
Abstract
Vascular endothelial growth factor (VEGF) is a secreted polypeptide and plays a pivotal role in angiogenesis in vivo. However, it also increases vascular permeability, and might exacerbate ischemic brain edema. The effect of this factor on the brain after transient ischemia was investigated in terms of infarct volume and edema formation, as well as cellular injury. After 90 minutes of transient middle cerebral artery occlusion, VEGF (1.0 ng/μL, 9 μL) was topically applied on the surface of the reperfused rat brain. A significant reduction of infarct volume was found in animals with VEGF application (P < 0.001) at 24 hours of reperfusion as compared with cases with vehicle treatment. Brain edema was significantly reduced in VEGF-treated animals (P = 0.01), and furthermore, extravasation of Evans blue was also decreased in those animals (P < 0.01). Terminal deoxynucleotidyl transferase-mediated dUTP-biotin in situ nick end labeling and immunohistochemical analysis for 70-kDa heat shock protein showed an amelioration of the stainings at 24 and 48 hours after reperfusion with VEGF treatment, which indicated reduction of neuronal damage. These results indicate that treatment with topical VEGF application significantly reduces ischemic brain damage, such as infarct volume, edema formation, and extravasation of Evans blue, and that the reductions were associated with that of neuronal injury.
Angiogenesis, the formation of new blood vessels, occurs in a wide range of physiologic and pathologic processes. In the normal condition, it occurs during development of the vascular system and in the corpus luteum (Cao et al., 1996). Under pathologic processes, uncontrolled angiogenesis sometimes accounts for the severe disability in some disorders such as diabetic and prematurity retinopathies (Folkman and Shing, 1992; Smith et al., 1997). In addition, neoplastic cells depend exclusively on nutrients and oxygen from newly formed tumor vessels (Folkman and Shing, 1992). Therefore, reduction of the new vessel formation might be an innovative means of therapy in these disorders (Hammes et al., 1996). On the other hand, in diseases such as limb ischemia and myocardial infarction, the ischemic tissue is often dependent on the collateral blood flow (Isner et al., 1995). In animal models of hind limb ischemia and myocardial infarction, administration of angiogenic factors potentiates collateral formation and reduces tissue injury (Banai et al., 1994; Takeshita et al., 1994 a, 1994b; Bauters et al., 1995). Thus, potentiating new vessel formation can be an important means of therapy. In brain infarct, however, little has been elucidated about the role of angiogenesis. It was demonstrated that the number of new vessels in the ischemic penumbra correlated with longer survival in patients of stroke (Krupinski et al., 1994). Thus, an active angiogenesis occurring in the brain infarct area may be beneficial for the tissue under ischemia. However, it is still uncertain whether therapeutic angiogenesis is useful for reducing ischemic injury in the brain.
Many factors are known to induce angiogenesis, and these include a variety of polypeptide growth factors (Yoshimoto et al., 1996). Among the polypeptide angiogenic factors, vascular endothelial growth factor (VEGF) has gained much attention because it is a specific mitogen for endothelial cells and, furthermore, is a secreted glycoprotein (Leung et al., 1989; Shweiki et al., 1992; Wang et al., 1996). These characteristics suggest a pivotal role of VEGF in angiogenesis in vivo. In human, four different isoforms exist by alternative splicing; these are VEGF206, VEGF189, VEGF165, and VEGF121 (Leung et al., 1989; Bacic et al., 1995). In normal rat brain, VEGF165 is the most abundant form and VEGF121 is the next, whereas VEGF206 is not detectable (Bacic et al., 1995). Isoforms VEGF165 and VEGF121 were also induced by focal ischemia in rat brain (Kovacs et al., 1996; Hayashi et al., 1997). Therefore, VEGF165 and VEGF121 may play more important roles in ischemic brains. As VEGF plays a pivotal role in new vessel formation, it could be used for therapeutic angiogenesis in ischemic diseases (Banai et al., 1994; Takeshita et al., 1994 a, 1994b; Bauters et al., 1995). However, the fact that VEGF increases vascular permeability and might exacerbate brain edema may make it difficult for a therapeutic use of this factor in cerebral infarction. Therefore, it is important to investigate how exogenously applied VEGF influences the brain during ischemia.
A part of ischemic neuronal injury is associated with the apoptotic process, which is detected with terminal deoxynucleotidyl transferase-mediated dUTP-biotin in situ nick end labeling (TUNEL) (States et al., 1996), and some growth factors are known to inhibit the apoptotic process of injured cells (reviewed by Nikolics et al., 1996). On the other hand, 70-kDa heat shock protein (HSP70) is a well-known molecular chaperon and a marker of injury that is induced under stressful conditions such as ischemia, and induction is reduced with cell protective agent (reviewed by Gething and Sambrook, 1992; Lee et al., 1994; Abe et al., 1997). To see the overall effect of VEGF, we studied how the application of VEGF influences the volume of brain infarct, edema formation, and extravasation of Evans blue (EB) caused by transient middle cerebral artery (MCA) occlusion. In addition, modification of TUNEL and HSP70 stainings were also examined semiquantitatively to investigate the effect of VEGF administration on neuronal cellular damage.
MATERIALS AND METHODS
Surgical preparation
Adult male Wistar rats (SLC, Shizuoka, Japan) weighing 250 to 280 g were used for the experiments. The animals were anesthetized with an intraperitoneal injection of 40 mg/kg pentobarbital, and positioned in a stereotaxic operating apparatus. A 2-mm diameter burr hole was carefully made at 3 mm dorsal and 5 mm lateral to the right from the bregma using an electric dental drill, avoiding traumatic brain injury. Dura mater was preserved at that time. The location of the burr hole was in the upper part of the right MCA territory. Then the incision was closed, and the animals were allowed to free access to water and food at ambient temperature (21°C to 23°C).
On the next day at about 24 hours after the drilling, the rats were lightly anesthetized by inhalation of a 69%/30% (v/v) mixture of nitrous oxide/oxygen and 1% halothane using a face mask. A midline neck incision was made and the right common carotid artery was exposed, and then inhalation of anesthetics was stopped. When the animal began to regain consciousness, the right MCA was occluded by insertion of 4-0 surgical nylon thread with silicone coating through the common carotid artery as in our previous reports (Nagasawa and Kogure, 1989; Abe et al., 1992). Using this technique, the tip of the thread occludes the origin of the right MCA. The reliability of producing successful strokes is almost complete in this model (Koizumi et al., 1986). During these procedures, body temperature was monitored with a rectal probe, and maintained at 37° ± 0.3°C using a heating pad. The surgical incision was then closed and the animals were allowed to recover at ambient temperature. After 90 minutes of MCA occlusion, CBF was restored by removal of the nylon thread.
Just after restoration of CBF, the dura mater under the burr hole was carefully removed, and a small piece (8 mm3) of Gelfoam (Upjohn, Kalamazoo, MI, U.S.A.), presoaked in 9 μL of either vehicle (artificial CSF consisting of (in mEq) Na+ 130, K+ 4, Ca2+ 3, Cl− 109, and lactate 28) or a solution containing 9 ng of recombinant human VEGF165 (Becton Dickinson Labware, Bedford, MA, U.S.A.) in 9 μL of vehicle, was placed in contact with the surface of the cerebral cortex, as our previous report (Abe et al., 1997). The Gelfoam was buried in the skull bone. The surface of the skull bone was then covered with vinyl tape, and the head skin incision was closed. It is reported that application of growth factor in Gelfoam is as effective as chronic infusion (Otto et al., 1989). The above operations were performed in a sterile fashion. Sham-operated control animals underwent burr hole surgery, exposure of the common carotid artery without MCA occlusion, and placement of Gelfoam presoaked in vehicle. The experimental protocol and procedures were approved by the Animal Committee of Tohoku University School of Medicine.
Quantitative analysis of infarct volume
The animals treated with vehicle (n = 5) or VEGF (n = 6) application were deeply anesthetized with diethyl ether at 24 hours after the restoration of CBF. After decapitation, the brains were quickly removed, and coronal sections of 2-mm thickness were prepared. The fresh brain slices were immersed in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC, Wako Chemical, Sendai, Japan) in normal saline at 37°C for 30 minutes, and then fixed in 10% phosphate-buffered formalin at 4°C. The TTC-stained brain slices were photographed using color film, and the volume of an infarct was calculated using NIH Image version 1.62.
Evaluation of brain edema
The animals treated with vehicle (n = 6) or VEGF (n = 6) application were deeply anesthetized with diethyl ether at 24 hours after the restoration of CBF. They were decapitated, and their cerebral cortices of right MCA territory were quickly taken. After the wet weight of each sample was determined, they were dried in an oven at 110°C for 24 hours and the dry weight was recorded. The water content was calculated as [(wet – dry weight)/wet weight] × 100% (Abe et al., 1988). Sham-operated control animals (n = 5) were also obtained.
Quantitative evaluation of Evans blue extravasation
After topical application of vehicle (n = 4) or VEGF (n = 4), 2% EB (Wako Chemical) in saline was intravenously injected (4 mL/kg). At 24 hours after the CBF restoration, the animals were anesthetized and the brains were perfused with saline through the left cardiac ventricle at 110 mm Hg pressure until colorless perfusion fluid was obtained from the cervical veins. Then the animals were decapitated under deep anesthesia with diethyl ether, and the extravasated EB in the brain was determined according to our previous report with slight modification (Kawagoe et al., 1992). Briefly, the cerebral cortex of the right MCA territory was taken and soaked overnight in a 5× volume of 1 mEq KOH at 37°C. The alkaline solution was then neutralized by adding 1.5 volumes of 5 N phosphoric acid, and 4.5× acetone was added. After vigorous shaking for a few seconds, samples were centrifuged at 1,800 g for 20 minutes. Absorbance of the extracted supernatant was measured by spectrophotometer (UV-240, Shimizu, Kyoto, Japan) at 620 nm. A control group which underwent sham-operation was also obtained (n = 4). The quantitative calculation of the dye content was based on external standards in the brain-soaked solution.
TUNEL staining
At 24 or 48 hours after the vehicle or VEGF application (n = 6 in each group at 24 hours, and n = 5 at 48 hours), the animals were deeply anesthetized with diethyl ether and decapitated. The brains were quickly removed, frozen in powdered dry ice, and then stored at −75°C. Sham-operated control animals were sacrificed at 24 or 48 hours after the exposure of carotid artery (n = 2, in each group).
Coronal sections of 10-μm thickness at the caudate level were cut on a cryostat at −20°C and collected on glass slides coated with poly-lysine. In accordance with our previous report (Abe et al., 1997), TUNEL study was performed using a kit (TACS in situ apoptosis detection kit #4810-30K, Trevigen, Gaithersburg, MD, U.S.A.), which detects double-strand breaks in genomic DNA with diaminobenzidine. The sections were counterstained with methyl green.
In this study, the number of positively stained cells were counted in 32 pixels of 0.10 mm2 by light microscopy with 100× magnification; 16 pixels were from the cerebral cortex at the boundary zone of the MCA and the anterior cerebral artery, and the other pixels were that of the MCA and the posterior cerebral artery. The total number of positive cells in these pixels was expressed as cells/mm2.
Immunohistochemical analysis for HSP70 expression
The sections used for this study were obtained from the same brains that were prepared for the TUNEL staining study. Sham-operated controls were also obtained. Immunostaining against HSP70 was performed according to our previous report (Lee et al., 1994). In brief, the sections were fixed in ice-cold acetone for 10 minutes and air-dried, followed by a rinse in phosphate-buffered saline. After blocking with 10% normal horse serum for 2 hours, the slides were washed in phosphate-buffered saline and incubated with a mouse monoclonal antibody against HSP70 (RPN1197, Amersham, London, U.K.) at 1:200 dilution in 10% normal horse serum and 0.3% Triton-×100 for 20 hours at 4°C. Endogenous peroxidase activity was quenched by exposing the slides to 0.3% H2O2 and 10% methanol for 20 minutes. The slides were then washed and incubated for 3 hours with biotinylated anti-mouse IgG (Vectastain, PK-6102, Vector Laboratories, Burlingame, CA, U.S.A.) at 1:200 dilution in phosphate-buffered saline containing 0.0018% normal horse serum. Subsequently they were incubated with avidin-biotin-horseradish peroxidase complex (PK-6102, Vector Laboratories) for 30 minutes, and then developed using diaminobenzidine as a color substrate. The reaction was stopped by washing in distilled water. Hematoxylin was used for nuclear staining.
The results of staining were categorized into four grades: no staining (−), slightly stained (±), moderately stained (+), and densely stained with nucleus staining (2+). The number of immunopositive cells in each staining grade was also counted in accordance with the TUNEL study described above, and evaluated semiquantitatively.
Statistical analysis
All data are expressed as means ± SD. The two-tailed unpaired Student's t test was used for each evaluation.
RESULTS
Quantitative analysis of infarct volume
Ninety minutes of MCA occlusion caused large infarcts of the lateral cortex and the underlying caudoputamen. The location of the burr hole was on the upper part of the infarcted cortex.
The mean infarct volume of the six VEGF-treated rats was 18.4% ± 2.1% of total volume of the ipsilateral hemisphere, and 33.2% ± 4.2% in rats with vehicle application. Application of VEGF produced a significant reduction in infarct volume (P < 0.001).
Evaluation of brain edema
The water content in the cerebral cortex of the right MCA territory was 80.4% ± 0.4% in the sham-operated group (n = 5). In the vehicle-treated animals, the water content was 84.9% ± 2.1% (n = 6; P < 0.01 versus the sham-operated group). On the other hand, it was 81.5% ± 0.8% in the VEGF-treated group (n = 6; P < 0.05 versus the sham-operated group, and P = 0.01 against the vehicle-treated group). Thus, transient MCA occlusion resulted in brain edema formation in the cerebral cortex of the right MCA territory. However, application of VEGF significantly reduced the brain water content. These results are summarized in Fig. 1.
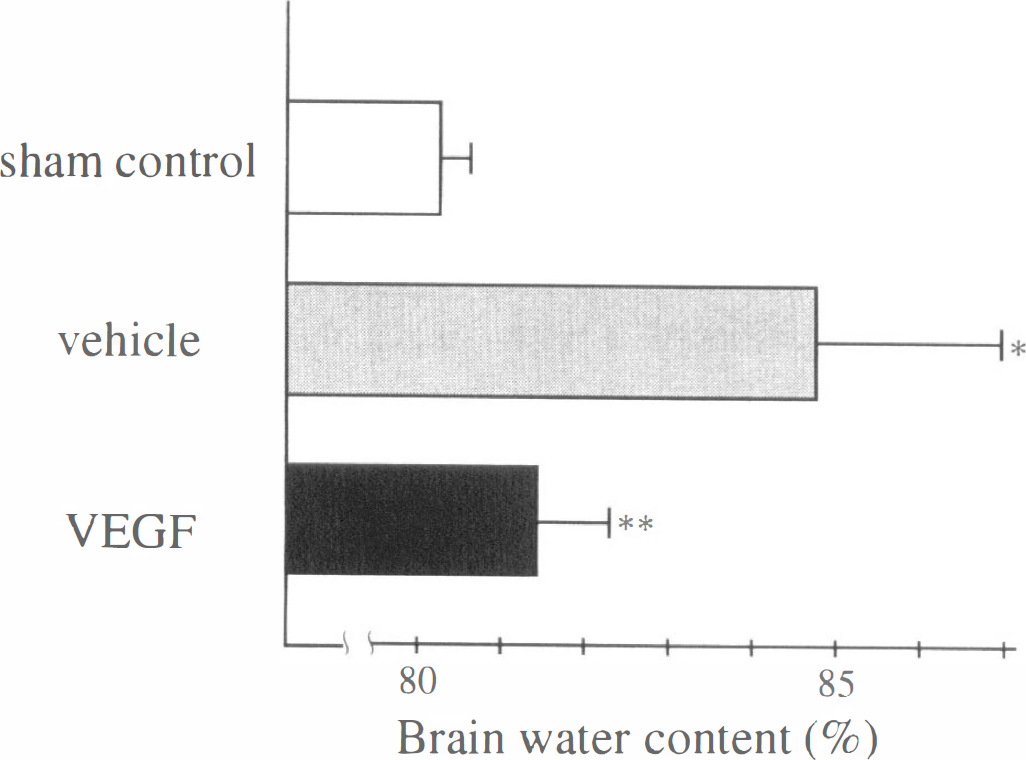
Brain water content in cerebral cortices of the middle cerebral artery territory at 24 hours after reperfusion. Compared with sham-operated brains (80.4% ± 0.4%, n = 5), vehicle-treated brains showed much increased water content (84.9% ± 2.1%, n = 6; * P < 0.01 versus sham-operated group). In brains with vascular endothelial growth factor (VEGF) application, the increase of water content was significantly lessened, though it was still higher than that of sham-operated brains (81.5% ± 0.8%, n = 6; ** P < 0.05 versus sham-operated group and P = 0.01 versus vehicle-treated group.)
Quantitative evaluation of Evans blue extravasation
In the sham-operated group, extravasated EB was 1.0 ± 0.1 μg/g tissue (n = 4). In the vehicle-treated brain, however, a great deal of EB was extravasated after transient ischemia and reperfusion. The EB content was 15.0 ± 2.1 μg/g tissue in the cerebral cortex of the right MCA territory (n = 4; P < 0.01 versus sham-operated group). On the other hand, topical application of VEGF significantly reduced the EB extravasation, and the dye content was 8.8 ± 1.3 μg/g tissue (n = 4; P < 0.01 versus vehicle-treated group and sham-operated group). These results are shown in Fig. 2.
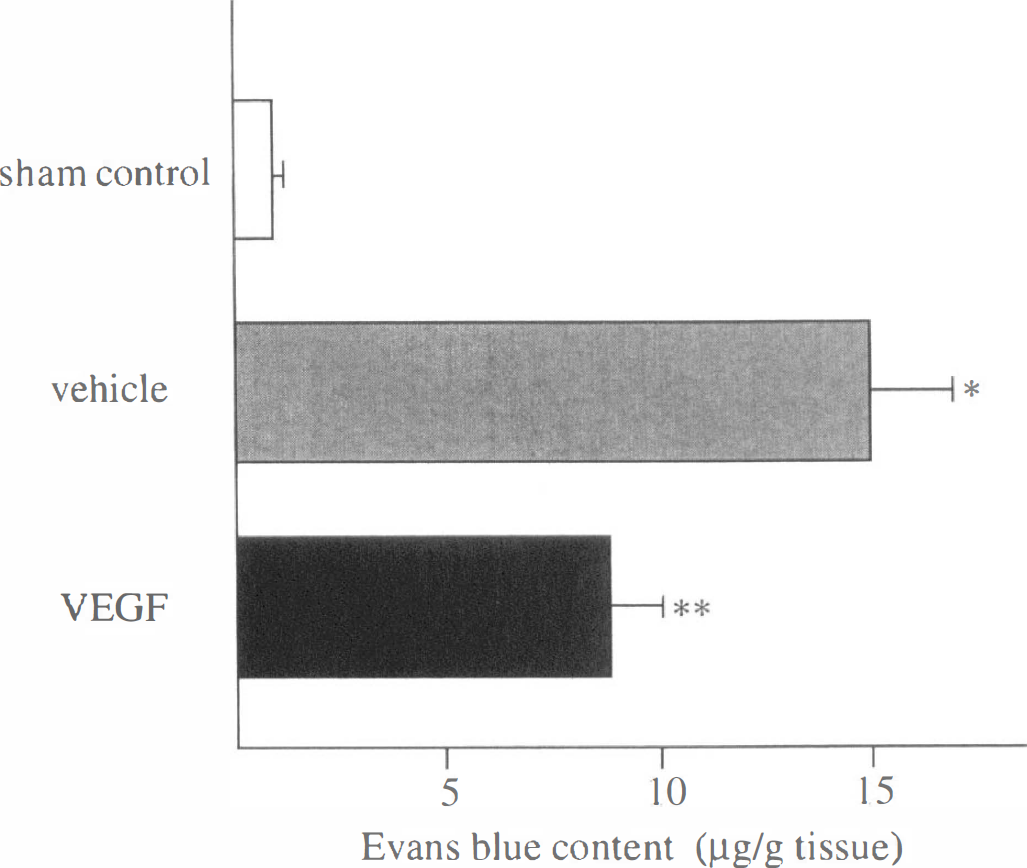
Evans blue content in cerebral cortices of middle cerebral artery territory at 24 h after reperfusion. Compared with sham-operated brains (1.0 ± 0.1 μg/g tissue, n = 4), ischemic brains with vehicle application showed much increased dye content (15.0 ± 2.1 μg/g tissue, n = 4; * P < 0.01 versus sham-operated group). However, treatment with VEGF significantly reduced the dye content (8.8 ± 1.3 μg/g tissue, n = 4; ** P < 0.01 versus sham-operated group and vehicle-treated group.) VEGF, vascular endothelial growth factor.
TUNEL staining
Cells with double-strand DNA breaks were not detected by TUNEL staining in sham-operated brains (Fig. 3a). However, a strong staining for TUNEL was present in a moderate to large number of cells at 24 hours of reperfusion in the vehicle-treated group (Fig. 3b), which became more conspicuous at 48 hours after reperfusion (Fig. 3c). The TUNEL-positive stained cells were distributed in the cerebral cortex and dorsal caudate of the MCA territory, but were not found in other areas of the ipsilateral hemisphere or the contralateral side. The staining was exclusively found in the nuclei of neuronal cells. As shown in Fig. 3 application of VEGF reduced TUNEL-positive cells both at 24 (Fig. 3d) and 48 hours (Fig. 3E) after CBF restoration.
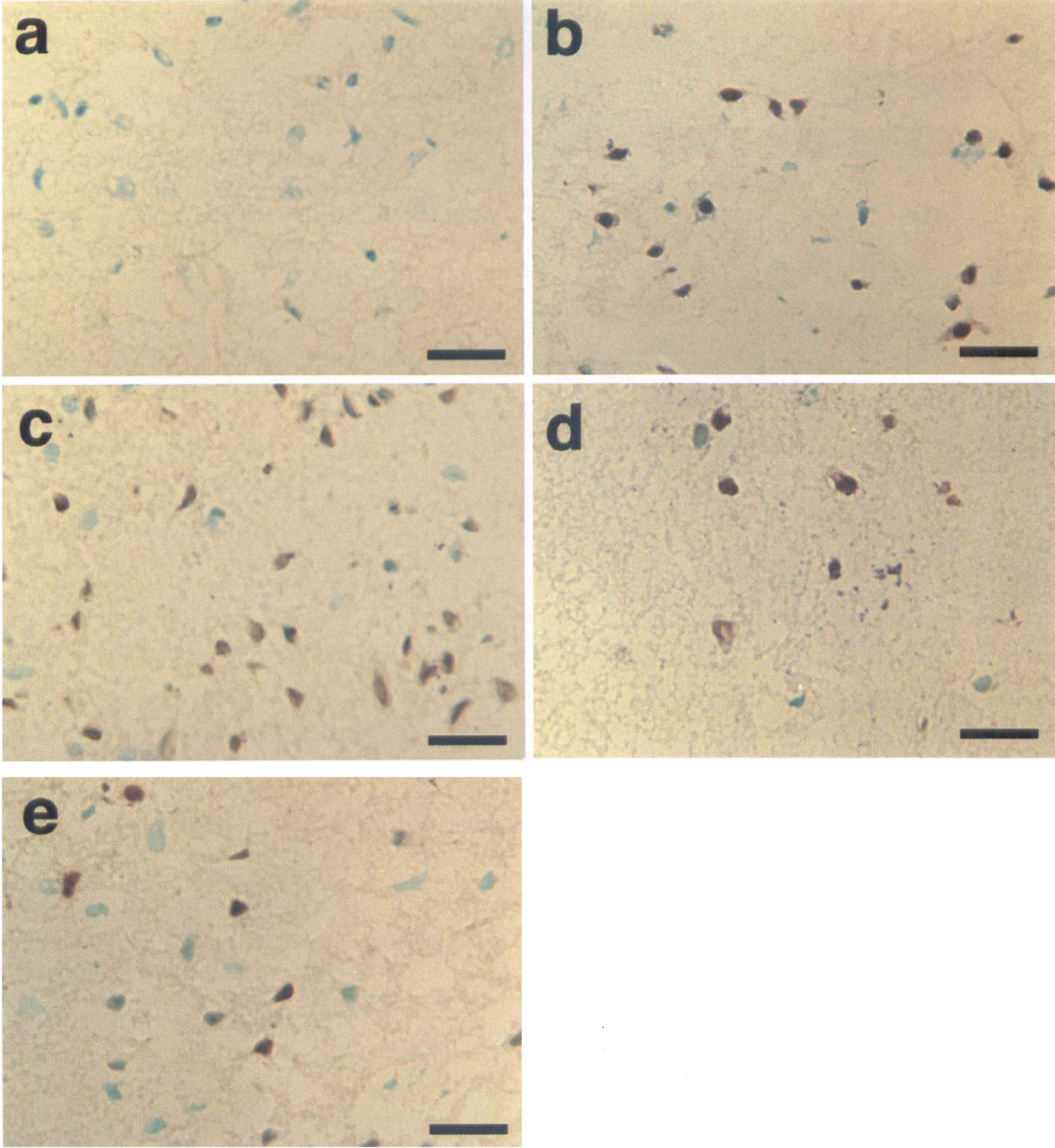
Representative photographs of cerebral cortices of the occluded middle cerebral artery area stained with terminal deoxynucleotidyl transferase-mediated dUTP-biotin in situ nick end labeling (TUNEL). Sham-operated brains showed no positive cells
In order to evaluate these results semiquantitatively, the number of positively stained cells was counted in the cerebral cortex at the boundary zone. In the brains with vehicle application, the number of positively stained cells was 46.4 ± 7.2 cells/mm2 at 24 hours (n = 6), and 68.1 ± 13.6 cells/mm2 at 48 hours after reperfusion (n = 5). However, it was 32.8 ± 8.5 cells/mm2 at 24 hours (n = 6) and 40.7 ± 12.5 cells/mm2 at 48 hours (n = 5) in the brains with VEGF application (Fig. 4). Apoptotic cells were substantially decreased with VEGF application, and the difference was statistically significant both at 24 and 48 hours (P < 0.05).
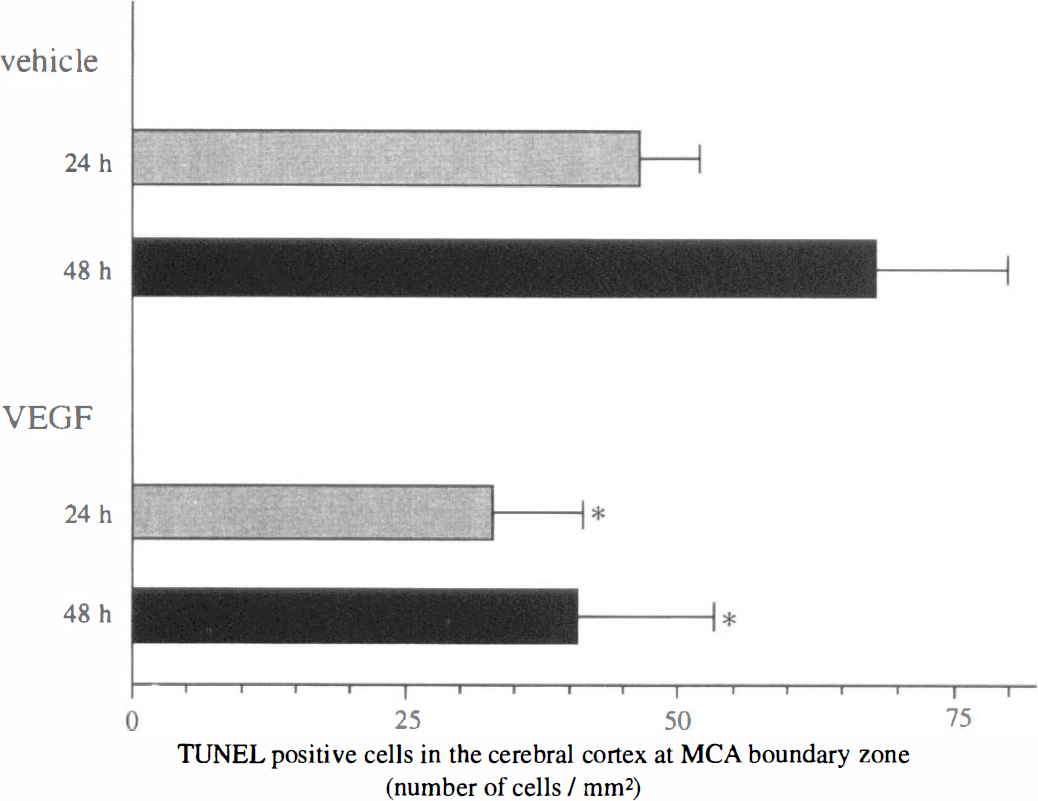
The number of TUNEL-positive stained cells in the cerebral cortex at the boundary zone of the MCA territory and the anterior cerebral artery or the posterior cerebral artery territory. A large number of cells were stained in the vehicle-treated group both at 24 (n = 6) and 48 hours (n = 5). By treatment with VEGF, the number of positively stained cells was substantially decreased both at 24 (n = 6; * P < 0.05) and 48 hours (n = 5; * P < 0.05). MCA, middle cerebral artery; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin in situ nick end labeling; VEGF, vascular endothelial growth factor.
Immunohistochemical analysis for HSP70 expression
HSP70 was not detected in the brains of sham-operated animals (Fig. 5a). In the brains of vehicle-treated animals, however, the neuronal cells in the cortex at the margin of the MCA territory were densely stained, both at 24 (Fig. 5b) and 48 hours (Fig. 5c) after CBF restoration. It is also noteworthy that not only the cytoplasm but also the nuclei was stained in some neurons (Fig. 5b and c). The staining was slightly reduced in the VEGF group compared with vehicle, both at 24 (Fig. 5d) and 48 hours (Fig. 5e) after reperfusion. In addition, the nuclei were not stained in these groups (Fig. 5d and e).
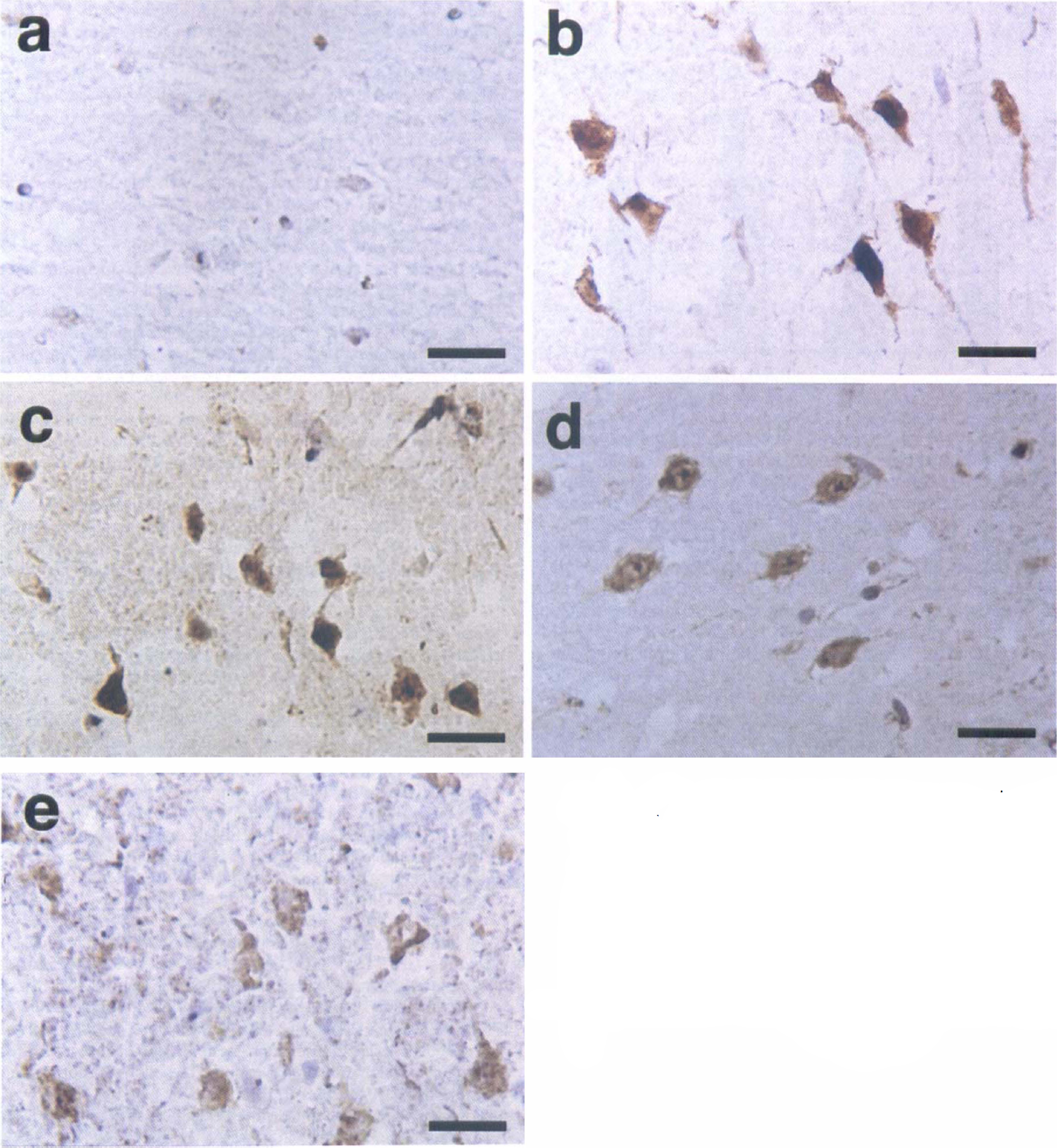
Representative photographs of immunohistochemistry for 70-kDa heat shock protein (HSP70). In sham-operated brains
Also in this study, the results were evaluated semiquantitatively by counting the number of positively stained cells in each grade. The results are summarized in Fig. 6. In the brains with VEGF application, the number of neurons with dense immunoreactivity was lower than that with vehicle application both at 24 hours (n = 6 in each group; P < 0.05) and 48 hours (n = 5 in each group; statistically not significant). The number of moderately stained cells was also lower in the VEGF-treated group (P < 0.05 at 24 hours, and statistically not significant at 48 hours). In contrast, the number of neurons with slight immunoreactivity was larger in the VEGF-treated group (P < 0.05 at 24 hours, and P < 0.01 at 48 hours), suggesting that the injury became relatively mild with VEGF treatment.
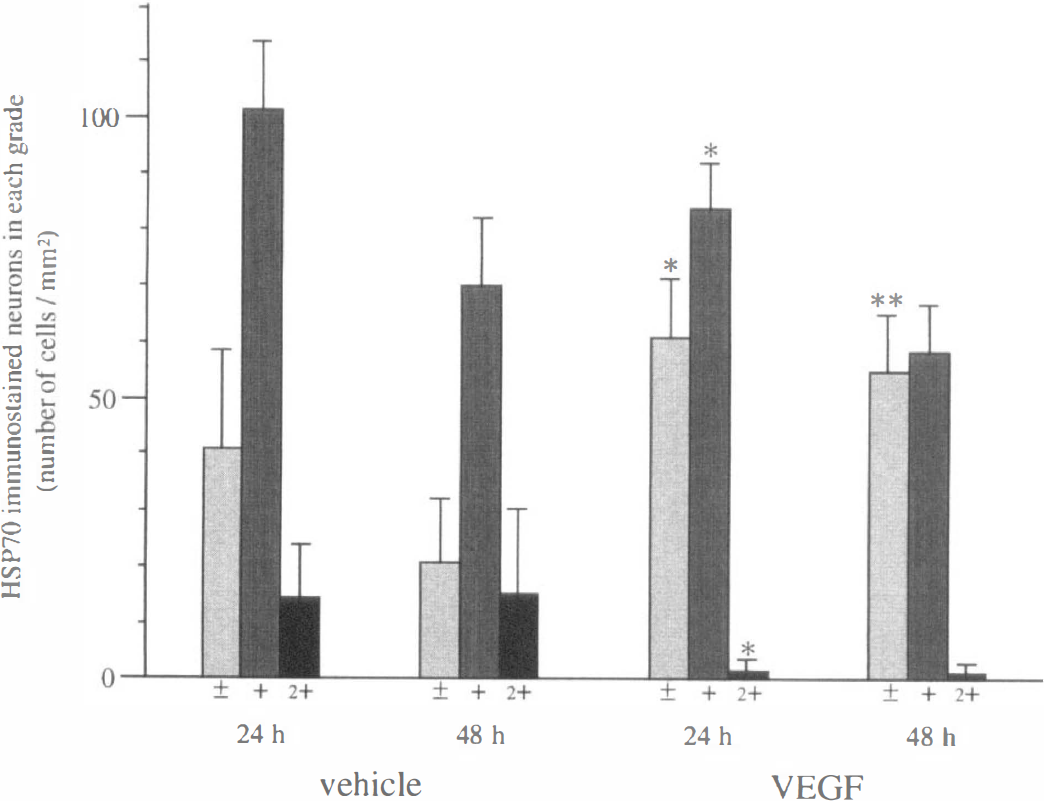
The number of neurons of each staining grade in the cerebral cortex at the boundary zone of the middle cerebral artery territory and the anterior cerebral artery or the posterior cerebral artery territory. Treatment with VEGF significantly increased slightly stained neurons both at 24 (n = 6 in each group; * P < 0.05) and 48 hours (n = 5 in each group; ** P < 0.01). On the other hand, the numbers of neurons with moderate and dense staining at 24 hours were significantly decreased with VEGF treatment (* P < 0.05). At 48 hours, neurons with moderate or dense staining were substantially decreased, but the differences were not statistically significant. VEGF, vascular endothelial growth factor.
DISCUSSION
Vascular endothelial growth factor, also known as vascular permeability factor, is a specific mitogen for endothelial cells and has the potency to increase vascular permeability (Connolly et al., 1989; Leung et al., 1989; Shweiki et al., 1992). Although VEGF gene expression is constitutive in some organs (Monacci et al., 1993; Cao et al., 1996), it is also induced by ischemia in various types of cells (Hashimoto et al., 1994; Pierce et al., 1995). It plays a pivotal role in inducing new vessel formation in tissues suffering from ischemia, and therefore, in animal models of myocardial infarction and hind limb ischemia, it can reduce ischemic injury mainly by potentiating collateral blood flow (Banai et al., 1994; Takeshita et al., 1994a, 1994b; Bauters et al., 1995). In the normal brain, it is expressed in the ependyma, choroid plexus, and granule cells in the cerebellum (Monacci et al., 1993), but its role is not well characterized. Previous studies demonstrated that VEGF was induced in glial cells (Ijichi et al., 1995), endothelial cells (Fisher et al., 1995), and pericytes (Nomura et al., 1995) under hypoxic conditions in vitro, and in glial cells, neurons, and infiltrated macrophages in a permanent MCA occlusion model of rat brain (Kovacs et al., 1996), though its exact role in brain infarct remains to be clarified. Thus, it has not been elucidated whether VEGF application is effective for brain infarct or not. In the present study, we investigated the effect of exogenously applied VEGF on the injured rat brain by transient MCA occlusion.
We employed a transient MCA occlusion model with nylon thread in accordance with previous reports from our laboratory (Nagasawa and Kogure, 1989; Abe et al., 1992). In this model, less than 1 hour of ischemic period produces only reversible neuronal damage, though 3 hours of ischemia produces secondary damage to the anterior cerebral artery-perfused area. Therefore, we selected 90 minutes of ischemia. We applied growth factor by placement of Gelfoam in contact with the surface of the cerebral cortex, in accordance with our previous report (Abe et al., 1997). Our preliminary study with immunohistochemical analysis revealed that exogenously applied VEGF permeated the cerebral cortex of the ischemic core and penumbra. Thus, VEGF could have reached arterioles in the pia mater and brain parenchyma, which are the targets of this factor. The possibility that it spread into the cerebrospinal fluid in the subarachnoid space and had an effect on the ischemic injury could not be excluded. However, immunohistochemical analysis mentioned above revealed that permeation was the major route of VEGF to reach its target.
By quantitative analysis with TTC staining, we demonstrated that the infarct volume was significantly reduced by topical application of VEGF. Because the variability in the extent of damage is small in this model (Koizumi et al., 1986; Nagasawa and Kogure, 1989), the difference of infarct volume should be attributed to application of VEGF, rather than diversity of anatomic variation. The dose of VEGF we chose was 9.0 ng (1.0 ng/μL), because its permeability-potentiating effect reached a plateau at 10 ng in a previous study with intradermal injection (Heiss et al., 1996). This dose was 150 to 500 times higher than the dose for a half-maximal mitogenic response in cultured endothelial cells (Cao et al., 1996), so the concentration should have been sufficient for inducing new vessel formation in the brain. However, the proliferation of endothelial cells usually begins at several days after ischemia, and furthermore, new vessel formation takes days to months (Chen et al., 1994; Krupinski et al., 1994). Thus, the neuroprotective effect of VEGF against ischemia seems to be related to a certain mechanism other than potentiating angiogenesis. The precise target of VEGF on which it has neuroprotective influence is, until now, uncertain. One possibility is that VEGF reduces brain injury through protecting the vascular system against ischemia or reperfusion injury. Vascular endothelial growth factor plays an essential role in maintaining or restoring the integrity of endothelial cells (Tsurumi et al., 1997). A previous study suggested that VEGF prevented capillary obliteration and protected endothelial cells from apoptotic cell death (Alon et al., 1995). It was also demonstrated that VEGF relaxed vascular smooth muscle cells through nitric oxide production (Ku et al., 1993). In this model, regional CBF is restored slowly after removal of nylon thread, and remains at a decreased level for several hours (Nagasawa and Kogure, 1989). Thus, relaxation of vascular smooth muscle cells by VEGF might improve blood flow after thread removal and contribute to the protection of brain tissue from ischemic injury. It is reported that the areas in which neurons tend to survive longer are the same as those demonstrated to be highly angiogenic (Krupinski et al., 1997).
In spite of the effect of VEGF to increase vascular permeability (Connolly et al., 1989), we found significant reduction of brain water content by VEGF application in the brain after transient ischemia (Fig. 1). The results in this study seems to be contradictory to the fact that VEGF plays a key role for the brain edema associated with brain tumor. However, there exist many differences between brain edema associated with brain tumor and that with brain infarct. For example, dexamethasone is effective for reducing brain edema with brain tumor, but not for the treatment of ischemic brain edema (Fishman, 1982). On the other hand, dexamethasone inhibited VEGF gene expression and reduced the ability of VEGF to increase vascular permeability (Bruce et al., 1987). These data suggest that VEGF is not mainly involved in ischemic brain edema formation. In the relatively early phase, ischemic brain edema formation is mainly caused by cytotoxic edema rather than vasogenic edema (Betz and Coester, 1990). It is reported that disruption of sodium transport through sodium-potassium ATPase, which occurs in a condition of energy failure, is the cause of cell swelling and cytotoxic edema (Betz et al., 1989). One possible mechanism of VEGF in the reduction of ischemic brain edema is, therefore, that it reduced the infarct volume by protection of endothelial cells, and thus reduced water content, without exacerbating vasogenic edema. The present study revealed that topical application of VEGF reduced EB extravasation significantly (Fig. 2). Evans blue binds with albumin and moves along with it in vivo (Kawagoe et al., 1992). Together with the previous finding that VEGF application through the carotid artery did not cause albumin extravasation (Criscuolo et al., 1990), it is probable that VEGF protected the blood-brain barrier rather than destroyed it. This amelioration of blood-brain barrier injury may be attributable to the protective effect of VEGF on endothelial cells.
The TUNEL staining was also decreased by VEGF application (Figs. 3 and 4). As this staining detects cells undergoing not only apoptosis but also necrosis (States et al., 1996), it cannot be concluded that VEGF prevents apoptosis in ischemia. Receptors for VEGF, i.e., flt and flk, are expressed exclusively in endothelial cells and not in neuronal cells (Cao et al., 1996; Gitay-Goren et al., 1996). Therefore, the decrease of TUNEL-positive stained neuronal cells is a secondary effect of VEGF in neuronal cells. In the CNS, HSP70 is induced by various injuries, and the degree of the stress and its induction is proportional (Gonzalez et al., 1989). In the present study, immunoreactivity for HSP70 was decreased in the VEGF-treated group (Figs. 5 and 6). In addition, VEGF-treated brain showed HSP70 immunoreactivity only in the cytoplasm of neurons, though vehicle-treated brain showed it in both the cytoplasm and nuclei of neurons (Fig. 5). Because HSP70 in the cytosol migrates into the nucleus during severe stresses and prevents protein malfolding in the nucleus (reviewed by Gething and Sambrook, 1992), the lack of immunoreactive HSP70 in nuclei (Fig. 5D and E) may also support the reduction of ischemic damage by VEGF application.
A previous report suggested that a subpopulation of neuronal cells stained with TUNEL or HSP70 immunohistochemistry were essentially different, and that only 5% to 7% of the cells were overlapping (States et al., 1996). In that experiment, most of the neurons stained with TUNEL eventually died, though most of the HSP70-expressing neurons did not. Recently we demonstrated marked reduction of TUNEL-stained neurons by topical application of glial cell line-derived neurotrophic factor in rat brain after transient ischemia, with only slight reduction of immunoreactive HSP70 induction (Abe et al., 1997). In the current study, however, modification of both TUNEL and HSP70 staining by VEGF application occurred to a relatively slight degree. This difference may be because glial cell line-derived neurotropic factor is a neurotrophic factor and protected neuronal cells directly, but the protective effect of VEGF is not direct on neuronal cells.
We demonstrated that exogenously applied VEGF reduced the brain infarct size, edema formation, and blood-brain barrier destruction after transient MCA occlusion. Furthermore, it reduced neuronal cell damage caused by ischemia. In the future, treatment with VEGF might become an effective means of therapy for ischemic stroke.