
Abstract
Aquaporin-9 (AQP9) is a new member of the aquaporin family of water-selective channels mainly expressed in liver and testis, presenting the characteristic of also being permeable to various solutes, particularly lactate. Recent data have shown the presence of AQP9 on tanycytes in the rat brain. In the current study, the authors show the expression of AQP9 in astrocytes in the mouse brain and changes in its expression after cerebral ischemia. Indeed, in control mouse, the AQP9 immunolabeling is present on astrocytic processes bordering the subarachnoid space and ventricles. The labeling also is observed on astrocytes in the white matter, hippocampus, hypothalamus, and lateral septum. After focal transient ischemia, an increase of the immunolabeling is detected on astrocytes in periinfarct areas. This AQP9 distribution study in mouse brain suggests a role of AQP9 in water homeostasis in the central nervous system. Furthermore, the overexpression of AQP9 on astrocytes surrounding an ischemic lesion suggests that AQP9 may also play a role in the regulation of postischemia edema and, in view of its permeability to monocarboxylates, in the clearance of lactate from the ischemic focus.
Keywords
Aquaporins (AQP) are integral membrane proteins that serve as selective pathways for water transport across the plasma membranes of many tissues and cell types. Ten AQPs have been identified, each with a distinct distribution, in the kidney, liver, lung, eye, and brain. Of the AQPs cloned so far, only two have been detected in the brain; these are AQP1, which in the adult is restricted to the choroid plexus and AQP4, which is expressed on ependymal cells and on astrocytes throughout the brain (Venero et al., 2001). The AQP9 gene was cloned in 1998 both in the rat and in the human (Ishibashi et al, 1998; Tsukaguchi et al., 1998). Although AQP9 mRNA has been detected in astrocytes (Tsukaguchi et al., 1998), the presence of the protein in brain was only recently demonstrated by Elkjaer et al. (2000); this study showed AQP9 immunolabeling on tanycytes in the mediobasal hypothalamus in rat. Interestingly, AQP9 is highly permeable to water and various solutes, including carbamides, polyols, purines, pyrimidines, urea, and monocarboxylates (Tsukaguchi et al., 1998; Ko et al., 1999).
AQP4 has been implicated in a number of potential roles, such as regulation of water homeostasis, which is important for physiologic functioning of the central nervous system (CNS) (Venero et al., 2001). Furthermore, brain edema plays a critical role in a wide variety of CNS pathologic conditions, including trauma, stroke, tumors, infections, and metabolic disorders (Klatzo, 1994). Recent in vivo studies have demonstrated a role of AQP4 in the development of brain edema after ischemic stroke (Manley et al., 2000; Taniguchi et al., 2000).
To further clarify the regional and cellular distribution of AQP9 and its possible regulation under pathophysiologic conditions, the authors performed immunocytochemical exploration of AQP9 expression in normal mouse brain, as well as after transient focal cerebral ischemia. AQP9 expression on astrocytes in various regions of the mouse brain was found. The authors also showed that, after transient focal cerebral ischemia, AQP9 overexpression develops on reactive astrocytes in the border to the infarct. These results suggest that AQP9 is involved in brain water homeostasis and brain edema formation.
MATERIALS AND METHODS
Animal experimental procedure
All animal experiments were conducted in accordance with guidelines of the cantonal veterinary service. Control male B6CF1 mice (20 to 25 g) were anesthetized with 2.5% halothane and then decapitated.
For transient focal ischemia, male B6CF1 mice were anesthetized with 2.5% halothane (induction) and maintained under 1% halothane in 30% oxygen and 70% nitrous oxide with a face mask. In all animals, regional cerebral blood flow (rCBF) was measured by laser–Doppler flowmetry with a flexible probe fixed on the skull (1 mm posteriorly and 6 mm laterally from the bregma) starting before onset of ischemia until 10 minutes after reperfusion. Focal cerebral ischemia was induced by middle cerebral artery occlusion by introducing a silicone-coated 8–0 filament from the common carotid artery into the internal carotid artery and advancing it (Huang et al., 1994). The filament was removed after 25 minutes to allow reperfusion. Rectal temperature was controlled and maintained at 36.5°C ± 0.5°C with a temperature control unit and a heating lamp during the anesthesia period. Animals were killed 48 hours after reperfusion. The brains were frozen in liquid nitrogen vapor and sectioned on a cryostat. Infarcted areas were quantitated on 20-μm hematoxylin and eosin–stained coronal 750-μm distant cryostat sections. Infarction volume was calculated directly by multiplying the sum of infarct areas by 750 μm (Huang et al., 1994), or indirectly using the following formula: contralateral hemisphere (mm3) – undamaged ipsilateral hemisphere (mm3) (Swanson et al., 1990). The difference between direct and indirect infarct volumes is likely to correspond to the brain swelling.
Immunoblotting
For Western blotting, liver, testis and cerebral hemisphere were frozen and then gently homogenized on ice in 10 mmol/L Hepes pH 7.6, 42 mmol/L KCL, 5 mmol/L MgCl2, 1 % sodium dodecyl sulfate (SDS), 1 mmol/L phenylmethylsulflonyfluoride (PMSF), 1 mmol/L ethylene-diamine-tetraacetic acid (EDTA), 1 mmol/L ethylene Glycol-bisaminoethyl-ether-tetraacetic acid (EGTA), 1 mmol/L dithiotreitol, 1.5 mmol/L pepstatin, 2 mmol/L leupeptin, 0.7 mmol/L aprotinin. The homogenate was centrifuged in an Eppendorf centrifuge at 4000 g for 5 minutes and supernatant was sonicated during 30 seconds. Twenty micrograms of total protein per lane for liver and testis and 40 μg and 100 μg for brain were subjected to SDS polyacrylamide gel electrophoresis in 12% Tris-Glycine gels and were transferred to the polyvinylidene fluoride membrane. Nonspecific binding was prevented by incubating the membrane in TBST (10 mmol/L Tris pH 8.0, 150 mmol/L NaCl, 0.05% Tween 20) with 5% nonfat powdered milk. The blot was probed with a rabbit polyclonal antibody against AQP9 (1/500; Chemicon, Temecula, CA, U.S.A.) in TBST with 3% nonfat milk for 4 hours at room temperature. After washing in TBST, the blot was incubated with a horseradish peroxidase conjugated secondary antibody against rabbit IgG for 2 hours at room temperature, washed in TBST, and processed with an enhanced chemiluminescence (ECL) Western blotting detection system (Calbiochem, Darmstadt, Germany). The blots then were exposed to film for 30 seconds (hyperfilm ECL; Amersham Pharmacia Biotech Europe, Uppsala, Sweden).
Immunohistochemistry
Serial 20-μm-thick coronal sections were fixed with paraformaldehyde (PAF) 4% in phosphate-buffered saline (PBS) during 2 hours at room temperature and then rinsed 3 × 10 minutes in PBS. All immunolabeling studies were performed in PBS containing 0.1% Triton × 100 and 1% bovine serum albumin. After each incubation, sections were washed in PBS 3 × 10 minutes.
For single immunolabeling, the sections were first incubated overnight at 4°C with 300 μL anti-AQP9 (1/200, Chemicon). They then were incubated for 2 hours at room temperature with a biotinylated secondary rabbit antibody, rinsed, and exposed for 1 hour to the avidin-biotin complex (ABC, Vectastain kit) solution before application of DAB (diaminobenzidine; Biosys kit; Biosys, Kangwon-do, Korea). Sections then were rinsed in PBS and dehydrated in successive ethanol 70%, 95%, 100% and xylene solutions. Sections then were coverslipped with Eukit medium.
Sections for double immunofluorescence labeling were incubated with a mixture of two primary antibodies AQP9 (Chemicon, 1/200) and glial fibrillary acidic protein (GFAP; Sigma 1/400; St. Louis, MO, U.S.A.) or AQP9 (Chemicon, 1/200) and MAP-2 (Sigma, 1/400) overnight at 4°C and then with a mixture of two secondary antibodies anti-Rabbit coupled to CY3 (Sigma 1/300) and anti-mouse coupled to fluorescein isothiocyanate (FITC; Jackson, 1/100) for 2 hours at room temperature. After double immunofluorescence, sections were coverslipped with the antifading medium Vectashield (Vector). Controls were performed by omitting either one or both primary antibodies. All controls gave negative results with no detectable labeling.
The single immunolabeling coupled to DAB staining were observed and photographed under optical microscopy (Leica, Wetzlar, Germany). Immunofluorescent preparations were examined under confocal laser scanning microscope (Leica). The image from the double-labeled preparations under the CLSM were processed using at 488 nm (FITC) and 568 nm (CY3). The epifluorescence of FITC and CY3 were successively recorded through separate channels.
To quantitate the increase in expression of AQP9 after ischemia, the cells labeled by the anti-AQP9 antibody were counted in 6 different fields (422 μm × 338 μm) in the border of the infarct and in matching contralateral fields in 3 animals. The number of positive cells per field was compared by a paired t-test and results were expressed by mean ± SD.
RESULTS
Distribution of the AQP9 immunolabeling in mouse brain
The authors first tested the AQP9 antibody on mouse liver sections and found an immunohistochemical localization of AQP9 on hepatocytes (Fig. 1A), consistent with previously published data in rat (Elkjaer et al., 2000). Specificity of the antibody was confirmed by immunoblotting of liver, testis (20 μg), and brain (40 μg and 100 μg) homogenates, revealing a band of the expected molecular weight of ∼30 kDa (Fig. 1B). In addition, higher molecular weight bands were seen consistent with dimers of ∼60 kDa (Fig. 1B).
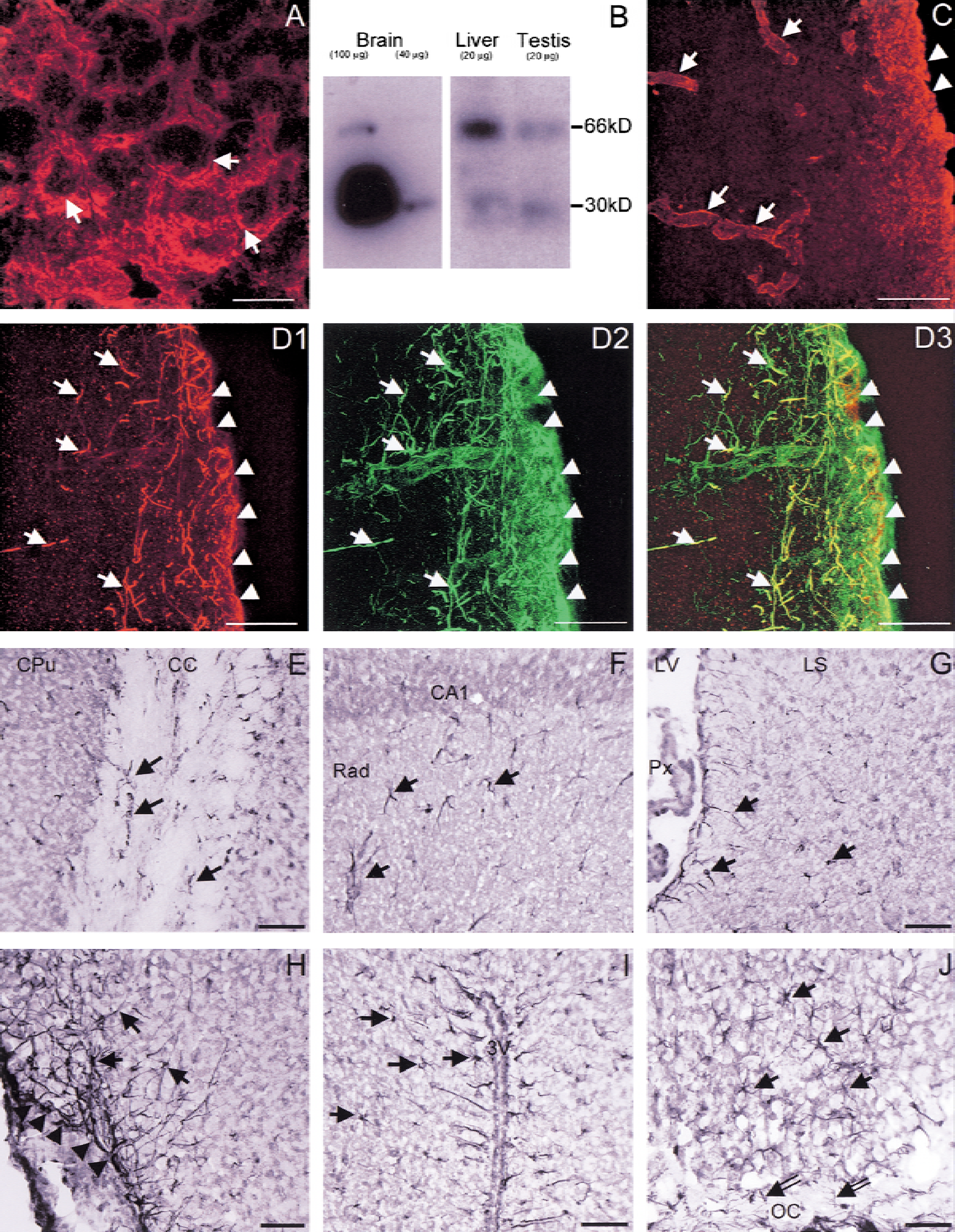
Immunocytochemical analysis of AQP9 expression in liver and brain of mice.
In the mouse brain, AQP9 labeling colocalized with GFAP-positive profiles suggesting an astrocytic expression (Fig. 1D). Double immunolabeling with AQP9 and GFAP revealed the presence of AQP9 labeling on astrocytic processes and cell bodies (Fig. 1D). In contrast with AQP4 (Fig. 1C) (Badaut et al., 2000a), AQP9 staining was not observed around all blood vessels (Fig. 1D). There was no colocalization of AQP9 with the neuronal marker MAP-2, suggesting that neurons do not express AQP9.
AQP9 labeling was observed on astrocytes of glia limitans and around the ventricles (Fig. 1). There was no AQP9 expression in other deep cortical layers in control mice. In contrast with the rat brain (Elkjaer et al., 2000), no AQP9 labeling was observed on the ependymal cells. White matter tracts (corpus callosum (Fig. 1E), anterior commissure, and optic chiasm) exhibited a marked AQP9 staining around neuronal processes. The AQP9 immunolabeling was also present in the hippocampus (Fig. 1F), in the septal nuclei (Fig. 1G), and in various hypothalamic nuclei, particularly in the supraoptic (Fig. 1H), the paraventricular (Fig. 1I), and the suprachiasmatic nuclei (Fig. 1J).
Expression of AQP9 after transient focal brain ischemia
AQP9 labeling was examined after 25 minutes of transient focal cerebral ischemia and 48 hours of reperfusion. During ischemia, rCBF was reduced to 7% ± 3% (mean ± SD) of baseline (n = 3) and increased to 68% ± 10 % of baseline during reperfusion. This procedure resulted in an infarct size of 88 ± 22 mm3 (n = 3). Calculated indirectly, infarct volume was 39 ± 5 mm3 (n = 3).
After ischemia, AQP9 labeling was markedly induced in regions bordering the infarct observed on adjacent sections with hematoxylin and eosin staining. The number of positive cells labeled by AQP9 antibody was 19 ± 9 cells per field in the infarct border (green dots in Fig. 2) versus 2 ± 3 in the contralateral side (P < 0.001). Regional distribution of AQP9 labeling was observed around the infarcted area, mainly in the cortex, in the ventral pallidum and in various nuclei of the amygdala (Fig. 2A to 2D). Double labeling demonstrated a colocalization of AQP9 and GFAP staining (Fig. 2E), whereas there was no colocalization with MAP-2.
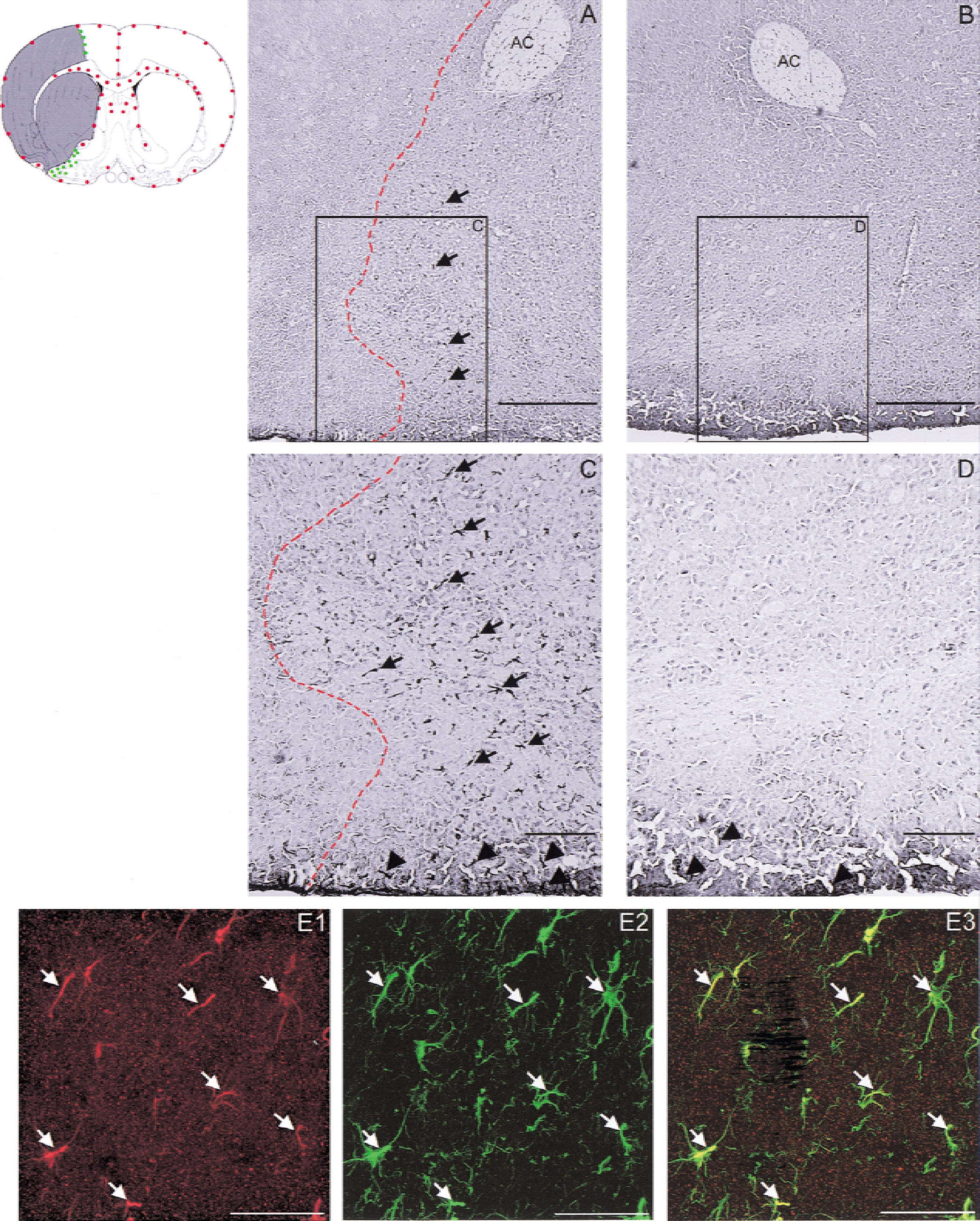
Immunohistochemical studies of AQP9-expression after transient focal ischemia in mouse brain. The schematic illustration of a coronal section of the mouse brain indicates the distribution of the AQP9 labeling in normal condition (red dots) and the AQP9 induction (green dots) in the periinfarct region (gray shaded area). Photomicrograph of AQP9 staining in the border zone of the ischemic lesion, which is on the left of the dotted line
DISCUSSION
Two AQPs have been described in the CNS. The AQP1 subtype is present on the choroid plexus and participates in cerebrospinal fluid formation. The predominant water channel in the brain is the AQP4 subtype, which shows a marked distribution in periventricular areas. AQP4 has been ascribed several roles within the CNS. Recent data support that AQP4 water channels are implicated in K+ buffering, brain water homeostasis, central osmoreception, volume transmission, cerebrospinal fluid formation, and brain edema evolution (Venero et al., 2001). Expression of the AQP9 subtype in the brain recently has been reported (Elkjaer et al., 2000) showing AQP9 immunolabeling on tanycytes of the mediobasal hypothalamus and ependymal cells lining the ventricles of normal rats. In the current study, the authors demonstrate the presence of AQP9 protein in normal mouse brain, as well as an increase of its expression after transient focal cerebral ischemia.
Results show an AQP9 labeling on astrocytes in the periventricular parenchyma, as well as on astrocytes of the glia limitans, bordering the subarachnoid space. As for AQP4, this localization suggests a role in regulating water homeostasis between cerebrospinal fluid and brain parenchyma.
The current study also shows that AQP9 is expressed on astrocytes in several other brain regions. Obvious AQP9 labeling was found in white matter tracts—that is, in the corpus callosum, the anterior commissure, and optic chiasm, as well as the hippocampus, septum, and several hypothalamic nuclei. This regional distribution of AQP9 is similar to that of AQP4 (Venero et al., 2001), indicating that AQP9 may be involved in the control of plasma osmolality by the CNS and extracellular water homeostasis.
Control of plasma osmolality involves the hypothalamic magnocellular nuclei (Bourque et al., 1997). The expression of AQP9 seen in magnocellular hypothalamic nuclei, suggests that, similar to AQP4 (Nielsen et al., 1997; Badaut et al., 2000a), AQP9 may participate in plasmatic osmotic sensing and favor water flux down the osmotic gradient.
Control of extracellular water homeostasis depends on water redistribution after the osmotic gradients. Neuronal activity is characterized by net ion fluxes (mainly K+) and neurotransmitter release, that induce modifications of the extracellular osmotic pressure (Dietzel et al., 1982; Andrew and MacVicar, 1994). K+ and water transport are tightly coupled and K+ efflux during neuronal activity represents a challenge to cell volume control. Such modifications can perturb neuronal signaling; it has been proposed that astrocytic AQP4 may play a critical role in extracellular water homeostasis (Wen et al., 1999; Badaut et al., 2000b; Venero et al., 2001). The authors show that AQP9 expression is present not only around microvessels, like AQP4, but also on astrocytic processes and cell bodies without contact with vascular structures. Based on this cellular distribution of AQP9, it can be hypothesized that AQP9 is involved in extracellular water homeostasis by favoring high transmembrane water fluxes to ensure an appropriate neuronal environment of the extracellular space. Astrocytic AQP9 may play a buffer role around active neurons and axons.
Using the model of transient middle cerebral artery occlusion in mouse (Huang et al., 1994), the authors further studied AQP9 expression in ischemic stroke. Overexpression of AQP9 was observed in the infarct borderzone, mainly in the cortex, ventral pallidum, and nuclei of the amygdala on reactive astrocytes. A similar overexpression of water channels at the border of an infarct has been reported after rat focal brain ischemia for AQP4-mRNA (Taniguchi et al., 2000). Vizuete et al. (1999) showed that the regulation of AQP4-mRNA expression correlates with the disruption of the BBB and hypertrophy of astrocytes. Manley et al. (2000) showed that mice deficient in AQP4 have decreased brain swelling compared with wild-type mice in two brain edema models, acute water intoxication and ischemic stroke. These previous reports suggest that AQP4 may contribute to edema formation. In contrast with rat, the authors have not found AQP4 overexpression in the infarct border in the mouse (data not shown). AQP9 overexpression in the infarct border suggests that AQP9 may participate in water movements and in edema formation in mouse brain.
The AQP9 permeability to lactate and water may be important under pathologic conditions such as brain ischemia. Interestingly, AQP9 permeability to lactate increases 4-fold when pH decreases to 5.5 (Tsukaguchi et al., 1998). Lactic acidosis during ischemia may increase the permeability of AQP9 and enable uptake of the excess lactate by the astrocytes. AQP9 could favor lactate clearing from the extracellular space under pathological condition. Lactate movement into the astrocytes is likely to be accompanied by a rapid water flux involving AQP9 and AQP4.
In conclusion, this study reports the demonstration of astrocytic AQP9 expression in the mouse brain. The distribution of this new water channel suggests that it may play a role in brain water homeostasis. Its permeability to metabolites, such as lactate, also suggests a role in brain metabolism. The observation of AQP9 overexpression, after focal transient brain ischemia, in reactive astrocytes in the periinfarct border zone indicates that AQP9 may also play a key role in water homeostasis and in lactate buffering in ischemic stroke. AQP9 could participate in the edema formation after brain ischemia. Pharmacologic and genetic manipulations should allow to further characterize its roles.