelial cells and simple in vivo models
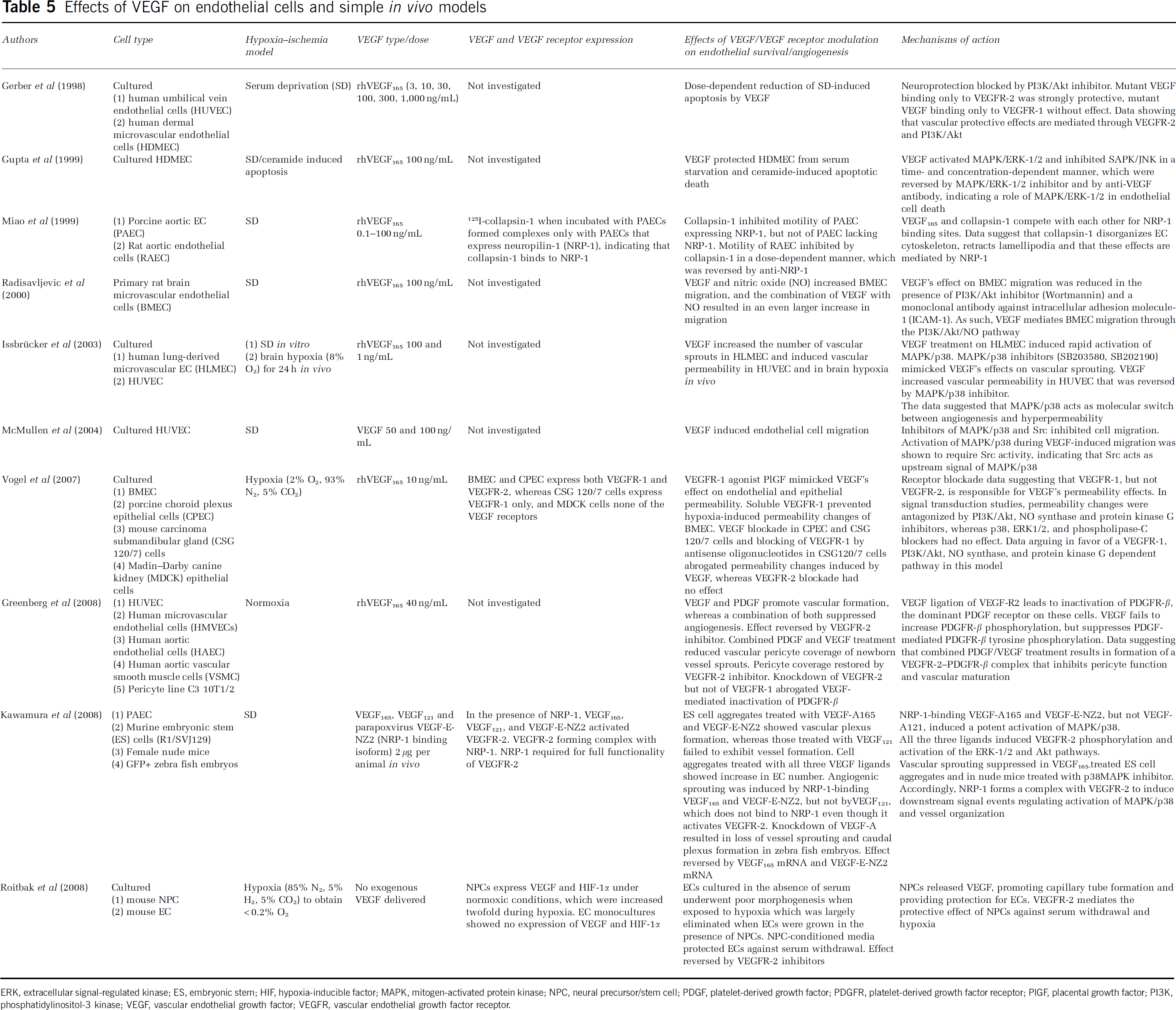
ERK, extracellular signal-regulated kinase; ES, embryonic stem; HIF, hypoxia-inducible factor; MAPK, mitogen-activated protein kinase; NPC, neural precursor/stem cell; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; PlGF, placental growth factor; PI3K, phosphatidylinositol-3 kinase; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Most studies agree that VEGFR-2 mediates the angiogenic and blood–brain barrier disturbances of VEGF in vitro and in vivo (Ferrara et al, 2003). Lack of binding of VEGF to VEGFR-2 induces severe disturbances in endothelial growth and vascular permeability. As a consequence, VEGFR-2−/− mice show a severe failure in vascular development that results in death in utero between days 8.5 and 9.5 (Shalaby et al, 1995; Ferrara et al, 2003). Endothelial cells exposed to serum withdrawal show poor capillary tube formation, which is reversed by VEGF, however, only as long as VEGFR-2 is functional (Roitbak et al, 2008) (Table 5). The fact that VEGFR-1, in addition to VEGFR-2, may have a role in blood–brain barrier permeability was recently suggested (Vogel et al, 2007). Using porcine, murine, and canine endothelial or epithelial cells, in which various deactivation strategies were tested (see Table 5), the authors described a role for VEGFR-1 in the permeability changes of VEGF (Vogel et al, 2007). Future studies will have to confirm this finding.
Downstream of the receptor level, signal factors PI3K/Akt (Kilic et al, 2006a; Vogel et al, 2007) and ERK-1/2 (Issbrücker et al, 2003; Vogel et al, 2007) have been evaluated as candidates to distinguish the angiogenic and permeability-inducing effects of VEGF. In the human carcinoma submandibular gland and lung-derived microvascular endothelial cells, VEGF increased the phosphorylation state of the signal factors Akt (Vogel et al, 2007) and ERK-1/2 (Issbrücker et al, 2003) in vitro (Table 5). Deactivation of PI3K/Akt (Kilic et al, 2006a; Vogel et al, 2007), but not of ERK-1/2 (Vogel et al, 2007), by pharmacological inhibitors abolished the blood–brain barrier disturbances of VEGF in vitro and in vivo (Table 5). Unfortunately, the PI3K/Akt pathway is involved both in angiogenic (Radisavljevic et al, 2000) and neuroprotective (Kilic et al, 2006a) functions of VEGF. It is therefore not a good candidate for modulating the permeability of VEGF.
Besides PI3K/Akt, the stress kinase MAPK/p38 has previously been suggested to represent a switch between the angiogenic and permeability-promoting effects of VEGF (Issbrücker et al, 2003). In lung-derived microvascular endothelial cells in vitro and in brain hypoxia in vivo, the authors showed that VEGF increases the phosphorylation of this kinase (Table 5). It is noteworthy that pharmacological MAPK/p38 inhibition blocked the blood–brain barrier disturbances of VEGF without antagonizing its angiogenic effects (Issbrücker et al, 2003). Unfortunately, in models of mouse submandibular gland carcinoma cells, the authors were recently unable to confirm their earlier finding (Vogel et al, 2007) (Table 5). Submandibular gland carcinoma cells are biologically distinct from endothelial cells. Additional studies are required to further define the role of p38 in regulating blood–brain barrier integrity.
Other strategies examined in the context of VEGF-induced blood–brain barrier breakdown included the inhibition of NO synthase and phospholipase-C. Nitric oxide synthase inhibition antagonized the permeability changes of VEGF, whereas phospholipase-C blockade did not (Vogel et al, 2007) (Table 5). Similar to PI3K/Akt, NO synthases are involved in the induction of angiogenesis (see above). Therefore, they are not suitable as candidates for modulating VEGF's permeability. Taken together, the existing data point toward a close interplay between vascular growth and blood–brain barrier function, which raises the question whether cytosolic signaling pathways represent the right targets for dissecting the effects of VEGF on angiogenesis and blood–brain barrier integrity.
Tight junctions represent assemblies of transmembrane proteins that are linked to various cytosolic adaptor and regulatory proteins. Within the transmembrane complex, VEGF has recently been shown to reduce specific components in an almost selective manner, namely the proteins claudin-5 and occludin (Argaw et al, 2009), whereas the adaptor and regulatory proteins, such as zona occludens-1, junctional adhesion molecule, and cingulin, remained unchanged (Argaw et al, 2009). Claudin-5 and occludin are strongly expressed in brain microvessels, representing key constituents responsible for the blood–brain barrier properties of endothelial cells. As such, claudin-5 and occludin might represent attractive targets for therapeutic interventions counteracting the blood–brain barrier disturbances in the ischemic brain (Argaw et al, 2009).
Effects on brain plasticity and neurogenesis
A number of studies suggest that VEGF induces neurite outgrowth in vitro and in vivo (Pitzer et al, 2003; Rosenstein et al, 2003). The fact that VEGF promotes brain plasticity is supported by the role of VEGF's receptor NRP-1 as a receptor for axonal repulsion molecules belonging to the semaphorin family (He and Tessier-Lavigne, 1997). In fact, VEGF and semaphorins (in particular, collapsin-1/semaphorin-3D) compete with similar sites of the extracellular domain of NRP-1 (Miao et al, 1999). Collapsin-1 bound to neuropilin was shown to reduce lamellipodia motility in one study, resulting in cytoskeleton collapse, whereas VEGF attenuated collapsin-1-induced lamellipodia repulsion (Miao et al, 1999) (Table 5). Semaphorin-3A, another repellent guidance cue, was found to induce apoptosis again through NRP-1 in another study, which was abolished by VEGF, resulting in the survival, proliferation, and migration of neural progenitor cells (Bagnard et al, 2001) (Table 4). It is noteworthy that the cellular actions of semaphorin-3A required RTK activity, as the effects of semaphorin-3A were blocked after VEGFR-1 inhibition. The authors interpreted their findings as further proof of evidence that NRP-1 acts as a coreceptor for VEGFR-1.
Only few studies analyzed the effects of VEGF on neuronal plasticity after stroke. Using intracerebroventricular delivery of either recombinant VEGF or VEGF165-containing plasmids in rats, and using constitutively VEGF-overexpressing mice, both Sun et al (2003) and Wang et al (2007a, 2007c) reported that VEGF increased neurogenesis in the subventricular zone and striatum (Table 2). As neurogenesis was also accompanied by improved sensorimotor recovery, even when VEGF delivery was initiated as late as 24 h after stroke (Sun et al, 2003), the authors concluded that newly formed neurons might contribute to functional improvements. Whether and how VEGF influences the plasticity of preexisting neurons and remodeling of white matter tract systems is still unknown. Using anterograde tract tracers, neutralizing antibodies directed against the growth-repulsive Nogo-A protein (Wiessner et al, 2003) as well as bone marrow-derived stem cells (Liu et al, 2007) have recently been shown to promote corticospinal sprouting contralesional to a stroke under conditions of successful neurologic recovery. Whether VEGF influences the plasticity of white matter fiber systems and how this plasticity contributes to motor and non-motor recovery remain to be shown.
Translation from bench to bedside?
Tissue protection, new vessel formation, and brain plasticity are key features underlying the neurovascular remodeling induced by VEGF. All the three processes are closely linked, being orchestrated in a temporospatial manner. As both VEGF and its receptors, similar to other receptor proteins and their ligands with which they interact (e.g., PDGF, semaphorins), are regulated upon stroke, it is not surprising that the effects of VEGF strongly depend on the setting in which the growth factor is applied. The multifaceted effects of VEGF—promoting neuronal survival, angiogenesis, and brain plasticity at the same time—had nourished hopes that by the use of a single molecule it may be possible to promote neurologic recovery after stroke. As shown in the meantime, these pleiotropic effects in the brain predispose to side effects, specifically to brain edema. These data show that, when pleiotropic growth factors are used, solid insights into pathophysiological processes are required before patient trials are faced. This may not only be important for VEGF but also for other angiogenic factors, such as Epo, bFGF, and G-CSF (Ehrenreich et al, 2002; Ren and Finklestein, 2005; Schäbitz and Schneider, 2007), which are presently under clinical investigation.
From studies in peripheral artery occlusive disease and coronary artery disease, a lot of enthusiasm was generated more than a decade ago (Isner et al, 1996; Hendel et al, 2000) when it was found that by therapeutic angiogenesis it might also be possible to promote neurologic recovery after stroke. In coronary artery disease, experimental studies rapidly led to clinical trials, in which gene therapy approaches using intramyocardial or intracoronary delivery of VEGF cDNA plasmids or adenoviral VEGF vectors were tested. In these studies, VEGF delivery led to improvements in functionally relevant readouts, for example, coronary vascularization or ECG abnormalities in some (Mäkinen et al, 2002; Hedman et al, 2003; Stewart et al, 2006) but not in other (Rajagopalan et al, 2003; Kastrup et al, 2005) studies. In ischemic stroke, VEGF-induced permeability has precluded clinical studies in stroke until now. The question arises as to whether there are applications that might nonetheless be considered for VEGF in the stroke brain. To answer this question, a number of open issues still have to be addressed.
Open issues and future strategies
Among previous studies analyzing the effects of VEGF in the stroke brain, particularly few studies have assessed clinically relevant stroke models, which might translate into conditions in elderly patients in whom cerebral hemodynamics are compromised. Only one study assessed the effects of VEGF in a clinically relevant thromboembolism model (Table 3). All studies used young animals with otherwise intact vasculature. Most importantly, no studies tested experimental conditions, in which CBF was already compromised before stroke and no studies tested atherosclerotic or aged animals. Studies in animals with hemodynamic disturbances could mimic the clinical situation of the chronically hypoperfused brain, which is not a rare problem in clinical neurology. Importantly, only two studies have analyzed the hemodynamic consequences of VEGF treatment until now (Zhang et al, 2000; Wang et al, 2006). To date, there is still no convincing evidence that therapeutic angiogenesis indeed improves blood flow. Studies in elderly animals may be of interest, as HIF-1α expression and binding activity and VEGF expression are age dependently suppressed in smooth muscle cells, both under physiologic conditions and during hypoxia (Rivard et al, 2000).
Most studies in the past used acute treatment paradigms, delivering VEGF immediately or up to 7 days after stroke (Tables 2 and 3). Only four studies evaluated prophylactic treatments beginning 2 weeks or less before the stroke (Tables 2 and 3). No study analyzed how chronic treatment continuing over more than 1 week influences angiogenesis and plasticity. No study has so far assessed which receptor mediates the angiogenic and neuroprotective effects of VEGF after stroke. Postacute treatments are highly relevant with respect to future clinical developments, as, on the basis of permeability changes, it may be expected that the risk of brain edema and hemorrhagic transformation does not have a similar role in the postacute stroke phase. Such studies should contribute additional insights into the effects of VEGF on brain plasticity processes and how VEGF promotes motor and nonmotor neurologic recovery. Prophylactic studies might contribute insights into the question whether VEGF may be useful in stroke prevention, for example, under conditions of severe intracranial stenosis.
An alternative strategy for the delivery of the VEGF protein might be the modulation of VEGF expression, e.g., by cell-based therapies or by pharmacological compounds. The HMG (hydroxy-3-methyl-glutaryl)-CoA reductase inhibitor atorvastatin has previously been shown to upregulate VEGF after stroke, resulting in an enhanced neurologic recovery of treated animals (Chen et al, 2005). The observation that atorvastatin not only increased VEGF but also its receptor VEGFR-2, as well as brain-derived neurotrophic factor, suggests that statin induced more complex remodeling processes. In contrast to VEGF, atorvastatin is widely described in secondary stroke prevention. According to the SPARCL (Stroke Prevention by Aggressive Reduction in Cholesterol Levels) study, which is a phase III trial in patients with recent stroke or transient ischemic attack (Amarenco et al, 2006), atorvastatin does not exhibit any unfavorable side effects such as brain edema or hemorrhagic transformation. In animals, statins are usually administered at a dosage ∼10 times more than that of human patients (Chen et al, 2005; Kilic et al, 2005). Whether HMG-CoA reductase inhibitors also enhance angiogenesis and brain plasticity in human patients remains to be shown.
Conclusions
On the basis of existing literature and in view of the above-described open questions, future studies should clarify the following questions: (1) whether an increased vessel density translates into improvements in CBF in the hemodynamically compromised brain; (2) how VEGF influences brain plasticity and contributes to motor and nonmotor recovery; (3) what are the actions of VEGF not only in young animals with preserved vasculature, in which previous studies were performed, but also in aged animals and animals with preexisting atherosclerosis; and (4) whether the effects of VEGF can be mimicked by pharmacological compounds or by cell-based therapies. Only on the basis of such information can more definite conclusions be made with regard to whether the translation of therapeutic angiogenesis into clinics is promising.
Footnotes
Acknowledgements
The authors would like to acknowledge Mr Ayman El Ali for stimulating and helpful discussions. The technical assistance of Mr Jayson Oommen and Mr Aneesh Kumar in the preparation of the figures is also acknowledged.
The authors declare no conflict of interest.