
Abstract
Cerebral small vessel disease (SVD) is a major cause of stroke and dementia. Pathologically, three lesions are seen: small vessel arteriopathy, lacunar infarction, and diffuse white matter injury (leukoaraiosis). Appropriate experimental models would aid in understanding these pathologic states and also in preclinical testing of therapies. The objective was to perform a systematic review of animal models of SVD and determine whether these resemble four key clinicopathologic features: (1) small, discrete infarcts; (2) small vessel arteriopathy; (3) diffuse white matter damage; (4) cognitive impairment. Fifteen different models were included, under four categories: (1) embolic injuries (injected blood clot, photochemical, detergent-evoked); (2) hypoperfusion/ischaemic injury (bilateral common carotid occlusion/stenosis, striatal endothelin-1 injection, striatal mitotoxin 3-NPA); (3) hypertension-based injuries (surgical narrowing of the aorta, or genetic mutations, usually in the renin-angiotensin system); (4) blood vessel damage (injected proteases, endothelium-targeting viral infection, or genetic mutations affecting vessel walls). Chronic hypertensive models resembled most key features of SVD, and shared the major risk factors of hypertension and age with human SVD. The most-used model was the stroke-prone spontaneously hypertensive rat (SHR-SP). No model described all features of the human disease. The optimal choice of model depends on the aspect of pathophysiology being studied.
Introduction
Lacunar stroke accounts for approximately a quarter of ischaemic stroke and is usually caused by cerebral small vessel disease (SVD; Fisher, 1968; Lammie, 2002). SVD is also the most common cause of vascular dementia, and a major contributor to lesser degrees of age-related cognitive decline (Erkinjuntti et al, 2002; Jellinger, 2007). Radiologically, small discrete regions of lacunar infarction are seen that may be accompanied by more confluent areas of leukoaraiosis observed as high signal on T2-weighted magnetic resonance imaging (MRI). Despite its importance, there are few specific treatments that have been shown to have efficacy in SVD. Hypertension is the major risk factor present in over 80% of patients (Khan et al, 2007) and antihypertensive agents reduce progression of leukoaraiosis on MRI (Dufouil et al, 2005). However, few other treatments have been evaluated specifically in SVD rather than in stroke as a whole.
An important factor underlying the lack of specific treatments is poor understanding of the pathogenesis of disease and a lack of animal models in which to test potential therapies. Animal models have been widely used to study acute cerebral ischaemia—such as middle cerebral artery occlusion (MCAo)—but such models produce large areas of predominantly grey matter infarction and are unlikely to be representative of cerebral SVD for a number of reasons. First, the pathologic lesions in SVD are primarily in the white matter and deep grey matter nuclei, where ischaemic mechanisms may differ from those operating in cortical grey matter (Imai et al, 2001; Sarti and Pantoni, 2003). Second, lacunar infarcts in SVD are much smaller than MCAo infarcts. Third, an important component of SVD is chronic ischaemic damage, seen as radiologic leukoaraiosis, which cannot be studied in traditional models of acute ischaemia. Fourth, chronic arterial injury occurs in SVD and most acute ischaemic models involve acute insults in previously normal vessels. A useful model in SVD must describe at least some of the characteristic pathologic features of clinical disease: either the arterial changes or the brain parenchymal damage or both.
Neuropathologic, radiologic, and epidemiologic data suggest that there may be two major categories of SVD with different clinical prognoses (Fisher, 1968; Boiten et al, 1993; Khan et al, 2007). Firstly, microatheroma in the larger intracerebral arteries at the origins of the perforating vessels, or within the proximal perforating arteries, results in one or a few larger lacunar infarcts, in the absence of leukoaraiosis. Secondly, a diffuse arteriopathy affecting smaller perforating arteries results in multiple smaller lacunar infarcts and leukoaraiosis. Current understanding of how these features result in the pathologic consequences of cerebral SVD are outlined in Figure 1. Histologically, this second form of SVD is characterized by wall thickening, collagen deposition, degeneration of smooth muscle cells, and narrowing of the lumen, and is often associated with localized blood—brain barrier (BBB) dysfunction (Fisher, 1968; Lammie, 2002; Jellinger, 2007). The histologic changes associated with leukoaraiosis are localized oedema, damage to axons, myelin, and oligodendrocytes, and local activation of astrocytes and microglia (van Swieten et al, 1991; Fernando et al, 2006; Jellinger, 2007). In the second, leukoaraiosis-associated SVD subtype, baseline cerebral blood flow and autoregulation are reduced (Terborg et al, 2000; O'Sullivan et al, 2002) and several lines of evidence suggest that endothelial dysfunction plays a pivotal role (Wardlaw et al, 2003; Hassan et al, 2003; Fernando et al, 2006; see Figure 1). It has been hypothesized that this could cause damage by hypoperfusion (O'Sullivan et al, 2002) and/or increased BBB permeability (Wardlaw et al, 2003). Embolism has been proposed as a mechanism for cerebral SVD, but histopathology (Fisher, 1968) and transcranial Doppler ultrasound to detect circulating emboli suggest that this is an infrequent cause (Kaposzta et al, 1999).
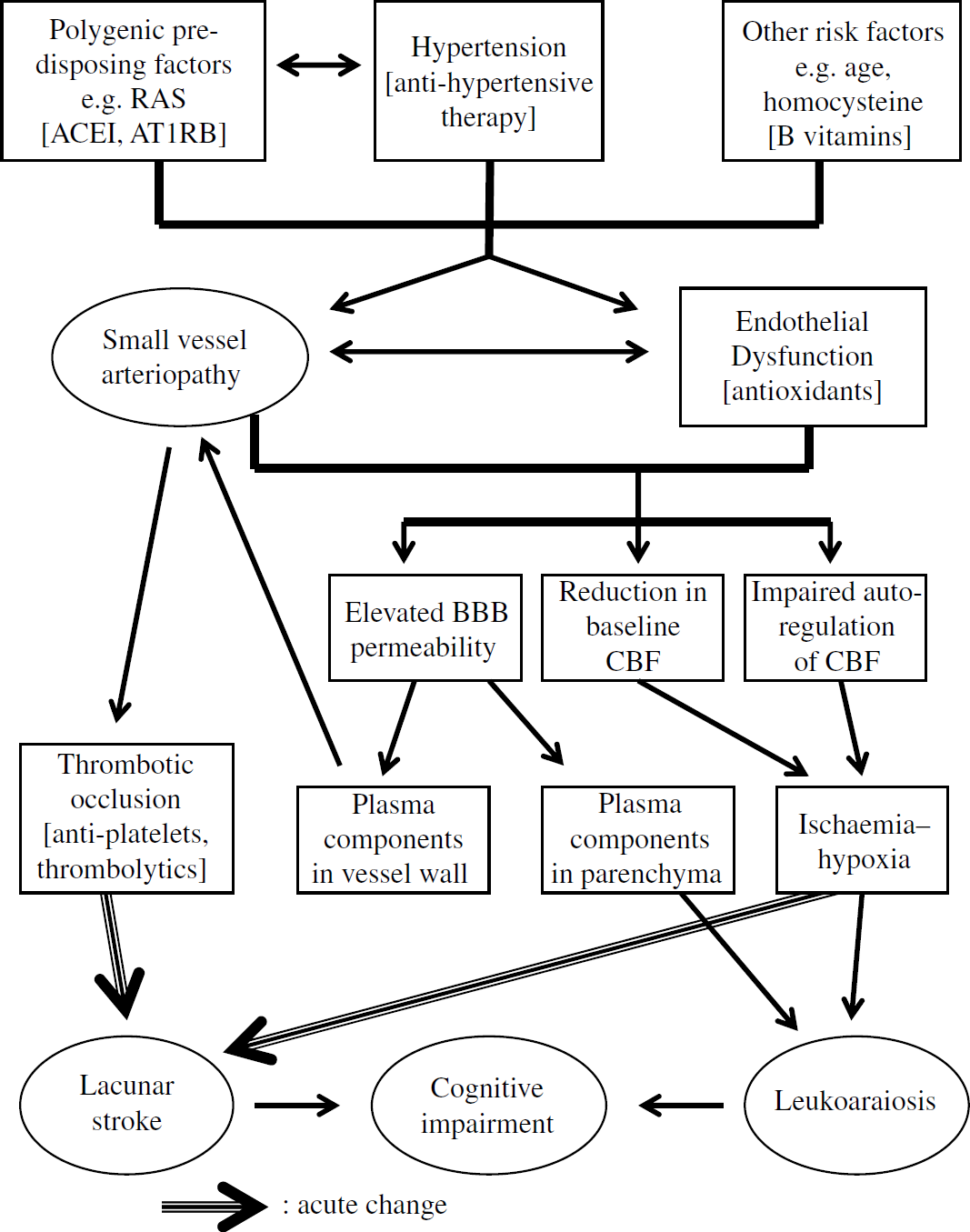
Current understanding of the pathogenesis of human cerebral small vessel disease. Heavy arrows indicate acute changes, whereas others indicate long-term effects. The key features considered in this review are contained in ellipsoids. For simplicity, the term ‘lacunar stroke’ is used to include both focal ischaemia and the acute motor symptoms of lacunar stroke. Interventions are given in square braces (e.g. [thrombolytics]). This schematic does not take into account monogenic forms of SVD, e.g. CADASIL. Abbreviations: RAS, renin-angiotensin system; ACEI, angiotensin converting enzyme inhibitors; AT1RB, angiotensin type 1 receptor blockers; BBB, blood-brain barrier; CBF, cerebral blood flow.
The different pathologic mechanisms underlying SVD/lacunar stroke suggest that one animal model is unlikely to describe all disease features. We performed a systematic review to identify potential models and to determine how well they described individual features of the disease.
Methods
The review protocol followed the Cochrane Handbook for Systematic Reviews of Interventions (version 4.2.6, Sept 2006: http://www.cochrane.org.uk/resources/handbook/). Using Medline, we searched publications in English for the following terms: (brain OR neur* OR cerebr*) AND (ischemi* OR ischaemi* OR stroke OR infarct OR cva) AND (in_vivo OR rodent OR rat OR mouse OR murine OR rabbit OR gerbil OR hamster OR pig OR cat OR dog OR primate OR monkey OR marmoset) AND (small_vessel OR lacun* OR svd OR lenticulostriat* OR lipohyalin* OR arteriolar OR leukoaraiosis OR tia).
Abstracts were viewed and the following exclusion criteria applied: (1) not an animal model; (2) not an
The initial search retrieved 986 publications (Set 1). After viewing abstracts, 943 were excluded and 43 included (Set 2). The bibliographies of these 43 papers and of all 24 review articles from Set 1 (see Supplementary Reference List S1) were hand searched, and a further 83 papers included, giving a total of 126 (Set 3). On hand searching
We excluded reports on MCAo (
Results
In the final dataset of 101 papers, 15 different experimental models were identified (Table 1). There were 5 reports on large non-human primates (
The animal models included

IHR, inducible hypertensive rat; 3-NPA, 3-nitropropionic acid; SHR-SP, stroke-prone spontaneously hypertensive rat.
Reference list for SHR-SP (Okamoto et al, 1975; Yamori et al, 1976; Yamori and Horie, 1977; Hart et al, 1980; Ogata et al, 1981; Coyle and Jokelainen, 1983; Sadoshima et al, 1983; Fredriksson et al, 1984, 1985; Tamaki et al, 1984; Werber and Heistad, 1984; Paschen et al, 1985; Tobian et al, 1986; Mayhan et al, 1987; Tagami et al, 1987; Baumbach et al, 1988; Mayhan et al, 1988; Fredriksson et al, 1988a, b ; Hajdu et al, 1991; Yang et al, 1991, 1993; Coyle and Feng, 1993a, b ; Baumbach et al, 1994; Richer et al, 1994; Yamaguchi et al, 1994; Linz et al, 1997; Minami et al, 1997; Blezer et al, 1998; Brosnan et al, 1999; Carswell et al, 1999, 2000a, b , 2004; Chillon and Baumbach, 1999; Marks et al, 2001; Sironi et al, 2001, 2004a, b , 2005; Kawashima et al, 2003; Banfi et al, 2004; Maguire et al, 2004; Kim-Mitsuyama et al, 2005; Liebetrau et al, 2005; Nagotani et al, 2005; Jesmin et al, 2007; Lee et al, 2007; Ballerio et al, 2007).
Methodological Characteristics of Included Studies
Methodological characteristics of included studies were assessed in terms of six domains, (Macleod et al, 2004; see Supplementary Tables S2 and S3). By virtue of our exclusion criteria, all studies were peer-reviewed publications (domain no. 1). Statement of controlled body temperature during general anaesthesia (domain no. 2) was rare in studies before 1990 and was irrelevant to some as they involved no general anaesthetic procedure. Random allocation of animals to experimental groups (domain no. 3) and use of masked observers to assess outcomes (domain no. 4) were generally rare, especially before 2000, but were more common in more recent studies. Most studies reported using a non-neuroprotective general anaesthetic agent where applicable (domain no. 5). The mildly neuroprotective anaesthesic agent, pentobarbitone, was used in 12 reports on SHR-SP (Sadoshima et al, 1983; Tamaki et al, 1984; Werber and Heistad, 1984; Mayhan et al, 1987, 1988; Baumbach et al, 1988, 1994; Hajdu et al, 1991; Yang et al, 1991, 1993; Chillon and Baumbach, 1999; Maguire et al, 2004) and in 4 other studies (Rosenberg et al, 1990; Toshima et al, 2000; Whitehead et al, 2005a, b ). The primate aortic coarctation studies used ketamine anaesthesia (Kemper et al, 1999, 2001; Moore et al, 2002), though this is unlikely to affect the data, given the prolonged pathogenesis of the lesions. Compliance with animal welfare regulations (domain no. 6) was almost never stated pre-1997, with a few exceptions (Rosenberg et al, 1990; Richer et al, 1994). In five studies the primary researchers were employees of a drug company (Linz et al, 1997; Ohta et al, 1997; Toshima et al, 2000; Maguire et al, 2004; Farkas et al, 2004). For some models, the included reports came from just one research group: detergent-induced emboli, photoactivated emboli, gerbil carotid coil; inducible hypertensive rat (IHR), R+/A+ mice, Notch-3 mice, Col4A1 mice, proteolytic enzyme injection, and murine leukaemia virus (MuLV)-infected neonatal mice.
Co-morbidities relevant to stroke—old age, hypertension, diabetes, or obesity—are listed in some previous methodological scoring (Macleod et al, 2004). In the studies we retrieved, hypertension was a co-morbidity present in primate-narrowed aorta, SHR-SP, IHR, and R+/A+ mice, and in one endothelin-1 (ET-1) injection study (Lecrux et al, 2008). In some studies, SHR-SP, IHR, and R+/A+ mice also had a high-salt diet. Aged animals (> half-normal lifespan) were used in the SHR-SP (Yamori and Horie, 1977; Hart et al, 1980; Ogata et al, 1981; Tagami et al, 1987) and Notch-3 transgenic mice studies (Ruchoux et al, 2003; Lacombe et al, 2005).
Summary of Outcome Measures
Outcome measures reported (Supplementary Tables S4, S5): (1) assessment of lesion extent (physical size or degree of brain injury); (2) histologic data including standard histologic staining, immunohistochemical, or electron microscopy studies; (3) MRI; (4) quantitative behavioural testing of motor deficits (e.g. Bederson scale, beam walking, Montoya staircase); (5) cognitive deficits in learning or memory (e.g. water maze, passive avoidance tests); (6) intervention studies. These were defined broadly to include not only drug treatments, but also dietary changes (Tobian et al, 1986), stage of the female reproductive cycle (Carswell et al, 2000a, b , 2004), and sympathetic denervation (Hart et al, 1980; Sadoshima et al, 1983; Werber and Heistad, 1984).
Outline of Individual Models
Embolic Models
Blood clot microemboli introduced by intracarotid injection of clot suspensions (maximal clot size 100 to 200 μm) produced acute lesions in adult rats (Kudo et al, 1982; Overgaard et al, 1992). These lay within the territories of MCA and anterior choroidal arteries, with signs of oedema and necrosis, progressing to a cystic cavity. Scattered small infarcts and micro-haemorrhages were seen in ∼25% of cases. Chronic changes to vessels and white matter were not seen.
Photoactivated thrombogenic agents deliver a shower of thromboemboli to a relatively restricted arterial territory (Watson et al, 1985; Ginsberg et al, 1987; Futrell et al, 1988, 1989). Again, the lesions were small cortical ischaemic foci, produced acutely by emboli that cleared within 24 h. Lesions progressed to form predominantly grey matter infarcts 0.1 to 2.0 mm in diameter, extending through all layers of the cortex. Endothelial damage was accompanied by rapid, temporary BBB breakdown within the irradiated region and in the penetrating vessels leaving it. In long term, damaged vessels exhibited myointimal smooth muscle proliferation (Dietrich et al, 1987a, b ). Cognitive dysfunction was evident at 2 days but not at 30 days after injury, as increased latency in a water maze task (Alexis et al, 1995). A sophisticated version of this model was recently used to assess brain vessel topology (Nishimura et al, 2006; Nishimura et al, 2007).
Intra-carotid injection of SDS detergent produced endothelial damage with subsequent thromboemboli, detectable in perforating arteries at 1 h, and multiple small infarcts in hippocampus, cortex, and thalamus at 3 days, with neuronal death and loss of myelin (Toshima et al, 2000).
For these three embolic models, behavioural deficits varied from mild to severe, roughly proportional to the degree of brain injury, with high mortality in the more severe groups (Kudo et al, 1982; Overgaard et al, 1992; Alexis et al, 1995). Among microembolic models, acute hyperglycaemia reduced infarct volume, whereas mannitol-evoked acute plasma hypertonicity had the opposite effect (Ginsberg et al, 1987). More recently, restoration of flow—indicative of clot dissolution—was achieved by haemodilution but not by the thrombolytic agent tPA (Nishimura et al, 2006). Lesion volume and neurologic deficit were improved by fasudil (administered immediately after injury), an inhibitor of the redox-sensing Rho kinase, which affects vascular smooth muscle cell migration (Toshima et al, 2000).
Hypoperfusion or Ischaemic Injury Models
Bilateral common carotid occlusion (‘2-VO’) in rats produced a chronic reduction in cerebral blood flow by 50% to 70%; (Ni et al, 1994; Wakita et al, 1995, 2002; Ohta et al, 1997; Ueno et al, 2002). Lesions were predominantly in white matter, with vacuolation of myelin, axonal damage, and demyelination in corpus callosum, internal capsule, and caudate-putamen. Lesions were apparent from 7 days after occlusion and were persistent (Wakita et al, 1995, 2002; Farkas et al, 2004), preceded by temporary BBB opening in white matter areas with collagen deposition in vessel walls (Ueno et al, 2002). Cognitive impairment was seen, with persistent learning deficit in Morris water maze and eight-arm radial maze tasks (Ni et al, 1994; Ohta et al, 1997). 2-VO-induced white matter lesions were reduced by the immunosuppressant cyclosporine A (administered daily after injury for up to 30 days), with reduced microglial and astrocyte activation in the affected regions (Wakita et al, 1995). White matter damage and learning deficits were also spared by the Cox-2 inhibitor nimesulide or by the metabolic enhancer idabenone (Ohta et al, 1997; Wakita et al, 1999).
Bilateral common carotid stenosis in gerbils, using a steel coil (inner diameter 0.25 mm) produced prolonged hypoperfusion, with ∼25% reduction in cerebral blood flow (Hattori et al, 1992; Kudo et al, 1993; Kurumatani et al, 1998). The insult relies on the gerbil's incomplete circle of Willis, and consequent lack of collateral supply. Neuropathologically, small necrotic foci (∼1 mm diameter) were produced in cortex and basal ganglia, accompanied by intense gliosis and small vessel proliferation (Hattori et al, 1992; Kudo et al, 1993). No vessel wall changes were reported. Patchy rarefaction of white matter—already sparse in gerbil brain—was seen in internal capsule, with myelin degradation and swelling of dendrites (Hattori et al, 1992; Kudo et al, 1993). These changes were seen after 8 or more weeks, accompanied by a learning deficit assayed in a passive avoidance paradigm (Kudo et al, 1993). This cognitive impairment appears to result from white matter lesions and varies with degree and duration of hypoperfusion.
Gerbils present logistic problems, not least the finding that many individuals have a circle of Willis that is not totally incomplete, with small communicating arteries (20 to 130 μm outer diameter) giving collateral supply to the forebrain (Seal et al, 2006). Recently, coil-mediated carotid stenosis was used in mice with similar results (white matter rarefaction, hippocampal lesions, impaired learning in passive avoidance tests), despite the differing brain vasculature (Shibata et al, 2007).
ET-1, a potent vasoconstrictor peptide, generated small infarcts when injected stereotaxically into rat striatum (Whitehead et al, 2005a, b ; Windle et al, 2006; Frost et al, 2006) or specifically into the internal capsule (Lecrux et al, 2008). ET-1 produced a locus of graded subcortical ischaemia, of approximately 1 h duration, forming a lesion ∼1 mm diameter, with abundant astrocytes, microglia, and inflammatory markers (Whitehead et al, 2005a, b ). For a few days, animals had a temporary deficit in motor function, assessed by Montoya staircase (Whitehead et al, 2005a, b ), foot-fault, or forelimb asymmetry tests (Frost et al, 2006; Lecrux et al, 2008). This was more pronounced when a combination of ET-1 and amyloid-β peptide was injected. Though no cognitive test has been applied, re-learning was required in the Montoya staircase, consistent with observed hippocampal damage (Whitehead et al, 2005a). No pathologic changes in blood vessels or white matter were reported. Injecting much higher doses of ET-1 generated a larger ischaemic lesion (Windle et al, 2006).
The anti-platelet drug triflusal (given orally for 8 days after injury) reduced functional deficits and inflammatory changes accompanying small ET1-induced infarcts (Whitehead et al, 2005b). Magnesium (MgSO4, 300 mg/kg, sc) administered 30 mins pre-ischaemia ameliorated the motor deficit after capsular lesions (Lecrux et al, 2008).
3-Nitropropionic acid (3-NPA), a mitochondrial toxin, produced selective neurodegeneration in the caudate-putamen (Beal et al, 1993; Borlongan et al, 1995; McCracken et al, 2001). We classified this ‘chemical ischaemic’ injury alongside hypoperfusion models. After systemic injection, damage to striatal neurons occurred within 24h, with injured oligodendrocytes, disrupted myelin, and axonal damage in the striatum and subcortical white matter (Beal et al, 1993; Borlongan et al, 1995; McCracken et al, 2001). Cortex, brainstem, and spinal cord were undamaged, and no vesselopathy reported. Behaviourally, animals developed dystonia in the hindlimbs, with paralysis at higher doses; some recovery of function was seen, though mortality was high (Beal et al, 1993). Animals treated for 1 month with low-dose 3-NPA had cognitive deficit in a passive avoidance test (Borlongan et al, 1995). Surgical and pharmacologic interventions suggest that the lesion is in part N-methyl-D-aspartic acid receptor mediated (Beal et al, 1993). Striatal mitotoxin challenge has also been applied to mice, with evidence of an oxidative stress mechanism (Kim et al, 2000; Cuthill et al, 2006).
Hypertensive Models
SHR-SP was obtained by selective breeding of spontaneously hypertensive Kyoto rats for stroke phenotype (Okamoto et al, 1975; Yamori et al, 1976; Yamori and Horie, 1977). SHR-SP developed progressive arterial blood pressure (ABP) increase during young adulthood, rising from normal levels at age 4 weeks to a plateau, mean ABP > 220 mm Hg by ∼20 weeks of age in males and 25 to 30 weeks in females (Yamori et al, 1976). The phenotype is polygenic and associated with overactivity of the renin-angiotensin system. Multiple cerebrovascular lesions of variable magnitude occur spontaneously, mainly in frontal, occipital, and parietal cortical areas (approximately 80%), the remainder primarily in striatum. Incidence of lesions is rare at < 12 weeks of age, but exceeds 80% by 30 weeks (Yamori and Horie, 1977; Tagami et al, 1987).
Histologic studies showed a vessel-based pathologic process with several features in common with human SVD (Ogata et al, 1981; Fredriksson et al, 1985, 1988
In keeping with wide variation in lesion size, SHR-SP showed a range of neurologic stroke-like behaviour, from mild paw paresis to paralysis/death. Adult SHR-SP showed learning deficits in the pre-stroke phase, relative to WKY background strain (passive avoidance test, three panel runway task), with worse performance after onset of stroke-like behaviour (Yamaguchi et al, 1994; Minami et al, 1997). Some lesions were associated with MRI changes, including diffuse T2-weighted hyperintensities (Yamaguchi et al, 1994; Blezer et al, 1998; Sironi et al, 2001, 2004
Numerous intervention studies have been performed in SHR-SP (see Supplementary Table S5). Beneficial effects of anti-hypertensive therapy—determined by longer survival and reduced stroke incidence—were achieved on long-term treatment with anti-adrenergic agents or the vasodilator hydralazine (Okamoto et al, 1975; Yamori and Horie, 1977; Hajdu et al, 1991). Sympathetic denervation (at 2 months) reduced the vessel wall thickening observed in deep brain parenchymal arterioles (but not in pial arterioles) of animals aged > 12 months (Hart et al, 1980). Inhibition of angiotensin signalling using angiotensin-converting enzyme (ACE) inhibitors improved survival (Hajdu et al, 1991; Richer et al, 1994; Blezer et al, 1998). Ramipril given from 4 weeks of age increased lifespan from 15 months to 30 months, equivalent to that of wild-type animals (Linz et al, 1997). ACE inhibitors also attenuated chronic brain vessel damage (Hajdu et al, 1991; Yang et al, 1993; Richer et al, 1994; Chillon and Baumbach, 1999; Liebetrau et al, 2005). Long-term treatment with perindopril (from 3 months of age) reduced wall thickening of pial vessels, whereas equivalent ABP reduction with propranolol did not, indicating a trophic action of angiotensin (Chillon and Baumbach, 1999). Similarly, angiotensin II type 1 receptor blockers, valsartan and candesartan, had beneficial effects on survival and lesion incidence—much more than equivalent ABP reduction with the calcium antagonist amlodipine (Sironi et al, 2004a; Kim-Mitsuyama et al, 2005). Lipid-decreasing therapy with a statin (from 4 to 6 weeks of age) reduced stroke incidence and suppressed inflammatory markers in brain vessels (Kawashima et al, 2003; Nagotani et al, 2005; Sironi et al, 2005). Survival was also improved by a high potassium diet, independently of ABP reduction (Tobian et al, 1986).
Notably, some therapies could reverse incident lesions. The ACE inhibitor enalapril, started after lesion detection on T2-weighted MRI, increased lifespan and reversed lesions (Blezer et al, 1998). Similarly, treatment with valsartan or with the anti-inflammatory agent pentoxyfilline, started after early lesions were detected by proteinuria, delayed appearance of brain lesions on MRI and greatly increased survival (Banfi et al, 2004; Sironi et al, 2004a).
IHR males carry the mouse renin gene (on the Y chromosome) under the control of an inducible promoter (Kantachuvesiri et al, 2001; Collidge et al, 2004). Induction of the transgene in adult life produced overactivation of the renin-angiotensin system and a degree of hypertension that could be regulated (by varying inducer concentration) rising to a steady state over ∼4 days. Vessel wall changes were observed in peripheral tissues—kidney, heart, and mesentery—with medial thickening and fibrinoid necrosis at 14 days, but no brain histopathology (Kantachuvesiri et al, 2001). To generate stroke lesions, adult rats in which the transgene had been induced were offered 0.9% saline in addition to drinking water (Collidge et al, 2004). Multiple small haemorrhagic lesions were reported (Collidge et al, 2004). By 14 days after induction, saline-offered IHR were in poor condition, with some mortality, and fluid intake risen to ∼200 mL per day (Collidge et al, 2004).
R+/A+ mice are double transgenic for human renin and human angiotensinogen (Baumbach et al, 2003; Iida et al, 2005; Wakisaka et al, 2008). Despite thickened cerebral arterial walls (Baumbach et al, 2003) these animals had only modest hypertension and normal lifespan but, if treated with a combined regimen of high salt diet and the nitric oxide synthase inhibitor L-NAME, they developed rapidly progressive hypertension, with 50% mortality within 8 weeks (Iida et al, 2005). Treated mice exhibited multiple haemorrhagic lesions (diameter ∼0.5 mm) distributed throughout the brain, especially within brainstem, accompanied by tremor and bradykinesia as the principal neurologic signs (Iida et al, 2005; Wakisaka et al, 2008). Treated animals also exhibited some small ischaemic lesions, with similar distribution but more sparse and distinct from the haemorrhagic foci (Wakisaka et al, 2008).
Aortic coarctation in monkeys was the only non-rodent model included. Surgical narrowing of the aorta (to ∼25% cross-section area) evoked a maintained reflex hypertension in large macaque monkeys, with ABP > 180/110 mm Hg (Hollander et al, 1977; Garcia et al, 1981; Kemper et al, 1999, 2001; Moore et al, 2002). This produced cerebrovascular lesions and some myocardial hypertrophy and retinopathy, but no obvious change in lifespan. Tiny infarcts were seen—discrete regions < 0.5 mm diameter—with loss of neurons and myelin (Kemper et al, 1999, 2001). These were most abundant in forebrain white matter (more rarely in periventricular white matter) and also scattered in cerebral cortex, hippocampus, striatum, midbrain, and cerebellum (Garcia et al, 1981; Kemper et al, 1999, 2001).
Lesions were often adjacent to a thickened blood vessel, though no atheroma, aneurysm, or haemorrhage were seen, and no vesselopathy resembling true SVD. Wall thickening of very small calibre vessels is a suggested mechanism, associated with BBB leakage (Garcia et al, 1981; Kemper et al, 1999, 2001). Diffuse white matter damage of the kind associated with leukoaraiosis was not observed (Kemper et al, 1999, 2001) but diffuse, non-infarcted regions of astrocyte and microglial clustering within white matter were present in some animals (Kemper et al, 2001), resembling white matter lesions in Binswanger's leukoencephalopathy. Hypertensive monkeys had progressive decline in executive function (assessed by a conceptual set-shifting task, adapted from the Wisconsin card sorting test) and impaired short-term memory and cognition (assessed by delayed recognition span), evident only after several months (Kemper et al, 2001; Moore et al, 2002).
Models Based on Damage to Blood Vessels
Notch3 transgenic mice express human mutant Notch-3, a morphogenic membrane receptor, under the control of a vascular smooth muscle-specific promoter (in addition to their own, murine Notch-3; (Ruchoux et al, 2003; Lacombe et al, 2005)). Mutant Notch-3 causes a rare autosomal dominant form of human SVD, CADASIL, characterized by leukoaraiosis and lacunar infarcts. Aberrant Notch-3 protein accumulates in vascular smooth muscle cells, and granular osmiophilic deposits are seen on electron microscopy. Transgenic mice exhibited granular osmiophilic deposits and Notch-3 accumulation in vascular smooth muscle cells, both at peripheral and cerebral sites, but only in aged animals (∼14 months). These signs were preceded by degenerative changes in vascular smooth muscle cells and endothelial cells, and some disruption of arterial wall integrity (from ∼10 months; Ruchoux et al, 2003). However, mice exhibited no stroke-like lesions or any other brain parenchymal changes, and may reflect the preclinical stage of CADASIL (Ruchoux et al, 2003; Lacombe et al, 2005). No abnormalities in cognitive or motor function were reported.
Col4a1 mice have a large deletion in type IV procollagen α1, a component of the basement membrane, that produced severely weakened vessel walls, particularly evident in small arteries (Gould et al, 2005, 2006). Col4a1 pups heterozygous for the mutation had cerebral haemorrhages when delivered by natural birth, with ∼50% mortality on postnatal day 1. Perinatal haemorrhage was avoided by surgical delivery, though mortality remained high. Animals that survived to adulthood all developed intracranial haemorrhage either multiple small intracerebral foci (∼0.5 mm) or a large subarachnoid haemorrhage. Focal lesions were usually located within the caudate-putamen. Reported as a model of SVD and lacunar stroke (Gould et al, 2006), it seems likely that the lesions result simply from weakened vessel walls, unable to support normal adult ABP, with small, branched penetrating arteries being especially susceptible. In humans, various point mutations in
Intracerebral injection of protease—such as collagenase or elastase—disrupted the extracellular matrix, and produced acute vessel weakness in adult animals (Rosenberg et al, 1990; Armao et al, 1997). Local injection into the caudate induced a rapid (∼10mins) rise in BBB leakage, followed by discrete haemorrhage (∼1 mm diameter) around the injection site, affecting cell bodies and local white matter tracts. These changes were associated with local oedema, necrosis, and inflammatory infiltration, resolution occurring within a few days, and cyst formation at 3 weeks (Rosenberg et al, 1990; Armao et al, 1997). Stroke-like behavioural deficits were evident ∼4 h after injection, but recovery almost complete by 7 days (Rosenberg et al, 1990). This paradigm is used as a model of intracerebral haemorrhage.
TR1.3 MuLV infection produced multiple small brain lesions in immature mice, because of viral damage within small arteries (Park et al, 1993, 1994; Murphy et al, 2004). Neonatal pups were injected intraperitoneally or intracerebrally with virus suspension on postnatal day 1 to 3 (animals become resistant after day 4). Neurologic signs appeared 8 to 18 days after infection—primarily tremors and seizures—with paralysis and death 1 to 2 days later (Park et al, 1993). Viral infection targeted vascular endothelial cells, and was widespread in many tissues, but lesions were predominantly within brain. Syncytial fusion of endothelial cells, with weakened vessel wall and BBB integrity produced multiple, discrete micro-haemorrhages throughout the brain, typically ∼1 mm diameter, affecting grey and white matter structures, with accompanying oedema. The immature state of the animals allowed only very rudimentary behavioural assessment, and no cognitive or intervention testing.
Discussion
We identified 15 models reflecting different aspects of human SVD. These will be discussed in terms of their relevance to SVD vesselopathy, leukoaraiosis, and lacunar infarct (see Table 2). An important distinction is between models that reflect the arterial lesions of SVD, and those that mimic the brain injury (leukoaraiosis, lacunar infarction; see Figure 1). No model represented all aspects of clinical disease (Table 2).
Do animal models exhibit clinicopathological features of human cerebral small vessel disease?
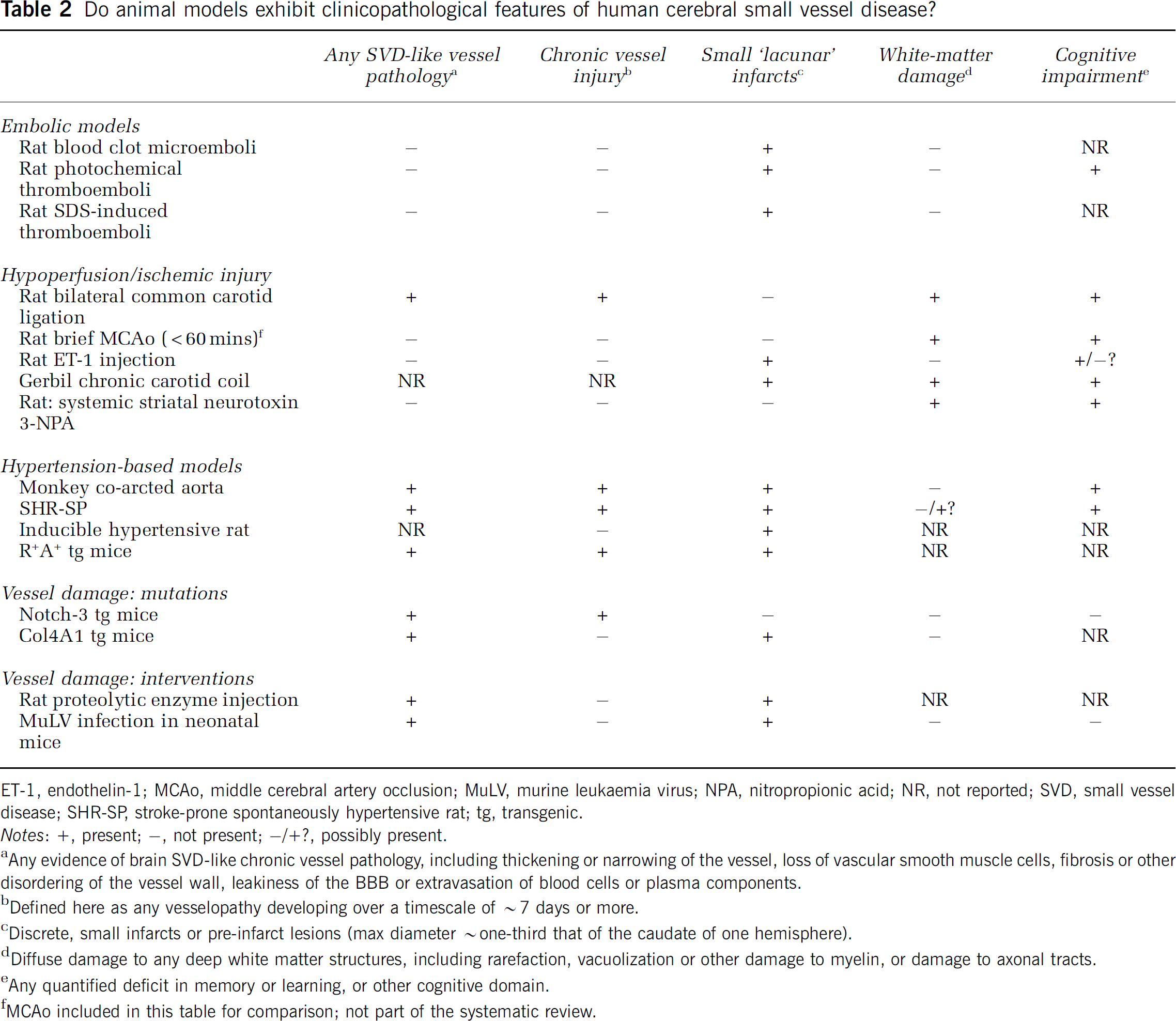
ET-1, endothelin-1; MCAo, middle cerebral artery occlusion; MuLV, murine leukaemia virus; NPA, nitropropionic acid; NR, not reported; SVD, small vessel disease; SHR-SP, stroke-prone spontaneously hypertensive rat; tg, transgenic.
Notes: +, present; -, not present; -/+?, possibly present.
Any evidence of brain SVD-like chronic vessel pathology, including thickening or narrowing of the vessel, loss of vascular smooth muscle cells, fibrosis or other disordering of the vessel wall, leakiness of the BBB or extravasation of blood cells or plasma components.
Defined here as any vesselopathy developing over a timescale of ∼7 days or more.
Discrete, small infarcts or pre-infarct lesions (max diameter ∼one-third that of the caudate of one hemisphere).
Diffuse damage to any deep white matter structures, including rarefaction, vacuolization or other damage to myelin, or damage to axonal tracts.
Any quantified deficit in memory or learning, or other cognitive domain.
MCAo included in this table for comparison; not part of the systematic review.
Models that Resemble SVD Arterial Vasculopathy
Histopathologic studies of SHR-SP revealed most of the cardinal signs of SVD: segmental wall damage, depletion of smooth myocytes, accumulation of fibrous tissue (e.g. collagen), wall thickening and luminal narrowing, local BBB leakage, and perivascular oedema (Ogata et al, 1981; Fredriksson et al, 1985, 1988
Transgenic animals are likely to be an increasingly rich source of SVD-like phenotypes, with vessel hypertrophy and endothelial dysfunction seen in several existing strains. For example, mice lacking endothelial nitric oxide synthase or CuZnSOD (the predominant isoform of superoxide dismutase in arterial walls) exhibit marked wall thickening in cerebral arterioles (Baumbach et al, 2004, 2006). TGF+ mice, overexpressing TGFΒ1 in brain astrocytes (Wyss-Coray et al, 2000), develop extensive changes in brain microvasculature from ∼4 months of age, including basement membrane thickening, endothelial abnormalities, and proliferation of perivascular astrocytes—but have apparently normal lifespan (Wyss-Coray et al, 2000; Tong et al, 2005).
In summary, a chronic insult appears to be required for SVD-like vesselopathy: SHR-SP, R+/A+ mice (probably), IHR (possibly), and hypertensive monkey (in some aspects). Also marked is the association with adult-onset hypertension. Hypertensive models (Table 2) all exhibit some prolonged vessel injury, preceding acute stroke events, in common with human SVD. Long-term effects of high ABP in animal models are unlikely to be simply due to tension-based damage in vessel walls. More likely, some pattern of blood pressure change—‘dips' (transient hypoperfusion), spasms, or failure in autoregulation—leads to chronic vessel injury. Trophic, non-pressor actions of the renin-angiotensin system may be involved. A drawback of the four hypertensive models (Table 2) is that the timing, magnitude, and precise location of stroke lesions can be predicted only approximately.
Models that Resemble Leukoaraiosis
In the rat 2-VO model and gerbil carotid stenosis, prolonged hypoperfusion produced diffuse white matter damage, to some degree resembling leukoaraiosis. Though MCAo was not included in our systematic analysis, white matter damage because of brief MCAo (up to 60 mins) may be relevant to leukoaraiosis. In subcortical white matter, rapid pathologic changes followed MCAo (Pantoni et al, 1996), with vacuolation of myelin and swelling of oligodendrocytes detectable at 30 mins, followed by swelling of astrocytes, such that regions of myelin pallor were clearly visible. The fact that diffuse white matter lesions were seen after 3-NPA intoxication or brief MCAo suggests that neither SVD-like vessel damage nor prolonged injury are required for diffuse white matter damage. More data are needed on small vessel damage after rat 2-VO or prolonged (> 8 week) gerbil and mouse carotid stenosis. Diffuse white matter lesions were not seen in any of the models based on vessel damage or embolic challenge (Table 2), though some exhibited white matter injury (proteolytic enzyme injection, endothelial MuLV infection).
Cognitive impairment was seen in all models where it was tested (7/15; see Table 2), with the exception of Notch-3 mice where no brain lesions occurr (Ruchoux et al, 2003). Diffuse white matter damage was always accompanied by cognitive deficit but, as expected, is not a requirement for it and some models showed cognitive impairment with no leukoaraiosis-like damage (e.g. hypertensive monkey, photochemical emboli).
In summary, diffuse white matter damage is seen in hypoperfusion-based injury models: either prolonged (rat 2-VO, carotid stenosis in gerbil or mouse, also SHR-SP); or acute (brief MCAo, 3-NPA injection).
Models that Resemble Lacunar Infarction
There are many ways to produce a small ischaemic lesion and, ultimately, a focal, lacune-like infarct. Our review yielded 12 such approaches (Table 2). All three embolic models can give small infarcts, because of thromboembolism or injected blood clot microemboli. There is a large early literature on embolic brain injuries in primates and larger mammals (see Garcia, 1984; Macdonald et al, 1995; Sarti and Pantoni, 2003). A study in large primates suggests that only ∼12% of small emboli (< 100 μm diameter) injected into the internal carotid artery reach deep penetrating arteries (Macdonald et al, 1995).
There is also a long history of focal infarct models based on vessel occlusion (Garcia, 1984; Ginsberg and Busto, 1989; Macdonald et al, 1995; Sarti and Pantoni, 2003). MCAo produces much larger infarcts than lacunar stroke, but modifications to give smaller, targeted lesions include surgical occlusion of small penetrating branches of the MCA, supplying the whisker barrel cortex in rats (Wei et al, 1995, 2001), or of medium-sized pial arteries as they enter the cerebral cortex (Hua and Walz, 2006). Both approaches produced small, focal lesions within a localized cortical territory. Striatal infarcts induced in baboons by surgically occluding multiple lenticulostriate penetrating arteries at their origin were intended to mimic a deep subcortical stroke (Yonas et al, 1981). This highly targeted injury nevertheless produced an infarct that extended throughout the caudate, putamen, and internal capsule, much more extensive than a clinical lacune. Similar surgical approaches produced more-or-less extensive, focal lesions in primates (Vajda et al, 1985) and dogs (Kuwabara et al, 1988; Guo et al, 1995). Occlusion of just one penetrating artery might have been more relevant to lacunar infarct. A recent study reported a small infarct in the internal capsule of pigs, produced by surgical occlusion of the anterior choroidal artery (Tanaka et al, 2008). Capsular infarcts (diameter ∼12 mm) were accompanied by motor functional deficits that recovered steadily up to 10 days after occlusion.
Stereotaxic injection of vasoconstrictor ET-1 gave acute, discrete lesions similar in scale to a human lacune in rat striatum (Whitehead et al, 2005a, b ; Windle et al, 2006; Frost et al, 2006) or internal capsule (Lecrux et al, 2008).
Hypertensive models displayed discrete, small lesions (Table 2), though not in every individual animal. Small, focal lesions are frequently seen in SHR-SP brains (e.g. (Yamori et al, 1976; Ogata et al, 1981; Fredriksson et al, 1985; Kawashima et al, 2003; Sironi et al, 2004b), distributed in cortex and striatum, usually with a haemorrhagic component. Similar lesions were seen in R+A+ transgenic mice (Baumbach et al, 2003; Iida et al, 2005; Wakisaka et al, 2008), possibly also in IHRs (Collidge et al, 2004). The focal infarcts in long-term hypertensive monkeys were histologically different, very small and non-cavitated (Kemper et al, 1999, 2001). Small infarcts that probably began as micro-haemorrhages were found in models of vessel damage because of enzyme injection (Rosenberg et al, 1990; Armao et al, 1997), Col4A1 mutation (Gould et al, 2006), or MuLV infection (Park et al, 1993), though not in Notch-3 mice, where vessel damage is milder (Ruchoux et al, 2003).
In summary, various insults can produce small ischaemic foci, some more ‘lacunar’ than others. Small, discrete infarcts can occur in the absence of SVD or leukoaraiosis-like white matter changes (e.g. ET-1 injection, hypertensive primate).
Intervention Studies: Comparison with Human Data
There are few studies in humans of interventions, specifically in SVD; most therapeutic trials have included all ischaemic stroke subtypes. Hypertension is a well-established risk factor for human SVD and lacunar stroke (Khan et al, 2007) and, in SHR-SP, numerous ABP-decreasing therapies were beneficial, including anti-adrenergic agents, ACE inhibitors and angiotensin II type 1 receptor blockers. The central role of the renin-angiotensin system in some models (SHR-SP, R+/A+ mice, and IHR) is in keeping with the clinical finding that ABP reduction by the ACE-inhibitor perindopril slows progression of leukoaraiosis (Dufouil et al, 2005), see Figure 1. Anti-platelet agents were beneficial in ET-1-evoked infarction (Whitehead et al, 2005a) and in SHR-SP spontaneous strokes (Banfi et al, 2004), in accord with a modest beneficial effect in human lacunar stroke. Lipid-decreasing statin drugs had a beneficial effect on stroke incidence and vessel damage in SHR-SP (Kawashima et al, 2003; Nagotani et al, 2005; Sironi et al, 2005), in keeping with the secondary preventive effect of statins in lacunar stroke.
Future Directions
There are at least three pathways for future development. First, genetic manipulation has great scope for designing new models of disease. Transgenic strains included here—such as R+/A+ mice or IHR—give promise of more information, as do milder Col4A1 strains, possibly corresponding to human single-site mutations. Crossing mild transgenic strains to produce double mutants may yield a new generation of models relevant to SVD. Second, future studies should consider aged animals, exposed to more prolonged arterial injury. Most previous experiments used young adults, with few comorbidities and a relatively acute insult. Human SVD develops over many years, and more prolonged, mild insults may be needed to model it—such as modest hypertension, borderline hypoxia, or partial BBB compromise. Finally, differences in brain size, length and structure of perforating arteries, and white matter/grey matter ratio, all make larger-brained species more relevant to human SVD. An interesting lacunar stroke model was recently reported in miniature pigs, producing a small infarct by surgical occlusion of the anterior choroidal artery (Tanaka et al, 2008). Pigs are long-lived mammals, with a gyrencephalic brain slightly larger than that of a macaque monkey.
Conclusions
No model describes all the features of human SVD—different models are of value in modelling different aspects of the human disease. The choice of model will depend on what research question is being asked. For example, interventions designed to reduce arterial damage may require a chronically hypertensive model, whereas studies of parenchymal tissue damage may call for a hypoperfusion model (leading to diffuse white matter lesions) or some form of highly localized subcortical ischaemia (to produce a lacunar infarct). To produce discrete focal lesions, many alternatives are open but those most resembling lacunar infarcts are SHR-SP (individuals with smaller lesions), R+/A+ mice, and ET-1 injections into discrete brain areas. To study leukoaraiosis-like white matter damage, potentially useful models—based on hypoperfusion—are rat bilateral carotid occlusion, gerbil, or mouse carotid stenosis, 3-NPA-induced striatal damage, brief MCAo, and possibly also the SHR-SP. To model SVD-like arteriopathy, SHR-SP appears closest to human SVD, particularly in aged animals. Vessel changes, resembling some features of SVD also occur in R+/A+ mice, IHR, and hypertensive primates—all prolonged hypertension states.
Disclosure/conflict of interest
The authors have no conflict of interest.
Footnotes
Acknowledgements
The authors thank Professor IM Macrae for helpful co mm ents on the article. AHH thanks Dr H Mulrooney for advice and discussions.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.