
Abstract
Erythropoietin (Epo) is gaining interest in various neurological insults as a possible neuroprotective agent. We determined the effects of recombinant human Epo (rhEpo, 5000 IU per kg bw) on brain edema induced in rats by traumatic brain injury (TBI; impact-acceleration model; rhEpo administration 30 mins after injury). Magnetic resonance imaging (MRI) and a gravimetric technique were applied. In the MRI experiments, the apparent diffusion coefficient (ADC) and the tissue T1 relaxation time were measured hourly in the neocortex and caudoputamen, during a 6 h time span after TBI. In the gravimetric experiments, brain water content (BWC) was determined in these two regions, 6 h after TBI. Apparent diffusion coefficient measurements showed that rhEpo decreased brain edema early and durably. Gravimetric measurements showed that rhEpo decreased BWC at H6 in the neocortex as well as in the caudoputamen. No significant differences in ADC, in T1, or in BWC were found between rhEpo treated-TBI rats and sham-operated rats. Our findings show that post-traumatic administration of rhEpo can significantly reduce the development of brain edema in a model of diffuse TBI. Further studies should be conducted to identify the biochemical mechanisms involved in these immediate effects and to assess the use of rhEpo as a possible therapy for post-traumatic brain edema.
Introduction
The pathophysiology of traumatic brain injury (TBI) is very complex—it induces excitotoxic damage, alterations in calcium homeostasis, free radical induced damage, inflammation, and mitochondrial dysfunction (Enriquez and Bullock, 2004). Various mediators are released, resulting in the occurrence of diffuse brain edema. The latter plays a major role in the worsening of the neurological status through the elevation of intracranial pressure and a concomitant impairment of brain perfusion (Unterberg et al, 2004). While numerous compounds have shown neuroprotective effects in experimental models of TBI, there is no pharmacological agent specifically aimed at blocking the progression of diffuse brain edema in the clinical setting, and therapy applied to patients with diffuse brain edema is still purely symptomatic (Narayan et al, 2002).
Among the agents with neurological tropism, erythropoietin (Epo) has been shown to confer neuroprotection and to reduce neuronal death in many in vivo and in vitro models of central nervous system (CNS) injuries (Bernaudin et al, 1999; Brines et al, 2000; Gorio et al, 2002; Grasso et al, 2002; Sakanaka et al, 1998; Siren et al, 2001). Also, improved neurological outcome and reduced lesion size were obtained in ischemic stroke patients treated with recombinant human Epo (rhEpo) (Ehrenreich et al, 2002). The mechanisms by which rhEpo exerts neuroprotective action are, however, incompletely understood. The overall expression of Epo receptors by neurons, astrocytes, and capillary endothelial cells elicited by hypoxia, ischemia (Bernaudin et al, 2000), or by nervous tissue injury (Grasso et al, 2005) supports the hypothesis of a well-organized Epo and Epo receptors network involved in the setting of an injury. While literature on the versatile properties of rhEpo in treating CNS disorders has flourished (Brines and Cerami, 2005; Grasso et al, 2004; Maiese et al, 2004), the effects of rhEpo after TBI have been sparsely studied. In a model of focal TBI induced by a blow delivered to the cortex, mice receiving rhEpo before or after TBI developed less necrosis at 10 days after injury than controls (Brines et al, 2000). In mice subjected to focal TBI, injection of rhEpo at 1 and 24 h after TBI induced a significant reduction of inflammation and of neuronal apoptosis, along with improved functional recovery (Yatsiv et al, 2005). Rats treated with rhEpo daily for 14 days after a focal TBI had an improvement in the performance of spatial memory tasks and increased neurogenesis in the dental gyrus (Lu et al, 2005). Since rhEpo could limit the production of tissue-injuring molecules (glutamate, radical oxygen species) (Morishita et al, 1997; Sakanaka et al, 1998) that are involved in the occurrence of diffuse brain edema, we hypothesized that rhEpo could reduce the development of post-traumatic brain edema. In a model of TBI resulting in early diffuse brain edema, rats receiving rhEpo after TBI were compared with non-treated TBI rats and to sham-operated rats. We used diffusion-weighted MR imaging to monitor the course of brain edema over 6 h after injury, and a gravimetric technique to measure brain water content (BWC) 6 h after the insult. We also tested T1 mapping as a possible approach to quantify regional BWC after TBI.
Materials and methods
Experimental Protocol
Male Wistar rats (350 to 400 g) were studied using magnetic resonance imaging (MRI) or a gravimetric technique. In both experiments, three groups of rats were studied: a first group of rats was subjected to TBI and a saline sham treatment (TBI-saline, n = 12 rats). A second group of rats was treated with rhEpo 30 mins after TBI (TBI-rhEpo, n = 12 rats). A third group was sham-operated rats, receiving either saline (n = 6 rats) or rhEpo (n = 6 rats), without TBI.
Preparation of animals was similar in all groups and conformed to the guidelines of the French Government (decree No. 87 to 848 of 19 October 1987, licenses 006683 and A38071) and to the European Guidelines for the Protection of the Vertebrate Animals (ETS No. 123, Strasbourg, 18 March, 1986). Anesthesia was induced and maintained with isoflurane during animal preparation. One percent lidocaine was injected subcutaneously for local anesthesia at all surgical sites. Each animal was placed in a stereotactic frame. A midline scalp incision was made and the skin and periosteum were retracted from the skull surface. A brass disc (10 mm in diameter and 3 mm thick) was then glued to the skull vault between the coronal and the lambdoid sutures. Catheters were inserted into the femoral artery and vein to monitor mean arterial blood pressure and to deliver drugs, respectively. Blood gases (PaO2 and PaCO2), arterial oxygen saturation of hemoglobin (SaO2), arterial pH (pHa) and hemoglobin content (tHb) were determined from less than 0.1 mL arterial blood samples (ABL 510, Radiometer, Copenhagen, Denmark). After tracheostomy, rats were mechanically ventilated with 60% air–40% oxygen using a rodent ventilator (SAR-830/P, CWE Inc., Ardmore, PA, USA). After completion of the surgical procedures, isoflurane was discontinued and anesthesia was maintained with intravenously administration of alpha-chloralose (80 mg/kg initially, followed by 26.7 mg/kg h) and pancuronium bromide (0.5 mg/kg initially, followed by 0.2 mg/kg h) in a normal saline solution at a rate of 2 mL/h throughout the study. The use of alpha-chloralose allowed to preserve functional metabolic coupling and to limit cerebral vasodilation (Silva et al, 1999). Isoflurane wash-out was allowed for more than 30 mins before measuring baseline physiological values with alpha-chloralose. Ventilation was adjusted to maintain PaCO2 at 30 to 35 mm Hg. Rectal temperature was maintained at 36.5°C ± 0.5°C by means of a heating pad placed under the abdomen.
Head trauma was induced according to the impact-acceleration model (Marmarou et al, 1994). The animal was placed prone on a foam bed within a Plexiglas frame and it was kept in place with belts. The injury was induced by dropping a 500 g mass through a vertical Plexiglas guide from a height of 1.5 m onto the metallic disc glued on the skull. Before applying the injury, the impactor was manually lowered onto the disc to ensure precise contact with it. Mechanical ventilation was transiently disconnected (< 10 secs) during the impact to prevent displacement of the tracheal tube. Rebound impact was prevented by sliding the Plexiglas frame away after the initial impact. After the impact, the metallic disc was removed and the scalp sutured. The animal was then installed in the MRI scanner to be monitored during the six ensuing hours. Criteria for exclusion from the study were the persistence, for 10 mins at least during the experiment, of out of range values of certain physiological parameters: mean arterial blood pressure < 80 mm Hg, PaO2 < 100 mm Hg, PaCO2 > 45 mm Hg, tHb < 80 g/L. The reference time (H0) corresponded to the TBI impact (TBI-rhEpo and TBI-saline) or equivalent time (sham-operated). Each group was administered (intravenously), at 30 mins after H0, either 0.5 mL of an isotonic saline solution, or 5000 IU/kg of Epoetin alfa (Eprex®, Janssen-Cilag Division Ortho Biotech, Issy-les-Moulineaux, France) diluted in 0.5 mL of saline solution. The sham-operated rats were treated similarly, but they were not submitted to TBI.
Magnetic Resonance Imaging Measurements
In a first series of experiments, MRI was performed at 7 T, in a horizontal bore magnet (Magnex Scientific Ltd, Oxford, UK) equipped with a 200 mT/m gradient system (12 cm inner bore diameter) and an SMIS console (SMIS Ltd, Guildford, UK). The rat was lying prone, its head secured via ear bars. A 79 mm internal diameter quadrature volume coil (Rapid Biomedical GmbH, Würzburg, Germany) was used for excitation and for detection. After radiofrequency coil matching and tuning, the magnetic field homogeneity was adjusted to obtain a half-height linewidth for water smaller than 0.3 part per million in the brain. A T1-weighted axial scout image was acquired to locate slices of interest underneath the metallic disc area. Five adjacent coronal slices (1.5 mm thickness) were then acquired in a multiple slice mode for diffusion weighted imaging, and one slice (1.5 mm thickness) co-localized with the central coronal image for T1 mapping.
Diffusion weighted images were acquired hourly from H1 to H6 using a T2-weighted spin-echo sequence (repetition time = 2000 ms; echo time = 80 ms; field of view = 30 × 30 mm2; acquisition matrix = 128 × 66; 2 averages) (Figure 1). In addition to a first image acquired without diffusion weighting, three images were acquired in the presence of diffusion sensitizing gradients applied in each of the three main directions (x, y, z) of the gradient system, respectively. Diffusion sensitization was obtained by applying the diffusion gradients symmetrically about the inversion RF pulse. Parameters of the diffusion sensitization were the following: δ = 9 ms, Δ = 20 ms, b = 500 s/mm2. Acquisition of the set of diffusion-weighted images lasted about 15 mins. T1 maps were also obtained hourly from H1 to H6, using an inversion recovery FLASH sequence (repetition time = 10 s; echo time = 3.2 ms; field of view = 30 × 30 mm2; matrix = 64 × 64; 4 averages) with 12 inversion times (TI) ranging between 100 and 6000 ms. The inversion pulse was specially nonselective to minimize in-flow contributions to the signal. Acquisition of each T1 map lasted approximately 5 mins.
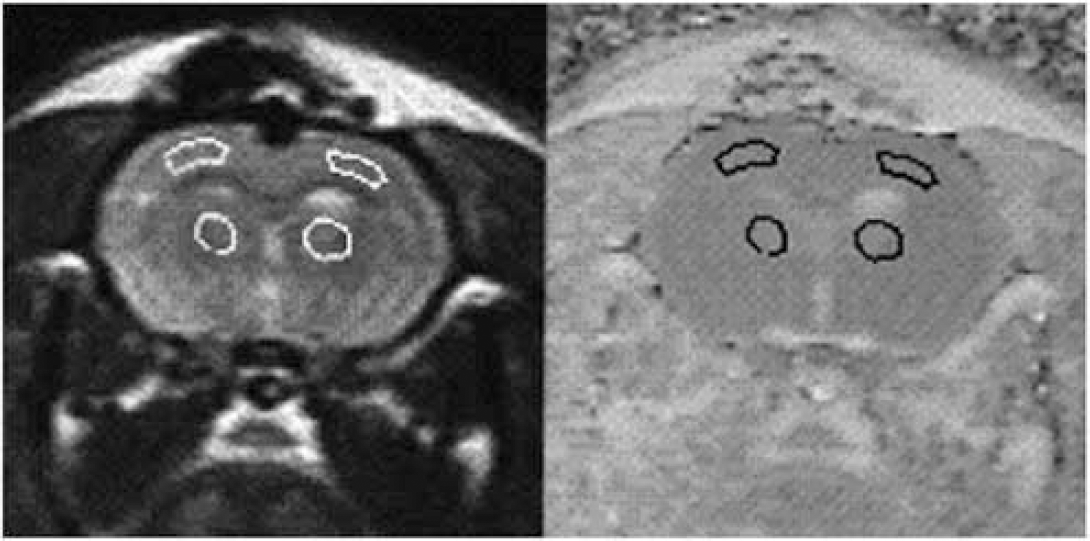
Representative T2-weighted (left) and diffusion-weighted (right) images, with the two regions of interest delineated in each hemisphere: neocortex and caudoputamen.
Apparent diffusion coefficient (ADC) and T1 maps were calculated on a Sunsparc workstation (Sun Microsystems Inc., Mountain View, CA, USA). The ADC maps were generated pixelwise by averaging the ADC values obtained for each of the three directions as follows:
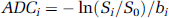
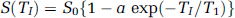
Gravimetric Measurements
In a second series of experiments, BWC was determined 6 h after TBI (or equivalent) in three groups of rats: TBI-saline (n = 12), TBI-rhEpo (n = 12), and sham-operated (n = 12). After the completion of a similar experimental protocol, rats were killed by an intravenous injection of a KCl solution. The brain was rapidly removed and placed in a rat brain matrix previously cooled in ice. Two, 2-mm thick, coronal slices were cut through the impact lesion. In each hemisphere, samples (20 to 40 mg each) were taken from the neocortex (n = 4) and from the caudoputamen (n = 3) and subjected to gravimetric analysis. The gravimetric technique determines the tissue specific gravity (SG), a parameter that is known to correlate with BWC (Marmarou et al, 1982). The technique uses a layered kerosene–bromobenzene linear column. After calibration of the column with 7.5 μL drops of anhydrous K2SO4 solutions with known SGs (1.050, 1.045, 1.040, 1.035, 1.030, and 1.025), brain samples were deposited in the column, and their SG noted 2 mins later. Samples containing gas bubbles or blood were excluded from the analysis. The BWC was then derived according to the equation applied to grey matter (Fatouros and Marmarou, 1999):
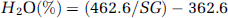
Statistical Analysis
Data are expressed as mean ± s.d. Analysis for statistical significance of changes during the 6 h time course was performed with two-way (group × repeated values) analysis of variance for repeated measurements (StatView SE program, SAS Institute Inc, Cary, NC, USA). Each value was compared with that obtained at the reference time (H0 for the physiological data, or H1 for the MRI measurements) using the Scheffé F test as a post hoc test (intragroup analysis). Comparisons between the groups of rats were subjected to factorial analysis of variance using the Scheffé F test as a post hoc test (intergroup analysis). Considering the diffuse nature of the insult, the results from the two hemispheres were averaged to provide a mean estimate for neocortex (or caudoputamen) values. Statistical significance was declared when P < 0.05.
Results
Magnetic Resonance Imaging Experiments
Physiological data are shown in Table 1. Temperature (H1), arterial pH (H6), and PaO2 (H1 and H6) decreased slightly over time in the three groups, with no intergroup differences. PaCO2 remained unchanged throughout the experiments. Significant interaction between groups and serial measurements was found for mean arterial blood pressure (F = 3.6; P = 0.01) and for the hemoglobin content (F = 3.1; P = 0.02). This was due, respectively, to mean arterial blood pressure increase at H6 in the TBI-rhEpo and in the sham-operated groups, and to hemoglobin decrease at H6 in the TBI-rhEpo group. The two TBI groups (TBI-saline and TBI-rhEpo) had comparable data at any time of the experiment. No MRI evidence for intracerebral hemorrhage was found at the site of impact in these two groups.
Physiological data in the three groups of rats having undergone the MRI experiments: TBI with saline (n = 12), TBI with rhEpo (n = 12), and sham-operated (n = 12)
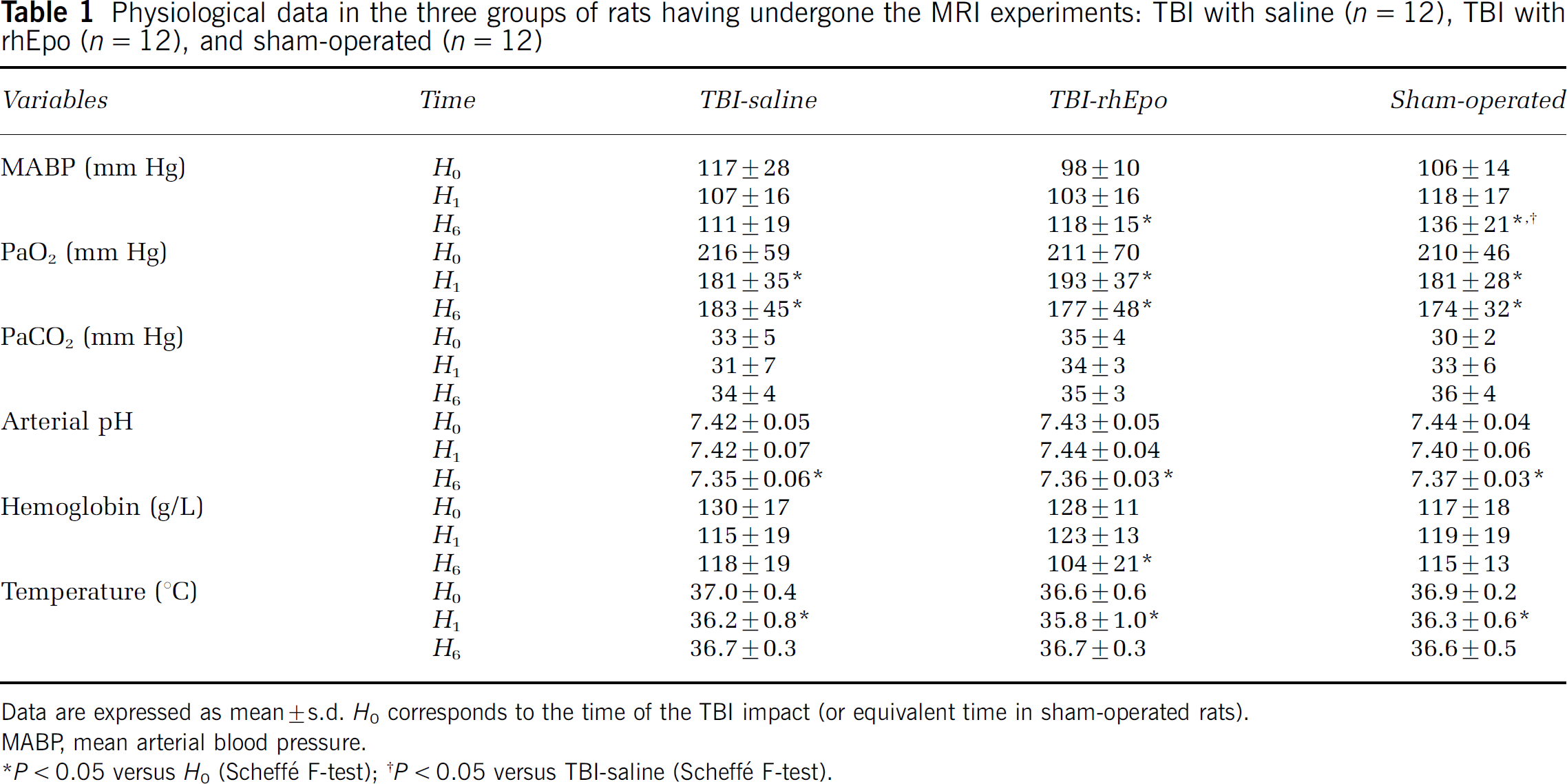
Data are expressed as mean ± s.d. H0corresponds to the time of the TBI impact (or equivalent time in sham-operated rats). MABP, mean arterial blood pressure.
P< 0.05 versus H0(Scheffé F-test);
P< 0.05 versus TBI-saline (Scheffé F-test)
Apparent diffusion coefficient measurements in the neocortex and in the caudoputamen were performed on all rats from the three groups. No significant ADC changes over time were found within each group of rats in the neocortex or in the caudoputamen (intragroup analysis). Apparent diffusion coefficient values in the neocortex were significantly lower in the TBI-saline group during the entire experiment: overall mean values of 0.61 × 10−3 ± 0.13 × 10−3 versus 0.73 × 10−3 ± 0.11 × 10−3 (TBI-rhEpo) and 0.71 × 10−3 ± 0.09 × 10−3 mm2/sec (sham-operated), respectively (P < 0.01). Similar differences were found for the ADC values in the caudoputamen: 0.60 × 10−3 ± 0.19 × 10−3 versus 0.74 × 10−3 ± 0.10 × 10−3 (TBI-rhEpo) and 0.79 ± 0.14 × 10−3 mm2/sec (sham-operated), respectively (P < 0.01). Intergroup analysis indicated significant differences between the TBI-saline group and the two other groups at the different measurement times of the experiment (Figures 2A and 2B). No differences in ADC were found between the TBI-rhEpo and the sham-operated groups.
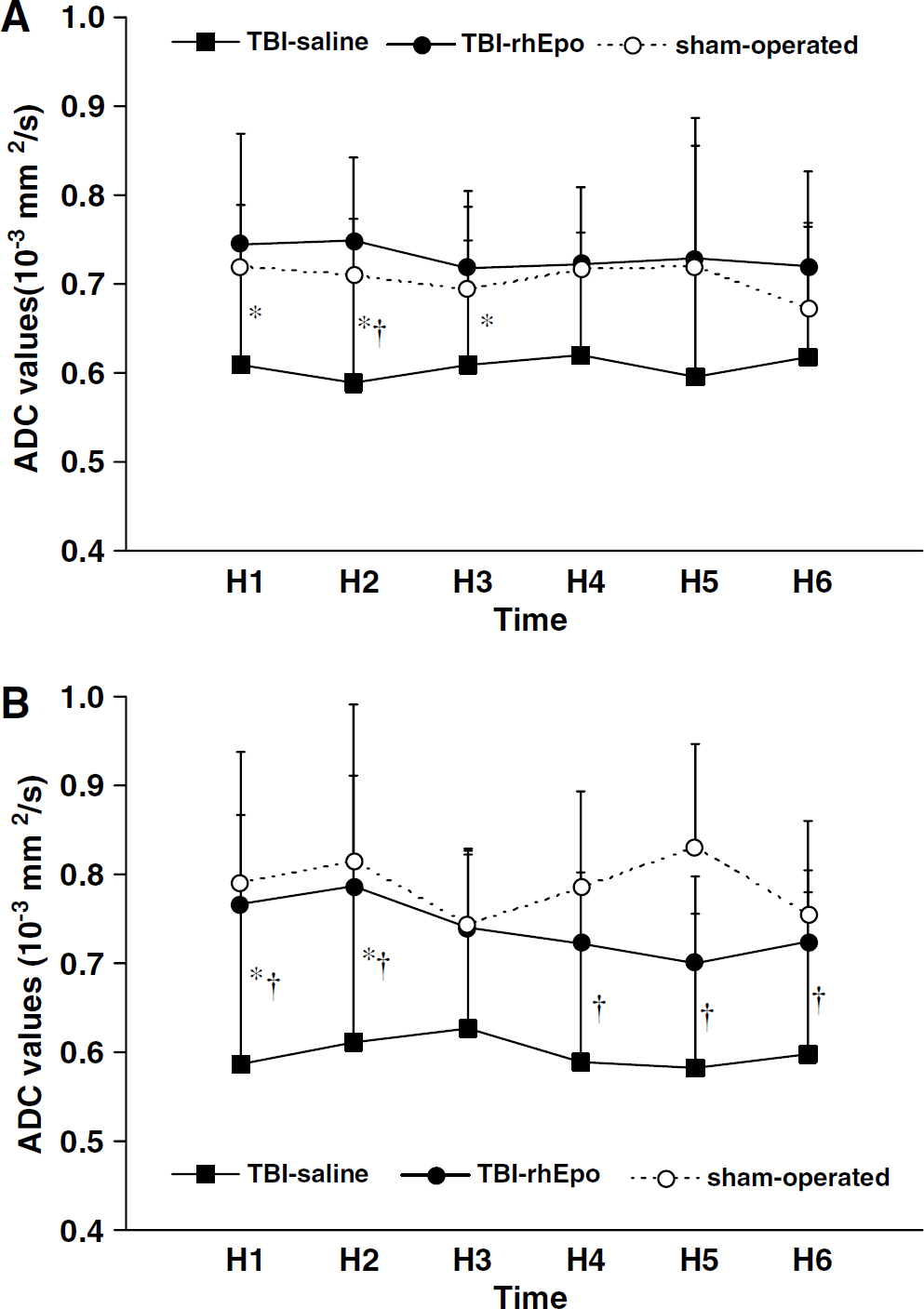
Time course of ADC values in neocortex (
T1 maps were obtained in 35 rats (one rat of the sham-operated group was excluded because of insufficient image quality). No significant T1 changes over time were observed within each group of rats, in the neocortex as well as in the caudoputamen. T1 values during the entire experiment were significantly longer in the TBI-saline group than in the TBI-rhEpo group in neocortex and in caudoputamen: 1821 ± 62 versus 1782 ± 85 and 1745 ± 119 versus 1676 ± 81 ms, respectively (P < 0.01) (Figure 3). No differences in T1 values were found between the TBI-rhEpo group and the sham-operated group. However, there were minor differences between the three groups when compared at the different measurement times of the experiment (intergroup analysis) (data not shown).
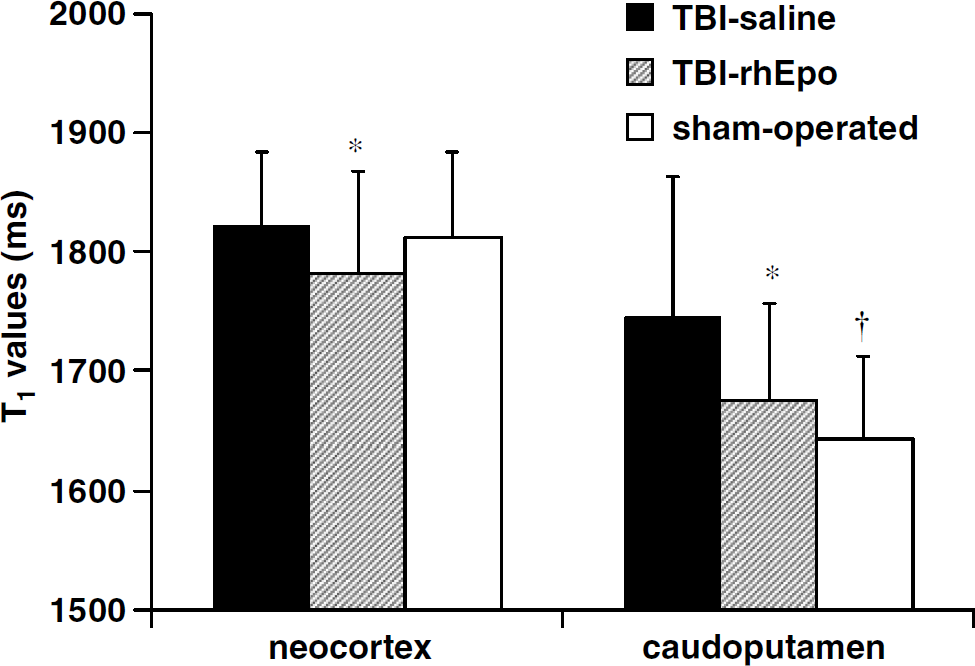
Quantitative T1 mean values in neocortex and in caudoputamen during the entire experiment in the three groups of rats: TBI with saline (n = 12), TBI with rhEpo (n = 12), and sham-operated (n = 11) (MRI experiments). Data are expressed as mean ± s.d. *P < 0.05 TBI-saline versus TBI-rhEpo; †P < 0.05 TBI-saline versus sham-operated.
Gravimetric Experiments
Specific gravity measurements were performed at H6 on 36 rats, 12 in the TBI-saline group, 12 in the TBI-rhEpo group, and 12 in the sham-operated group. No significant differences in the physiological parameters of these three groups were found between the two series of experiments, MRI versus gravimetric (data not shown). Brain water content measurements in neocortex and in caudoputamen were significantly lower in the TBI-rhEpo group than in the TBI-saline group: 78.2% ± 0.3% versus 79.1% ± 0.4% and 78.0% ± 0.3% versus 78.6% ± 0.5%, respectively (P < 0.01) (Figure 4). No differences in BWC were found between the TBI-rhEpo group and the sham-operated group.
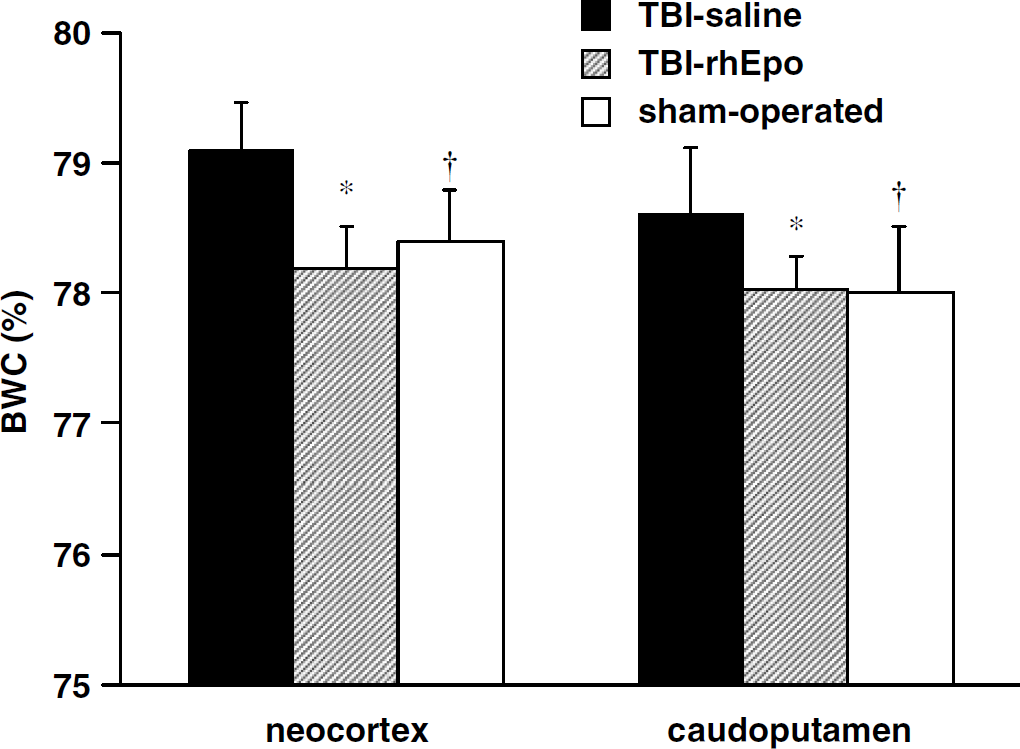
Brain water content in neocortex and in caudoputamen at H6 in three groups of rats: TBI with saline (n = 12), TBI with rhEpo (n = 12), and sham-operated (n = 12) (gravimetric experiments). Traumatic brain injury impact was delivered on H0 for TBI-saline and TBI-rhEpo groups. Data are expressed as mean ± s.d., *P < 0.05 TBI-saline versus TBI-rhEpo. †P < 0.05 TBI-saline versus sham-operated.
Discussion
This study shows that the administration, postinjury, of rhEpo can significantly alter the development of TBI-induced brain edema in rats. The effect of rhEpo is early and lasts at least 6 h, as shown by serial MR measurements of the ADC and of tissue T1, and by BWC measurements at H6. These parameters show consistent results in favor of a strong action of post-TBI administration of rhEpo against TBI-induced brain edema.
To investigate the effects of rhEpo following diffuse TBI, we used the rodent impact-acceleration model (Marmarou et al, 1994). In pilot experiments on spontaneously breathing rats (n = 42), we observed, after TBI, a rate of skull fractures of 5% and a mortality rate of 45% at animal awakening, in good agreement with the initial report (Marmarou et al, 1994). To prevent mortality, we maintained animals under anesthesia and mechanical ventilation during the course of the experiments. Impact-acceleration model does not induce subdural- and intraparenchymal hemorrhages, in contrast to the focal TBI models such as the fluid percussion model and the controlled cortical impact model. In addition, early diffuse injury of neurons, axons and microvasculature, astrocytic swelling in cortex, and massive diffuse axonal swelling in corpus callosum, thalamus, internal capsule, and brain stem were reported using impact-acceleration model (Foda and Marmarou, 1994). Diffuse brain edema, elevated intracranial pressure and brain hypoperfusion were reported as early as the first hour postinjury in anesthetized and ventilated rats (Barzo et al, 1997; Ito et al, 1996). Given these observations, we performed a longitudinal study of TBI-induced brain edema starting 1 h after trauma, with the neocortex and the caudoputamen as the sites of edema measurement.
We used diffusion-weighted MR imaging and T1 mapping over 6 h after TBI. Diffusion-weighted imaging is a well-documented technique to study changes in ADC values in various CNS insults and to indicate the nature of brain edema (Huisman, 2003). The reduction in brain ADC is thought to represent mainly cellular (cytotoxic) edema, while an ADC increase indicates mainly vasogenic edema resulting from blood–brain barrier (BBB) disruption. In the fluid percussion model (focal TBI), a significant decrease in tissue ADC was reported 1 to 2 h post-trauma (Albensi et al, 2000; Assaf et al, 1999). In the rodent impact-acceleration model (diffuse TBI), ADC measurements were less conclusive. An ADC increase up to 4 h post-TBI was reported along with an increased BBB permeability (Cernak et al, 2004). Others found no significant post-TBI ADC changes in normotensive and normo-ventilated rats (Ito et al, 1996). A biphasic response to injury was found with a transient ADC increase lasting less than 1 h after TBI (vasogenic edema), followed immediately by a prolonged and marked reduction in brain ADC (cellular edema) (Barzo et al, 1997). Although we cannot exclude a transient post-TBI increase (< 1 h) in the permeability of the BBB, the lower ADC measurements found from H1 on in the TBI-saline group compared with the sham-operated group are in line with the latter findings, suggesting cellular predominance of the post-TBI edema. Our ADC measurements thus indicate that the postinjury treatment with rhEpo can significantly reduce the cellular contribution to brain edema. Postinjury rhEpo treatment did not affect ADC of brain water in rats subjected to a stereotactic resection of the frontal lobe (Matchett et al, 2006). Whether the lack of response is due to the vasogenic nature of this model with BBB breakdown or to the limited area of damaged tissue is unclear.
To our knowledge, this study is the first to use T1 mapping to monitor the evolution of post-traumatic brain edema. T1 mapping has been successfully used to quantify BWC, and a linear relationship was obtained between R1 (= 1/T1) and 1/BWC following stroke (Fatouros et al, 1991; Lin et al, 2000). In a model of focal brain ischemia in rat, a significant increase in ischemic tissue T1 was reported, together with an ipsilateral decrease in brain ADC and BWC increase as measured with the gravimetric technique (Barbier et al, 2005). Our results are consistent with those findings, showing an increase in the tissue T1 in the TBI-saline group. They are also in line with the ADC results by showing a reduction in the tissue T1 values in rhEpo-treated TBI rats. This indicates a possible benefit of rhEpo on the occurrence of brain edema. The T1 results were however less clearcut than those of the ADC measurements (Figure 3). This could be due to brain water accumulation being more reduced after diffuse TBI than after occlusion of a terminal brain artery in rats: 0.5% to 1% in this TBI model (Cernak et al, 2004) versus 2% to 4% in models of ischemia (Barbier et al, 2005; Lin et al, 2000). Similarly, moderate brain water accumulation was reported in diffusely head-injured patients presenting brain swelling and concomitant elevated intracranial pressure (Marmarou et al, 2006).
We found strong differences in the ADC and BWC values between rhEpo-treated and saline-treated TBI rats, providing evidence for rhEpo affecting brain water accumulation after TBI. In both experiments, physiological parameters of the two groups of TBI rats (rhEpo versus saline) were comparable and maintained within normal ranges. The 0.5% to 1% rhEpo-induced reduction in BWC at H6 may significantly improve brain perfusion in closed head injury with low compliance (Frei et al, 1973). Although we could not address whether the drug had a persistent effect, these results are in line with those obtained in rhEpo-treated mice showing attenuation of cell apoptosis and tissue inflammation, and improvement of neurological outcome 3 to 14 days after focal TBI (Yatsiv et al, 2005). In the present study, ADC differences between treated and non-treated groups were found as early as the first hour post-TBI. Actually, rhEpo can rapidly cross the BBB since a significant increase in the concentration of Epo in cerebrospinal fluid has been found as soon as 30 mins after intraperitoneal administration of rhEpo at 5000 UI/kg (Brines et al, 2000). A peak serum level of 2000 ng/mL was obtained 15 mins after intravenous administration of a similar dose of rhEpo (Gorio et al, 2005). Despite that the exact mechanisms by which rhEpo mediates its effects across the BBB are uncertain, these findings might account for immediate changes in biochemical pathways involved in injured brain tissue after intravenous injection of rhEpo.
At present, we can only formulate conjectures to explain our main findings. While apoptotic and necrotic pathways after TBI and antiapoptotic effects of rhEpo in models of ischemia and of TBI have been well identified (Raghupathi, 2004; Siren et al, 2001; Yatsiv et al, 2005), such mechanisms need several hours after the insult to be effective. Similarly, neuroinflammatory response is another target for rhEpo (Brines et al, 2000; Yatsiv et al, 2005). Tissue accumulation of leukocytes, activation of glial cells, and/or cytokine production equally occur hours after TBI (Enriquez and Bullock, 2004). We should thus consider processes occurring within minutes after TBI, such as the release, by injured cells, of glutamate, lactate, proton, potassium, calcium, nitric oxide (NO), arachidonic acid, free oxygen radicals, histamine, and kinins (Unterberg et al, 2004). In response to the early release of glutamate into the extracellular space, glutamate uptake by astrocytes is enhanced through sodium-dependant transporters, leading to astrocyte swelling and intracellular calcium accumulation (Chen and Swanson, 2003). This ionic imbalance with a dramatic intracellular influx of sodium, calcium and/or, chloride may explain early glial swelling after injury (Kimelberg, 2004). Rapid and massive intracellular accumulation of free calcium was found within minutes after TBI (Nilsson et al, 1993). Therefore, the biological basis of an immediate beneficial effect of rhEpo could involve modulation of the glutamate release, restoration of ionic imbalance, and/or alteration in free oxygen radicals production, including NO. In cultured neurons subjected to chemical ischemia, activation of Epo receptors inhibited the exocytosis of glutamate (Kawakami et al, 2001). Pretreated neurons with rhEpo prevented glutamate-induced neuronal damage (Bernaudin et al, 1999; Morishita et al, 1997). In animals subjected to brain ischemia, post-treatment with rhEpo significantly reduced the NO toxicity to neurons, inhibited its tissue production, and reduced the formation of brain edema (Calapai et al, 2000; Sakanaka et al, 1998). Binding of Epo to its neuronal receptor triggers the activation of several Epo signal-regulated kinases and protein kinases (Jak2 and NF-κB), which in turn modulates damage because of the generation of glutamate, free radicals, and NO in cultured neurons (Digicaylioglu and Lipton, 2001). We are investigating whether the post-TBI administration of rhEpo results in an early activation (< 1 h after treatment) of such molecular pathways. There are yet concordant results to support an early benefit of post-traumatic treatment with rhEpo on the formation of brain edema.
Epoietin alfa was administered at a dose of 5000 UI/kg because this dose had been proven effective in various models of CNS injuries (Brines et al, 2000; Gorio et al, 2002; Siren et al, 2001; Yatsiv et al, 2005). Although a dose–response study of rhEpo has not yet been conducted in TBI, benefit increased with the dose of rhEpo in models of spinal cord injury (Gorio et al, 2002). The rhEpo dose used here is already higher than that currently recommended in the clinical setting, and concerns may be raised about the safety of rhEpo when administered for neuroprotection (Grasso et al, 2004). The development of fully neuroprotective analogs of rhEpo without any erythropoietic action should thus be of particular interest (Leist et al, 2004). If such analogs of rhEpo also appear beneficial in treating post-traumatic edema, their optimal therapeutic window and dose will need to be determined before using them in the clinical setting.
In conclusion, our findings add to the accumulating evidence of beneficial effects of Epo in various CNS insults by showing a significant reduction in brain edema after experimental TBI. Given the current lack of a specific pharmacological treatment aimed at blocking the development of brain edema in head-injured patients, these results offer new perspectives in the treatment of TBI and should prompt mechanistic as well as outcome studies on the potential use of rhEpo in TBI-induced brain edema.
Footnotes
Acknowledgements
The authors thank Myriam Bernaudin and Samuel Valable for helpful comments on the manuscript, and Jana Dunbar for her assistance to realize the TBI model. Epoetin alfa (Eprex®) was kindly provided by Janssen-Cilag Division Ortho Biotech, Issy-les-Moulineaux, France.