
Abstract
Molecular mechanisms of cerebral vasospasm after subarachnoid hemorrhage (SAH) include specific modes of cell signaling like activation of nuclear factor (NF)-kB and vascular cell adhesion molecules (VCAM)-1 expression. The study's hypothesis is that cisternal cerebral spinal fluid (CSF) from patients after SAH may cause Ca2+ oscillations which induce these modes of vascular inflammation in an in vitro model of human cerebral endothelial cells (HCECs). HCECs were incubated with cisternal CSF from 10 SAH patients with confirmed cerebral vasospasm. The CSF was collected on days 5 and 6 after hemorrhage. Cytosolic Ca2+ concentrations and cell contraction as an indicator of endothelial barrier function were examined by fura-2 microflurometry. Activation of NF-κB and VCAM-1 expression were measured by immunocytochemistry. Incubation of HCEC with SAH-CSF provoked cytosolic Ca2+ oscillations (0.31 ± 0.09 per min), cell contraction, NF-κB activation, and VCAM-1 expression, whereas exposure to native CSF had no significant effect. When endoplasmic reticulum (ER) Ca2+-ATPase and ER inositol trisphosphate (IP3)-sensitive Ca2+ channels were blocked by thapsigargin or xestospongin, the frequency of the Ca2+ oscillations was reduced significantly. In analogy to the reduction of Ca2+ oscillation frequency, the blockers impaired HCEC contraction, NF-κB activation, and VCAM-1 expression. Cisternal SAH-CSF induces cytosolic Ca2+ oscillations in HCEC that results in cellular constriction, NF-κB activation, and VCAM-1 expression. The Ca2+ oscillations depend on the function of ER Ca2+-ATPase and IP3-sensitive Ca2+ channels.
Introduction
After rupture of an intracranial aneurysm that causes subarachnoid hemorrhage (SAH), patients' prognosis is determined by a variety of pathophysiologic mechanisms like vascular inflammation, cerebral vasospasm (CVS), and ischemia (Claassen et al, 2002; Dorsch, 1995). Alterations of function in cerebral vascular endothelial cells seem to be involved in the pathophysiology of all these phenomena (Frijns et al, 2002). Endothelial cells are known to modulate immune defense, coagulation, permeability, and the contraction of the vascular smooth muscle cells and thus vascular tone (Faraci and Heistad, 1998; Iuliano et al, 2004). Many of the endothelial functions and intracellular signal transduction pathways are Ca2+ dependent (Tran et al, 2000).
Recently, cytosolic Ca2+ oscillations were described as a specific mode of intracellular signal transduction. The underlying mechanism was first described in the De-Young and Kaiser model (Jafri and Keizer, 1995) as a periodic Ca2+ release from the endoplasmic reticulum (ER) to the cytosol through activation of the inositol trisphosphate (IP3)-gated Ca2+ channel and Ca2+ reuptake into the ER by Ca2+-ATPase. Meanwhile Ca2+ oscillations are regarded as a universal mode of cell signaling that increases signal specificity, prevents desensitizing, and amplify signals in case of low-level stimuli (Dolmetsch et al, 1998). Ca2+ oscillations occur in many nonexcitable cells like T-cells, keratocytes, and endothelial cells (Kuebler et al, 2002; Paltauf-Doburzynska et al, 2000). Ca2+ oscillations can be induced in human umbilical vascular endothelial cells and human aortic endothelial cells by histamine, thrombin, bradykinin, ATP, or hydrogen peroxide (Hu et al, 1998; Paltauf-Doburzynska et al, 2000). The frequency of Ca2+ oscillations has been found to be 0.7 ± 0.1 per min in aortic endothelial cells stimulated by 100 μmol/L hydrogen peroxide and 0.3 per min by 1 μmol/L histamine (Hu et al, 1998).
The activation of NF-κB has been linked to Ca2+ oscillations (Dolmetsch et al, 1998). Nuclear factor (NF)-kB is a ubiquitous proinflammatory transcription factor that is activated by degradation of the inhibitory subunit IkB. It regulates the endothelial inflammatory response by inducing the gene expression of adhesion molecules like intercellular adhesion molecules, vascular cell adhesion molecules (VCAMs), chemotactic cytokines like interleukins (ILs) IL-1, IL-6, and IL-8 as well as tumor necrosis factor-α (Dolmetsch et al, 1998; Faraci and Heistad, 1998). However, VCAM-1 expression may also be induced by a variety of other mechanisms. (Chen et al, 1995; Keegan and Zamorano, 1998) VCAM-1 is a member of a family of leukocyte adhesion molecules that are expressed on the luminal surface of endothelial cells and are involved in monocyte adhesion to endothelial cells. This may result in the transmigration of T-cells and monocytes into the vessel wall (De Martin et al, 2000). In addition, both NF-κB and VCAM-1 have been shown to be involved in the pathogenesis of CVS after SAH (Kaynar et al, 2004; Polin et al, 1998; Zhou et al, 2007).
The intracellular Ca2+ concentration regulates the function of the endothelial barrier between blood and the surrounding tissue. Ca2+ is an activator of myosin light chain kinase, whose activation leads to phosphorylation of myosin light chains and subsequently to actin—myosin interaction. This interaction induces contraction of cells and an increased permeability of the endothelial cell layer (Tiruppathi et al, 2002).
After SAH, endothelial cells are exposed to the bloody cerebral spinal fluid (CSF) because of a direct CSF circulation from the subarachnoid space to the subendothelial layer (Ohta et al, 1992). Macromolecules diffuse within a few minutes to the endothelial cells (Uemura et al, 1987; Yamamoto et al, 1991). The effect of SAH-CSF on the function of cerebral endothelial cells is still unknown. To date, only one publication investigated the influence of erythrocyte lysate and bloody CSF on bovine endothelial cells and showed an adenosine-dependent rise of the cytosolic Ca2+ concentration (Zhang et al, 1996).
The goal of this study was to test the hypothesis whether cisternal CSF from patients who experience aneurysmal SAH may cause vascular injury that is initiated by intracellular Ca2+ oscillations and modulated by the activation of NF-κB and expression of VCAM-1 in an in vitro model of human cerebral endothelial cells (HCECs). In addition, it is hypothesized that Ca2+ oscillations lead to a contraction of HCEC and to an impaired barrier function.
Materials and methods
Cerebral Spinal Fluid and Patient Population
Samples of CSF were derived from a total of 10 SAH patients (male to female = 5:5) with a mean age of 58 ± 11 years (range 40 to 73 years). Patients were included when thick blood clots were seen in the basal cisterns, or intraparenchymal or intraventricular hemorrhage (Fischer 3 or 4) was present on their first computerized tomography scan. In addition, patients underwent surgery for clipping. At the end of surgery, a catheter was placed into the basal cerebral cisterns. Cisternal CSF was sampled on day 5 or 6 after hemorrhage. All patients included experienced global CVS which was confirmed by cerebral angiography on posthemorrhage day 9 ± 2.
For control studies, native CSF was sampled from patients who underwent myelography for the diagnosis of degenerative spinal diseases. In the control group the mean age of the patients was 63 ± 6 years (65 to 75 years).
The CSF from both SAH patients and controls was centrifuged (3,000 g for 10mins) and stored at −80°C upon analysis.
Culture of Human Cerebral Endothelial Cell
The HCEC (Cell Systems Corporation, Kirkland, WA, USA) were cultured in Cell Systems Complete Medium. This medium served for all experiments. The culture medium was changed every 48 h.
Ca2+ Measurement
The cytosolic concentration of Ca2+ was measured with the fluorescent dye fura-2 AM. Cells were incubated with 2.5 μmol/L of the acetoxymethyl ester of fura-2 for 60 mins. Fura-2 fluorescence was analyzed using a TILL Photonics imaging system (Gröfelfing, Germany). Illumination was induced by exposing the cells to an excitation light with a wavelength that alternated between 340 and 380 nm. Emitted light was detected at 510 nm. Fura-2 fluorescence was calibrated according to the methods described by Grynkiewicz et al (1985). Cells were exposed to 5 μmol/L ionomycin in modified 4-(2-hydroxyethyl)-1-piperazi-neethanesulfonic acid (HEPES) buffer containing either 3 μmol/L Ca2+ or buffer that was Ca2+ free with 500 μmol/L ethylene glycol tetraacetic acid to obtain the maximum (Rmax) and the minimum (Rmin) of the ratio of fluorescence (R), respectively. Intracellular Ca2+ concentration was calculated according to the following equation:
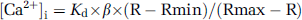
where Kd is the dissociation constant of fura-2 determined in intact cells and ᵝ is the ratio of the 380 nm excitation signals of ionomycin-treated cells at 5 mmol/L ethylene glycol tetraacetic acid and at 3mmol/L Ca2+.
Blockers of Endoplasmic Reticulum Ca2+-ATPase and Ca2+ Channels
In the present investigation, ER Ca2+-ATPase, IP3-sensitive Ca2+ channels, and ryanodine-dependent Ca2+ channels were selectively blocked by 50 μmol/L thapsigargin, 10 μmol/L xestospongin, and 10 μmol/L ryanodine, respectively. The optimal concentrations of the blockers were determined from dose—response curves (data not shown). Because xestospongin and thapsigargin are known to cause apoptosis and necrosis, it was tested whether the concentrations of ER blockers used may influence cell viability. Human cerebral endothelial cells were exposed to 50 nmol/L thapsigargin, 10 μmol/L xestospongin, 10 μmol/L ryanodine, and HEPES-buffered saline solution. Apoptosis and necrosis were evaluated by staining with Hoechst-3342 and propidium iodide as previously described (Kumar et al, 2007). For quantitative assay, a blind analysis of 180 to 250 nuclei from randomized 5 fields of view was applied.
Cellular Constriction
Cellular constriction was quantified by planimetrical analysis of the Ca2+ independent fluorescence intensity at 360 nm in parallel with the measurement of the fura-2 ratio. Before the addition of CSF (at baseline), the absolute value of cell-surface area was set at 100%. After the addition of CSF the cell-surface area was measured every 6 secs and expressed as a percentage of baseline.
Detection of Nuclear Factor-kB Activity and Vascular Cell Adhesion Molecules-1 Expression
Immunocytochemistry was used to measure activated NF-κB and VCAM-1 expression similar to the method described by Abdallah et al (2007). Human cerebral endothelial cell were stimulated with SAH-CSF in the absence or presence of xestospongin or thapsigargin. Control experiments were performed with native CSF or HEPES-buffered saline solution. After 60 mins stimulation, cells were fixed and permeabilized with ice-cold methanol, followed phosphate-buffered saline/bovine serum albumin blocking. Subsequently, the endothelial cells were incubated with antibodies increased against VCAM-1 or phospho-specific human pS 276 p65 subunit of the activated NF-κB (Ashikawa et al, 2002). The antibodies were obtained from Rockland Laboratories (Gilbertsville, PA, USA). After 60 mins, the endothelial monolayers were washed with phosphate-buffered saline before being incubated with a secondary Cy 2-conjugated goat anti-rabbit immunoglobulin G (1:200) overnight. On the following day, the cells were washed and mounted on coverslips. The fluorescence signal was analyzed by fluorescence microscopy (TILL Photonics) using TillvisION software. All steps were performed at room temperature.
Data Analysis and Statistics
Data are reported as mean ± s.d. or percentage of control. Comparisons among different treatment groups were analyzed with analysis of variance. A value of P < 0.05 was considered statistically significant. The effects of the blockers were tested by paired Students' t-test.
Results
Effect of Subarachnoid Hemorrhage-Cerebral Spinal Fluid on the Cytosolic Endothelial Ca2+ Concentration
Figure 1 shows the time course of the fura-2 ratio in a HCEC monolayer. The brightness of the micrographs is proportional to the Ca2+ concentration. The HCECs were incubated in modified HEPES buffer solution for 10 mins. Then the solution was replaced by CSF of SAH patients. Cytosolic Ca2+ oscillations appeared with a latency period of 62 ± 25 secs in all the 10 patients. The mean frequency was 0.31 ± 0.09 per min and the oscillations persisted during the whole experiment. Within the oscillatory peaks the cytosolic Ca2+ concentrations were 4.8-fold higher compared with the basal concentration (327 ± 98 versus 68 ± 12 nmol/L) (P < 0.001, n = 10). There was variability in the intracellular Ca2+ concentration both over time and across cells (Figure 1A). When the endothelial cells were incubated with native CSF, no Ca2+ oscillations occurred (Figure 1B). This finding was consistently reproduced in eight experiments.
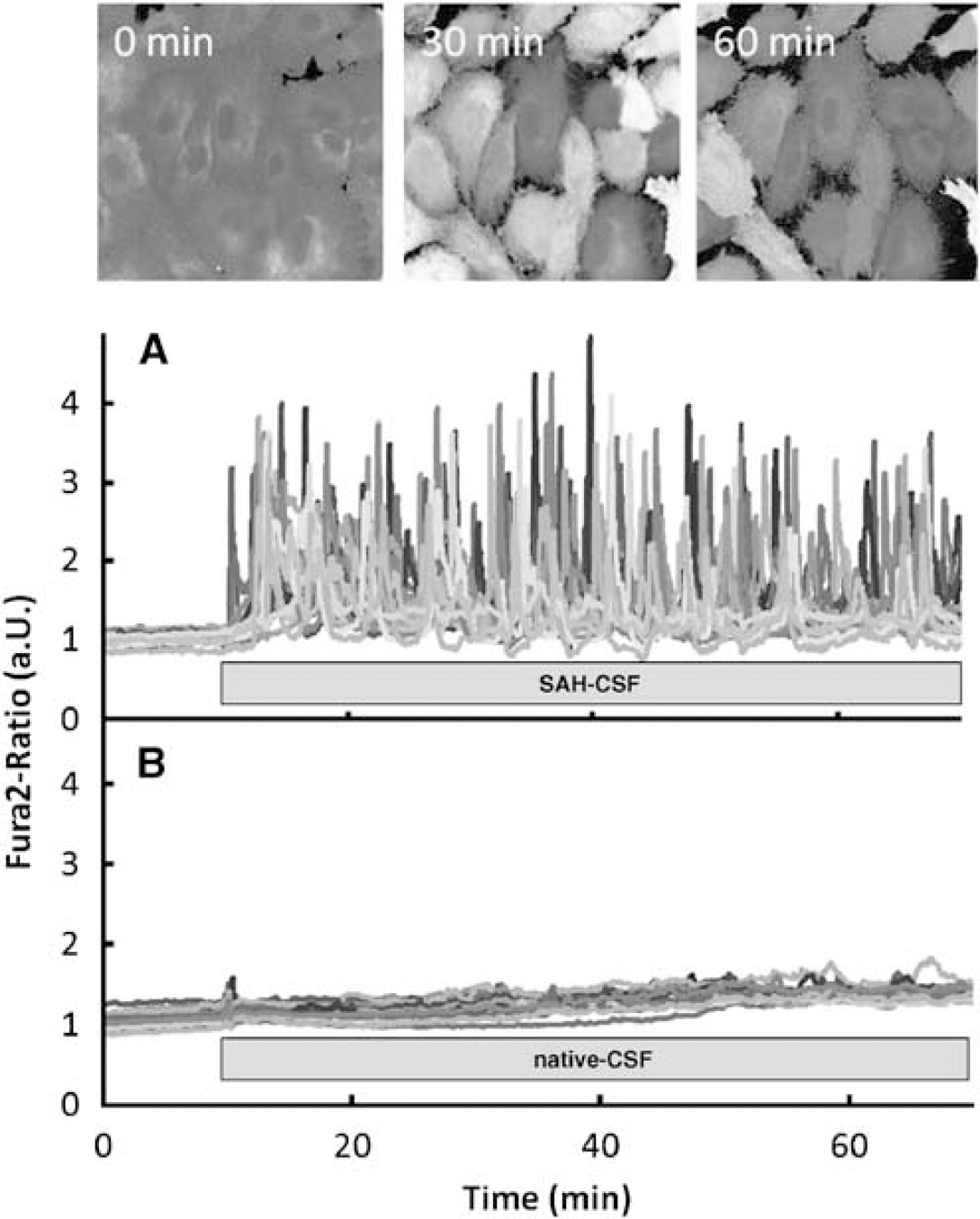
Ca2+ oscillation in fura-ratio pictures and Fura-2 Ratio recordings after exposing cells to CSF. Photomicrographs with fura-2 staining are shown at three different time points after starting the measurement. The brightness of pixels is directly proportional to the Ca2+ concentration within the pixel. CSF was added 10 mins after starting the experiment at 0 min. During the observation period, the cytosolic Ca2+ concentration is highly variable within and across cells. The maximum cytosolic Ca2+ concentration was reached periodically approximately every 3.2 mins, resulting in an oscillation frequency of 0.31 ± 0.02 per min. Each graph showing the fura-2 ratio (in arbitrary units, a.u.) represents the Ca2+ concentration of one recorded single cell. After an initial incubation for 10 mins the bath solution was replaced by CSF. (
The Origin of Subarachnoid Hemorrhage-Cerebral Spinal Fluid-Induced Cytosolic Ca2+ Oscillations
Under different physiologic conditions Ca2+ oscillations in endothelial cells originate from disturbances of ER Ca2+-channel function and Ca2+-ATPase activity. Ca2+-ATPase and IP3- or ryanodine-dependent Ca2+ channels are the primary candidates for the origin of Ca2+ oscillations. When thapsigargin was added to the SAH-CSF 20 mins after the onset of cytosolic Ca2+ oscillations, Ca2+ oscillations ceased immediately and almost completely. This was consistently reproduced in all five studies. Figure 2 shows the effect in a representative cell.
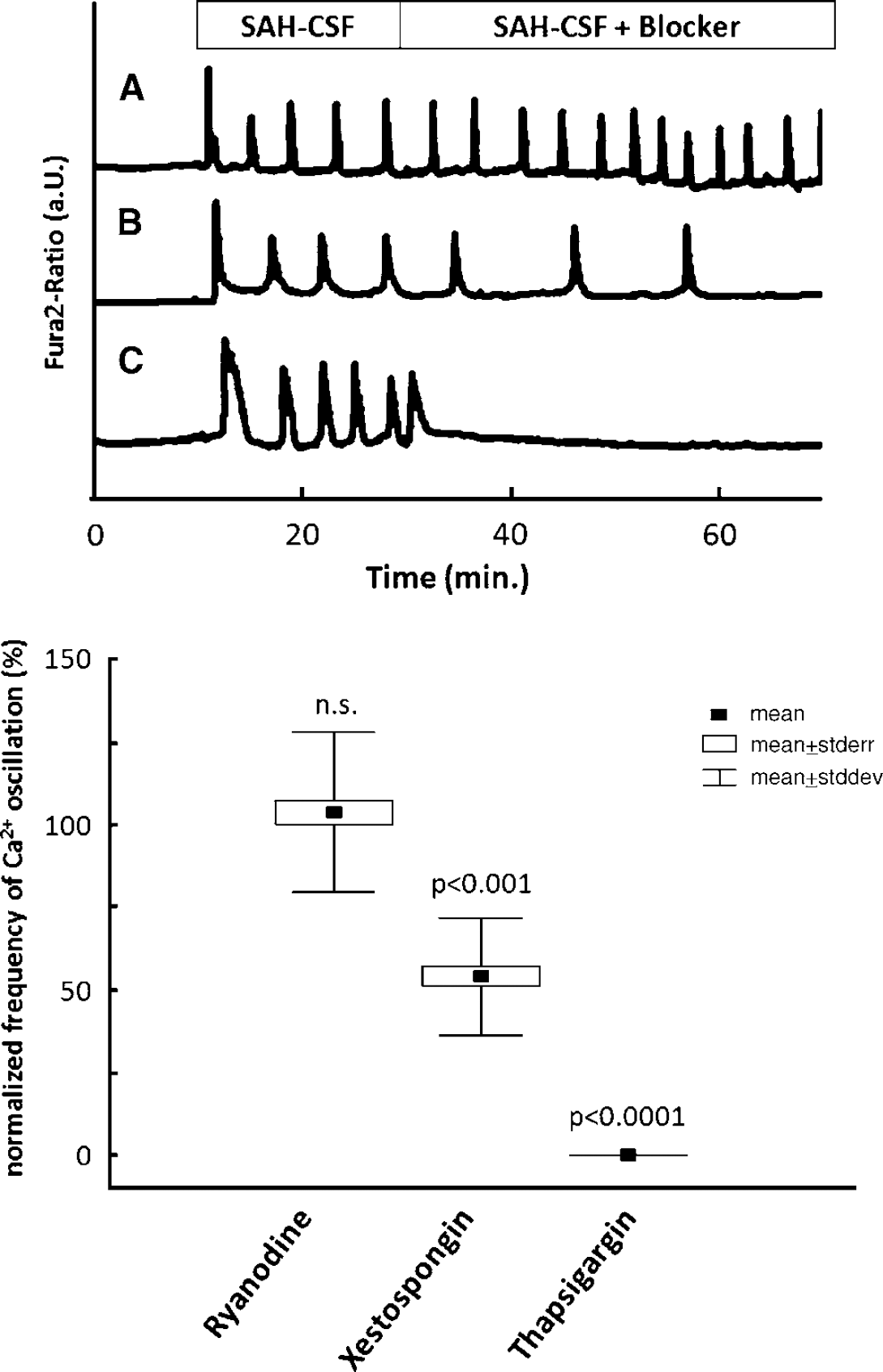
Effects of ryanodine, xestospongin and thapsigargin on Ca2+ oscillations. Ryanodine-dependent Ca2+ channels, IP3-dependent Ca2+ channels, and Ca2+-ATPase were blocked by ryanodine, xestospongin, and thapsigargin, respectively. All three inhibitors were added 20 mins after the onset of Ca2+ oscillations. (A—C) Each graph represents the cytosolic Ca2+ concentration recorded on a representative single endothelial cell. After the addition of thapsigargin to the SAH-CSF, Ca2+ oscillations ceased completely, whereas xestospongin reduced the frequency of Ca2+ oscillations significantly to 54% ± 18% of control. Ryanodine had no effect on the frequency of Ca2+ oscillations (A).
The addition of xestospongin to the SAH-CSF reduced the oscillation frequency significantly to 54% ± 18% (P < 0.001) of the control frequency, whereas the addition of ryanodine did not lead to a significant change in Ca2+ oscillation frequency (104%± 24%).
These results are not because of the toxicity of the blockers and consecutive cell death. In control experiments, where HCECs were exposed to blockers without SAH-CSF for 2 h, the rate of necrosis was 0.3%, 0.5%, and 0.3% for thapsigargin, xestospongin, and ryanodine, respectively. The rate of apoptosis was 0.1%, 0.12%, and 0.33% for thapsigargin, xestospongin, and ryanodine, respectively. In HCECs that were incubated in HEPES-buffered saline solution the rate of necrosis and apoptosis was 0.3% and 0.2%, respectively. The differences between groups were not statistically significant.
Cellular Constriction
When endothelial cells were exposed to SAH-CSF as shown in Figure 3, there was a rapid decrease in cell-surface area because of the shrinking of cells. Twenty minutes after the onset of the oscillations, the cell-surface area was reduced to 79% ± 5% of the area before the addition of SAH-CSF. Sixty minutes after the onset, the reduction was 71%± 6%. Native CSF did not result in a significant change in the surface area. When 10 μmol/L xestospongin or 50 nmol/L thapsigargin were added 20 mins after the stimulation of the cells by SAH-CSF, the cells expanded again. Forty minutes after addition of the blockers, the cell-surface area recovered to 87% ± 4% and 98%± 4% of control, respectively (Figure 3).
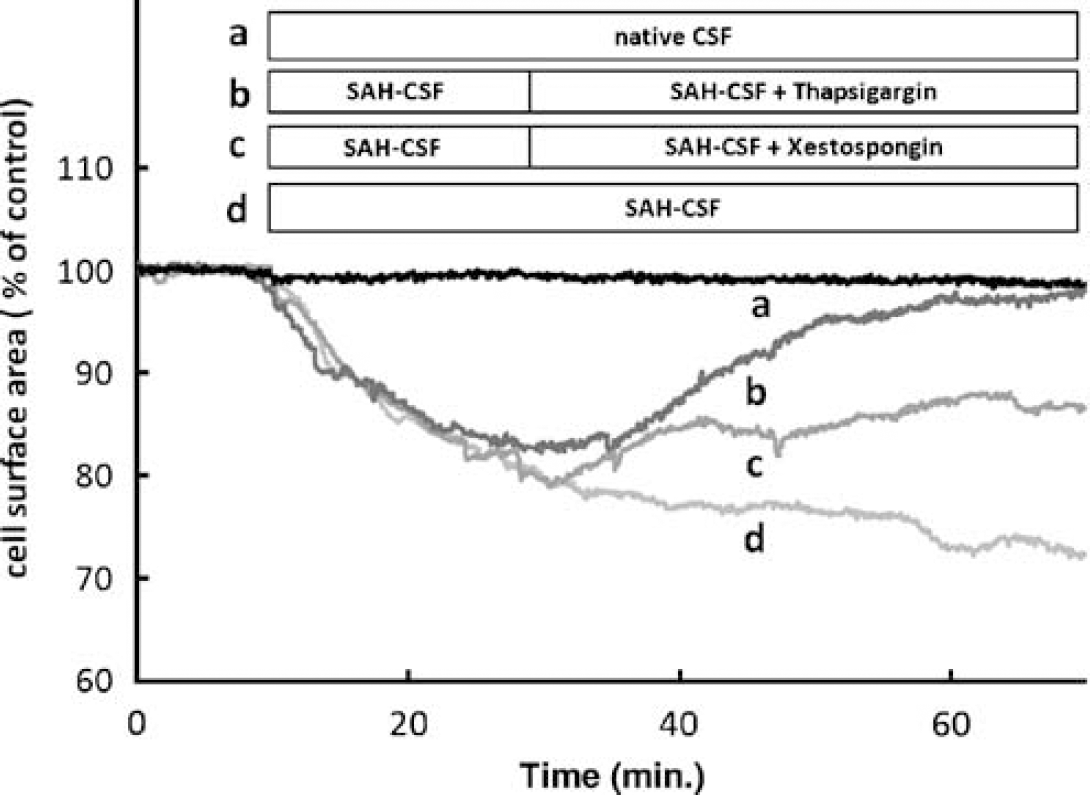
Gap formation. After replacement of the HEPES-bufferd saline solution by CSF, the cell-surface area shrinks to 71% ± 6% of control within 60 mins (d). Blocking the Ca2+ oscillations by xestospongin (c) and thapsigargin (b) led to a reexpansion of the cells. Forty minutes after the addition of the blockers, the cell-surface area reached 87% ± 4% and 98% ± 4% of control, respectively. When the cells were incubated in native CSF (a), the cell-surface area was nearly constant over the time.
Nuclear Factor-kB and Vascular Cell Adhesion Molecules-1
When activation of NF-κB in HCEC was measured after incubation with HEPES-buffered saline solution and native CSF, there was no significant difference between results with these substances. However, when HCECs were exposed to SAH-CSF, NF-κB activation (Figure 4) increased to 205% ± 28% of control (P < 0.001). This phenomenon could be dampened after preincubation of the cells with 10 μmol/L xestospongin or 50 nmol/L thapsigargin, which reduced the SAH-CSF-induced NF-κB activation to 163% ± 32% and 138% ± 28% of the basal activity, respectively. These results were statistically significant, as shown in Figure 5. After the same protocol, SAH-CSF induced VCAM-1 expression (Figure 4) to 181% ± 19% of basal fluorescence, whereas in the presence of xestospongin and thapsigargin VCAM-1 expression reached only 119% ± 24% and 129% ± 19% of control, respectively. All differences were statistically significant (P < 0.001; Figure 5).
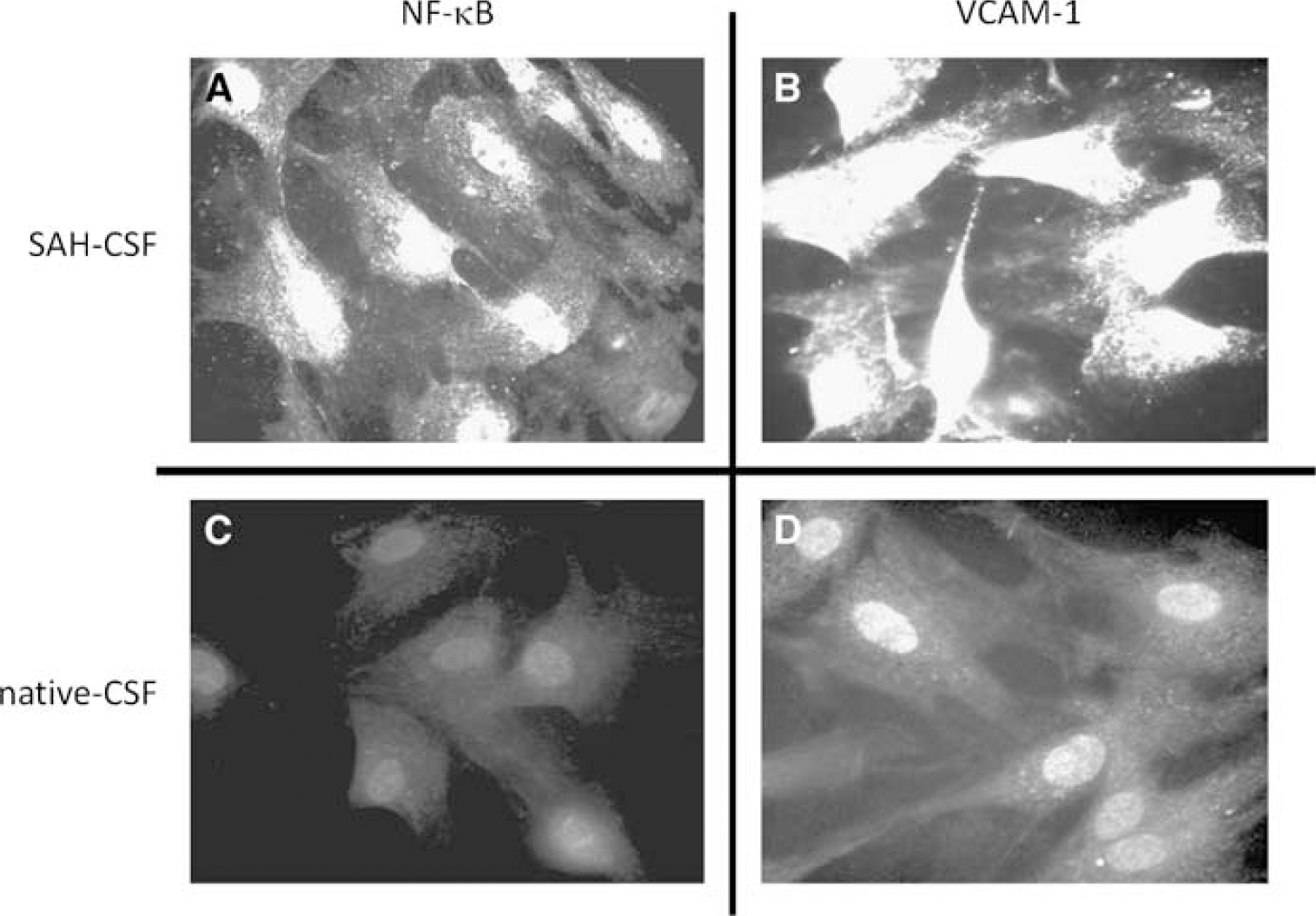
Immunocytochemistry. Human cerebral endothelial cells are labeled by antibodies against VCAM-1 or the phospho-specific human pS 276 p65 subunit of activated NF-κB. The cells were exposed to SAH-CSF, which caused VCAM-1 expression (
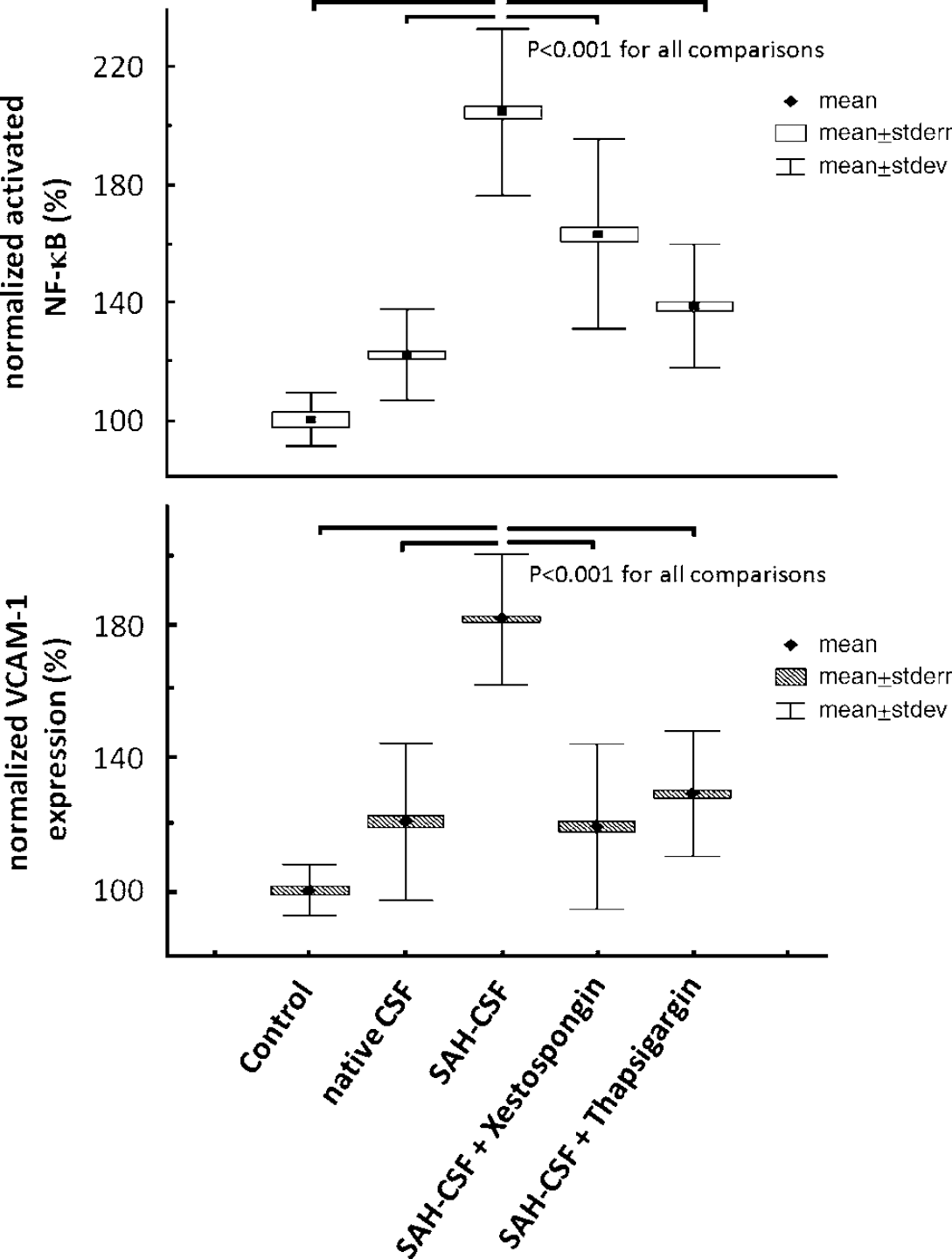
NF-κB activity and VCAM-1 expression. Immunofluorescence was used to detect activated NF-κB and expressed VCAM-1 in human cerebral endothelial cells after being incubated with HEPES-buffered saline solution and CSF, both native and after SAH. Note that CSF from SAH patients nearly doubled the activation of NF-κB. The addition of xestopongin and thapsigargin to CSF from SAH patients reduced the activation of NF-κB and VCAM-1 expression significantly in a close analogy to the reduction of Ca2+ oscillations.
Discussion
This study investigates the signaling cascade leading from SAH to a rudimentary endothelial inflammatory reaction after SAH (Figure 6). The principal findings are summarized in the following steps:
Subarachnoid hemorrhage-cerebral spinal fluid sampled from patients with confirmed CVS induces cytosolic Ca2+ oscillations in HCEC.
Ca2+ oscillations originate from the ER by repetitive uptake and release of Ca2+ by the ER Ca2+-ATPase and IP3-dependent Ca2+ channels.
Ca2+ oscillations cause reversible shrinking of HCEC, that is, impaired barrier function.
The Ca2+ oscillations activate proinflammatory NF-κB and lead to increased VCAM1 expression.
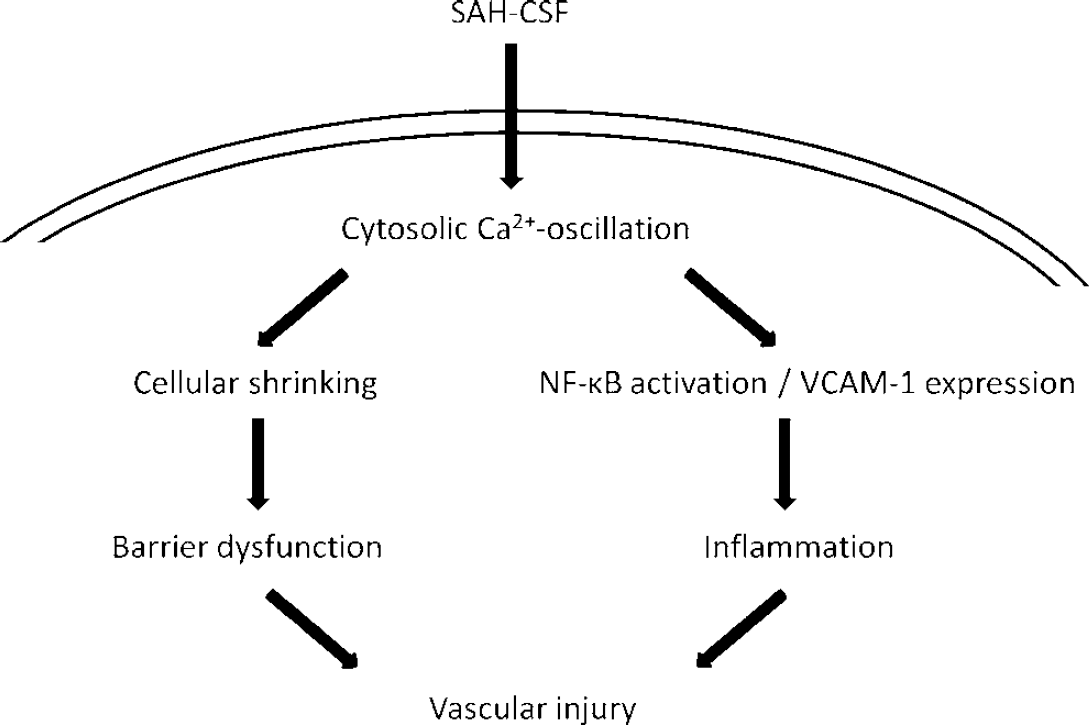
Scheme of SAH-induced vascular injury. In HCEC, SAH-CSF-induced cytosolic Ca2+ oscillations lead to two distinct pathways that both contribute to vascular injury. On one side impaired endothelial barrier function occurs as a result of cellular shrinking. On the other side activation of NF-κB and expression of VCAM-1 may be regarded as a rudimentary inflammatory reaction.
Mechanisms of Intracellular Ca2+ Homeostasis and Signal Transduction
The ER and mitochondria serves as intracellular Ca2+ stores. The main store is the ER, containing 75% of the intracellular Ca2+. The mitochondria contain the remaining 25% (Tran et al, 2000). There are a variety of mechanisms that influence intracellular Ca2+ homeostasis. Ca2+-ATPase primarily acts by pumping Ca2+ continuously into the ER lumen. Endoplasmic reticulum Ca2+ release is IP3 dependent (Jafri and Keizer, 1995). Inositol trisphosphate binds to specific receptors on the ER surface and activates IP3-gated Ca2+ release channels, which results in an increase in the cytosolic Ca2+ concentration. The IP3-gated Ca2+ channels are the predominant ER Ca2+ channels in nonexcitable cells. The ER ryanodine-dependent Ca2+ releasing channel is of little importance in endothelial cells. These channels have been most extensively characterized in muscle cells (Tran et al, 2000). In many cell types the IP3-gated Ca2+ channels are in a closed loop with the Ca2+-ATPase that is responsible for cytosolic Ca2+ oscillations.
The Role of Ca2+ Oscillations in Normal Physiology and after Subarachnoid Hemorrhage
Ca2+ oscillations trigger universal processes such as cell division, cell differentiation, and apoptosis (Miyakawa-Naito et al, 2003). The frequency or the amplitude or both of Ca2+ oscillation control the specificity of this signal (Dolmetsch et al, 1998).
Cytosolic Ca2+ osillations are observed in different phenotypes of endothelial cells. In aortic endothelial cells, Hu et al, 1998 reported a frequency of 0.7 ± 0.1 per min stimulated by 100 μmol/L hydrogen peroxide and 0.3 per min by 1 μmol/L histamine. As shown in the present paper, Ca2+ oscillations in HCEC occur at an average frequency of 0.31 ± 0.02 per min after exposure to CSF from SAH patients. This frequency is higher than 0.1 per min, which has been described as the cutoff frequency for the exponential activation of NF-κB (Dolmetsch et al, 1998).
Inositol trisphosphate-dependent Ca2+ oscillations propagate from one endothelial cell to another (Boitano et al, 1992). Vascular endothelial cells interfere with the surrounding smooth muscle cells through gap junctions. Thus, an increased endothelial cytosolic Ca2+ concentration may result in vessel constriction. This has been shown in an in vitro hamster model where a strong vasoconstriction was induced by an increase in endothelial Ca2+ concentration (Yashiro and Duling, 2000). The propagation of the Ca2+ signal was stopped by thapsigargin (Yashiro and Duling, 2003). The mechanisms involved in regulation of cytosolic calcium are dramatically different in endothelial and vascular smooth muscle cells, and effects in one cell type cannot be extrapolated to the other. It remains speculative whether Ca2+ oscillations are involved in the development of CVS.
The specific factors in SAH-CSF that caused Ca2+ oscillations in the present investigation are not entirely clear. Previous investigations suggested that oxygen radicals induce endothelial cytosolic Ca2+ oscillations by inhibiting the normal function of ER Ca2+ channels and pumps (Hu et al, 1998). Oxygen radicals directly attack the Ca2+-ATPase at the ATP-binding site (Xu et al, 1997) and inactivate the Ca2+ pump. In contrast, others hypothesize that free oxygen radicals activate phospholipase C. Phospholipase C phosphorylates inositoldiphosphate to IP3 and activates the IP3-dependent Ca2+ channels at the ER (Berridge, 1993). The sources of oxygen free radicals after SAH may be the perivascular oxyhemoglobine and the endothelial nicotinamide adenine dinucleotide phosphate-oxidase (Kim et al, 2002; Paravicini and Sobey, 2003). In addition, thrombin and ATP may also induce Ca2+ oscillations both alone and in concert. Both are proven to increase levels of IP3, which is linked to ER IP3-dependent Ca2+-channel activity (Glass and Bates, 2004; Tiruppathi et al, 2002). After SAH, CSF contains both substances (Tsurutani et al, 2003; Zhang et al, 1996).
The frequency of SAH-CSF-induced Ca2+ oscillations are in the range of frequencies that have been shown to activate NF-κB (Dolmetsch et al, 1998). In our experiments the frequency of oscillations was reduced with xestospongin and thapsigargin to 54%± 17% and 0% of control, respectively. In addition, a significant reduction of NF-κB activation was observed that was similar to the reduction of the frequencies of Ca2+ oscillations after xestospongin or thapsigargin addition. According to these results, it may be stated that SAH-CSF activates NF-κB, depending on the frequency of Ca2+ oscillations. These findings confirm the results of Hu et al who used xestospongin to reduce the frequency of Ca2+ oscillations when oscillations were induced by free oxygen radicals. As a result, a significant decrease of NF-κB activity was observed.
The activation of NF-κB is necessary for the induction of an inflammatory reaction. Activation of this factor leads to gene expression of intercellular adhesion molecule-1, VCAM-1, tumor necrosis factor-α, IL-1, IL-6, and IL-8. Tumor necrosis factor-α causes ET-1 gene expression (Dolmetsch et al, 1998; Faraci and Heistad, 1998). The expression of adhesion molecules and chemotactic cytokines can cause T-cells and monocytes to transmigrate into the vessel wall (De Martin et al, 2000). The perivascular-activated monocytes after SAH have been reported as one major source of ET-1 that can cause delayed CVS (Fassbender et al, 2000). There may be a possible link of CVS to SAH-induced Ca2+ oscillations through inflammation, because inactivation of NF-κB by pyrolidine dithiocarbamate or antisense oligonucleotids attenuates vasospasm in animal models of SAH (Ono et al, 1998; Zhou et al, 2007). In analogy to the reduction of Ca2+ oscillation frequency and NF-κB activation, xestospongin, and thapsigargin reduced the expression of VCAM-1. Ca2+ oscillations seem to be important in inducing vascular inflammation after SAH.
The Endothelial Barrier Function
The endothelial contractile mechanism is regulated through phosphorylation of the endothelial cell myosin light chain, which is controlled by Ca2+/ calmodulin-activated myosin light chain kinase. The cellular shrinking is Ca2+ dependent and subsequently leads to increased vessel permeability. The vessel permeability is proportional to the size of the intercellular space that opens up after cellular shrinking (Schnittler et al, 1990). According to our experiments, Ca2+ oscillations were responsible for the development of HCEC constriction. In control experiments with native CSF, no significant change in the intercellular space was observed. Human cerebral endothelial cell constricted when SAH-CSF-induced Ca2+ oscillations were present. In parallel with the blockade of the SAH-CSF-induced Ca2+ oscillations by thapsigargin and xestospongin, the cells reexpanded after the addition of blockers. These results are consistent with previous findings suggesting that plasmin and thrombin induce cellular constriction in cerebral vascular endothelial cells. The investigators assumed a Ca2+-dependent mechanism (Nagy et al, 1995).
After endothelial shrinking, vasoactive substances may diffuse directly from the lumen into the vessels' smooth muscle layer and cause vasoconstriction. This has been shown by Iuliano et al, 2004 in a primate animal model.
The increase of permeability in the blood-brain barrier has also been linked to the formation of brain edema in SAH models of both animals and humans (Xi et al, 2001). Together with VCAM-1, cellular constriction may facilitate the transmigration of monocytes into the vessel wall where these cells can generate one of the most potent vasoconstrictors that are endothelin-1 and others (Fassbender et al, 2000).
Conclusions
After SAH, CSF induces a vascular inflammatory response through Ca2+ oscillations in human cerebral endothelial cells. The oscillations depend on ER IP3-receptor-gated Ca2+ channels and ER Ca2+-ATPase activity. They lead to cellular constriction, NF-κB activation, and VCAM-1 expression.