
Abstract
Brain edema formation is one of the most important mechanisms responsible for brain damage after ischemic stroke. Despite considerable efforts, no specific therapy is available yet. Arginine vasopressin (AVP) regulates cerebral water homeostasis and has been involved in brain edema formation. In the current study, we investigated the role of AVP V1 and V2 receptors on brain damage, brain edema formation, and functional outcome after transient focal cerebral ischemia, a condition comparable with that of stroke patients undergoing thrombolysis. C57/BL6 mice were subjected to 60-min middle cerebral artery occlusion (MCAO) followed by 23 h of reperfusion. Five minutes after MCAO, 100 or 500 ng of [deamino-Pen(1), O-Me-Tyr(2), Arg(8)]-vasopressin (AVP V1 receptor antagonist) or [adamantaneacetyl(1), O-Et-d-Tyr(2), Val(4), Abu(6), Arg(8,9)]-vasopressin (AVP V2 receptor antagonist) were injected into the left ventricle. Inhibition of AVP V1 receptors reduced infarct volume in a dose-dependent manner by 54% and 70% (to 29±13 and 19±10 mm3 versus 63±17 mm3 in controls; P<0.001), brain edema formation by 67% (to 80.4%±1.0% versus 82.7%±1.2% in controls; P<0.001), blood-brain barrier disruption by 75% (P<0.001), and functional deficits 24 h after ischemia, while V2 receptor inhibition had no effect. The current findings indicate that AVP V1 but not V2 receptors are involved in the pathophysiology of secondary brain damage after focal cerebral ischemia. Although further studies are needed to clarify the mechanisms of neuroprotection, AVP V1 receptors seem to be promising targets for the treatment of ischemic stroke.
Keywords
Introduction
To date thrombolysis is the only clinically approved treatment for stroke. However, most patients do not benefit from this therapeutic option because of multiple exclusion criteria. Among those, the most important are late hospital admission exceeding the 3-h therapeutic window of current thrombolysis protocols and conditions associated with complications after reperfusion, for example, intraparenchymal bleeding or formation of postischemic brain edema (The National Institute of Neurological Disorders and Stroke, 1995). Despite considerable efforts, especially the pathophysiologic understanding of brain edema formation following reperfusion after cerebral ischemia is still so limited that specific clinical treatment options are not expected to be available within the near future.
Arginine vasopressin (AVP), also known as antidiuretic hormone (ADH) or argipressin, is a cyclic nonapeptide synthesized in the hypothalamus and released into the systemic circulation by the neurohypophysis (Antoni, 1993). Its primary action is the control of water and solute excretion in the kidney; however, AVP is also released in various parts of the brain where it may act as a neurotransmitter (Buijs, 1978; Riphagen and Pittman, 1986). In the central nervous system (CNS), AVP has multiple functions including regulation of brain water (Rosenberg et al, 1992; Bemana and Nagao, 1999), brain ion homeostasis (DePasquale et al, 1989; Cserr and Latzkovits, 1992), and regulation of cerebral microvascular resistance (Fernandez et al, 2001). The effects of AVP are mainly mediated via three seven-transmembrane G-protein-coupled receptors, namely V1a, V1b, and V2. The predominant type of AVP receptors in the CNS belongs to the V1 subtype (Brinton et al, 1984; Pearlmutter et al, 1988; Phillips et al, 1988); however, V2 receptors may also be expressed in the brain (Kozniewska and Szczepanska-Sadowska, 1990).
Cerebral ischemia induces AVP mRNA (Liu et al, 2000) and protein (Liu, 1992) expression, and increased AVP plasma levels have been reported in stroke patients (Barreca et al, 2001). Application of AVP exacerbates acute ischemic brain edema (Liu et al, 1991; Dickinson and Betz, 1992; Ikeda et al, 1997) and this exacerbation can be reduced by AVP antiserum (Liu et al, 1991) or by the AVP release inhibitor RU51559 (Ikeda et al, 1997). The role of AVP for postischemic brain edema formation was further supported by findings that brain water content was reduced in AVP-deficient rats 4 h after permanent middle cerebral artery occlusion (MCAO) (Dickinson and Betz, 1992) and by a study showing reduced hemispheric swelling of TTC-stained brain sections 48 h after permanent (embolic) focal cerebral ischemia in rats treated with a V1 receptor antagonist (Shuaib et al, 2002).
Despite the compelling evidence that AVP is involved in brain edema formation after permanent cerebral ischemia, studies investigating the role of AVP and its receptors under conditions relevant for patients undergoing thrombolysis, that is, cerebral ischemia followed by reperfusion, are missing yet. Furthermore, no information about the role of V2 receptors for the development of postischemic brain damage and edema formation is available to date.
Therefore, we performed the current study to investigate the role of AVP V1 and V2 receptors on infarct volume, brain edema formation, blood-brain barrier (BBB) integrity, and neurologic outcome in a model of transient focal cerebral ischemia.
MATERIALS AND METHODS
Animals
Male C57/BL6 mice weighing 22 to 28 g (Charles River, Sulzfeld, Germany) were used for the current study. The animals had free access to tap water and pellet food. Animal experiments were conducted in accordance with institutional guidelines approved by the Government of Upper Bavaria (Protocol Number 88-03).
Focal Cerebral Ischemia
Middle cerebral artery occlusion was induced by the intraluminal filament method as described previously (Plesnila et al, 2001, Plesnila et al, 2004; Kataoka et al, 2004). Briefly, animals were anesthetized with halothane (0.8% in 30%/70% oxygen/nitrous oxide) and the left common carotid artery (CCA) was exposed through a midline neck incision. A silicone-coated 8-0 monofilament was inserted into the internal carotid artery (ICA) and advanced towards the origin of the middle cerebral artery (MCA) until occlusion occurred. Middle cerebral artery occlusion was monitored by laser-Doppler fluxmetry (Perimed, Stockholm, Sweden). After 60 mins of MCAO, reperfusion was accomplished by withdrawing the intraluminal filament.
Physiologic Monitoring
In a parallel group of animals, the left femoral artery was cannulated and the mean arterial blood pressure (MABP) was recorded continuously from 30 mins before MCAO until 30 mins after reperfusion. Arterial blood gases (ABG) were measured in 60 μL arterial blood samples taken before ischemia and 15 mins after reperfusion. Laser-Doppler flux was continuously measured from 30 mins before MCAO until 30 mins after reperfusion.
Quantification of Infarct Volume
Twenty-four hours after MCAO, animals were deeply anesthetized with 4% halothane and killed by cervical dislocation. Brains were removed and frozen on powdered dry ice. Beginning at the middle of the olfactory bulb, 10-μm-thick coronal sections were collected every 750 μm throughout the brain. Sections from the resulting 12 levels were stained with cresyl violet and digitized using a digital camera connected to a microscope. The infarct area and the areas of the two hemispheres were quantified on each section by an investigator masked towards the treatment of the animals using an image analysis system (Olympus DP-soft, Munich, Germany). Data were expressed as contralateral hemisphere (mm3) minus undamaged ipsilateral hemisphere (mm3) to correct for brain edema. Infarct volume was calculated by multiplying the corrected infarct areas with the distance between sections as previously described (Plesnila et al, 2001).
Measurement of Brain Water Content
Twenty-four hours after MCAO, mice were killed and brains removed as described above. The pons and olfactory bulb were removed and the brains were weighted to obtain their wet weight (ww). Thereafter brains were dried at 110°C for 24 h for determining their dry weight (dw). Brain water content was calculated by using the following formula: (ww-dw)/ww × 100.
Evaluation of Blood-Brain Barrier Permeability
Blood-brain barrier permeability was evaluated by measuring Evans' Blue (EB) extravasation, as previously described by Uyama et al (1988). Briefly, mice received 4 mL/kg of a 2% EB solution by tail vein injection 30 mins after MCAO. Twenty-three hours after reperfusion animals were perfused transcardially with 100 mL saline solution to wash out intravascular EB. Thereafter, brains were removed and the hemispheres were separated and weighted. For extraction of EB from brain tissue hemispheres were placed in 2 mL 50% trichloroacetic acid (wt/vol) and homogenized by sonication. The homogenate was centrifuged at 10 000 r.p.m. for 20 mins and the supernatant was diluted 1:12 with ethanol. Evans' blue was measured at an excitation wavelength of 620 nm and an emission wavelength of 680 nm using a fluorescence spectrophotometer (Model 650-10S, Perkin-Elmer, Norwalk, OH, USA). By using a standard curve (100 to 500 ng/mL) EB concentrations were calculated and expressed as μg/g brain tissue.
Evaluation of Neurologic Deficits
Neurologic deficits were evaluated 24 h after MCAO as previously described (Plesnila et al, 2001) using a five-point scale: 0 = normal motor function, 1 = flexion of contralateral torso or forelimb on lifting by tail or failure to extend forepaw when suspend vertically, 2 = circling to either side while having normal posture at rest, 3 = loss of righting reflex, and 4 = no spontaneous motor activity.
Application of V1 and V2 Receptor Antagonists
The selective antagonists [deamino-Pen(1), O-Me-Tyr(2), Arg(8)]-vasopressin (V-1880; Lot # 053K13071; Sigma, St Louis, MO, USA) and [adamantaneacetyl(1), O-Et-d-Tyr(2), Val(4), Aminobuturyl(6), Arg(8,9)]-vasopressin (V-2381; Lot # 073K11151; Sigma, St Louis, MO, USA) were used for inhibition of V1 and V2 receptors, respectively (Manning et al, 1987). One hundred or 500 ng of the drugs in 2 μL saline were administered 5 mins after MCAO by intracerebroventricular administration (0.9 mm left, 0.1 mm posterior, and 3.1 mm deep relative to bregma). Control animals were subjected to MCAO and received 2 μL saline intracerebroventricularly. Sham-operated animals were anesthetized, not subjected to exposure of carotid arteries and MCAO and received 2 μL saline intracerebroventricularly.
Experimental Design
Infarct volume and functional outcome were investigated 24 h after MCAO in five different groups: (1) control (n = 8), (2) 100 ng V1 receptor antagonist (n = 7), (3) 500 ng V1 receptor antagonist (n = 7), (4) 100 ng V2 receptor antagonist (n = 7), and (5) 500 ng V2 receptor antagonist (n = 7).
Blood-brain barrier permeability was measured 24 h after MCAO in three different groups: (1) control (n = 7), (2) 500 ng V1 receptor antagonist (n = 7), and (3) 500 ng V2 receptor antagonist (n = 7).
Brain water content was measured 24 h after MCAO in four different groups: (1) sham (n = 6) (2) control (n = 7), (3) 500 ng V1 receptor antagonist (n = 7), and (4) 500 ng V2 receptor antagonist (n = 7).
The MABP and ABG were assessed in three separate experimental groups (n = 4 each): (1) control, (2) 500 ng V1 receptor antagonist, and (3) 500 ng V2 receptor antagonist.
Animals were randomly assigned to the different treatment groups and the investigator who performed animal surgery was masked towards the treatment of the animals.
Statistical Analysis
All result are presented as mean±s.d. Differences between groups were evaluated using ANOVA on ranks followed by Tukey's or the Student-Newmann-Keuls test as post hoc analysis (SigmaStat 2.0, Jandel Scientific, Erkrath, Germany). Differences were considered significant at P<0.05.
RESULTS
Physiologic Parameters
Cerebral ischemia alone or together with intracerebroventricular injection of AVP V1 or V2 receptor antagonists did not influence MABP, PaCO2, PaO2, pH, or blood glucose levels (Table 1).
Cerebral blood flow (rCBF) was reduced to less than 15% of baseline in each group and was not different between groups (data not shown). Drug application did not have any influence on intra- or postischemic blood flow (Figure 1).
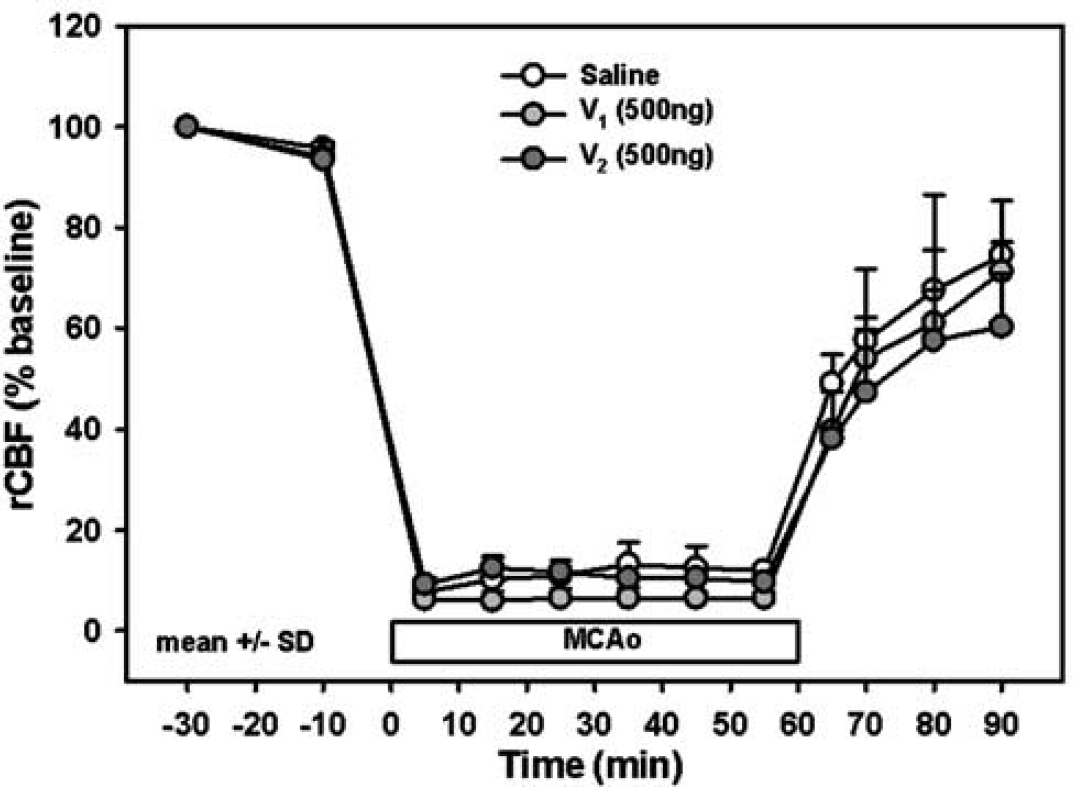
Regional cerebral blood flow (rCBF) before and during MCAO, and after reperfusion in mice receiving saline, AVP V1, or AVP V2 receptor antagonist (500 ng) (n = 4 each) 5 mins after MCAO.
Physiologic parameters 30 mins before and 90 mins after MCAO of animals treated with saline, AVP V1, or AVP V2 receptor antagonist intracerebroventricularly 5 mins after MCAO (n = 4 each)
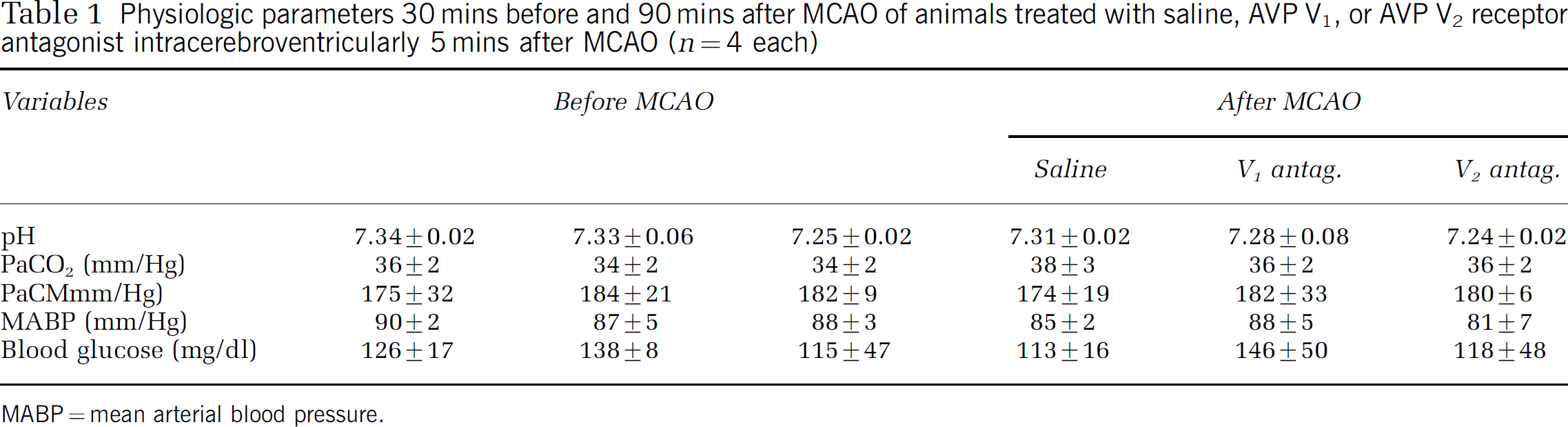
MABP = mean arterial blood pressure.
Plasma sodium was measured by tail vein puncture in normal mice (0, 2, and 24 h) and in mice subjected to cerebral ischemia (before, 2, and 24 h after initiation of MCAO). Plasma sodium concentrations were elevated after cerebral ischemia in all experimental groups most likely because of dehydration as a result of the reduced food and water intake after the insult. Treatment with AVP V1 or V2 receptor antagonists (100 or 500 ng intracerebroventricularly), however, had no additional effect on plasma sodium concentrations (Figure 2).
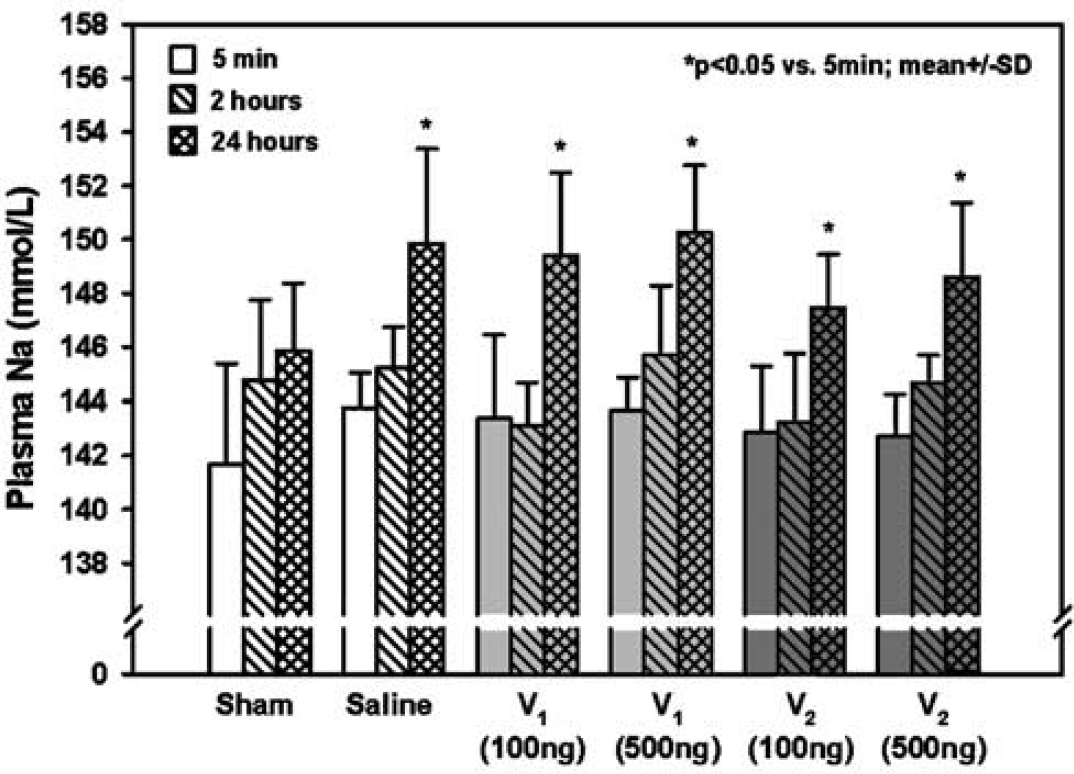
Plasma sodium concentration at different time points in control mice and in animals subjected to MCAO with internal carotid artery injection of saline, AVP V1, or AVP V2 receptor antagonist (100 or 500 ng) (n = 5 to 7 each).
Effect of V1 and V2 Receptor Antagonists on Infarct Volume
The mean infarct volume in saline-treated control animals was 64±17 mm3. Treatment with an AVP V1 receptor antagonist at doses of 100 and 500 ng administered 5 mins after MCAO resulted in a reduction of cerebral infarction volume by 54% and 70%, respectively (29±13 and 19±10 mm3, respectively; P<0.001) (Figure 3A). Administration of a V2 receptor antagonist at doses of 100 and 500 ng had no significant effect on infarct volume (59±11 and 45±15 versus 64±17 mm3 in control animals). Neuroprotection exerted by inhibition of AVP receptors was mainly seen in the cortex, which represents the penumbra in the currently used experimental stroke model (Plesnila et al, 2004), while almost no protection was observed in the infarct core, that is, the basal ganglia (data not shown). These findings are also reflected by the data showing that V1 receptor-mediated neuroprotection is mainly seen in the posterior part of the MCA territory where cortical, that is, penumbral, tissue predominates (Figure 3B).
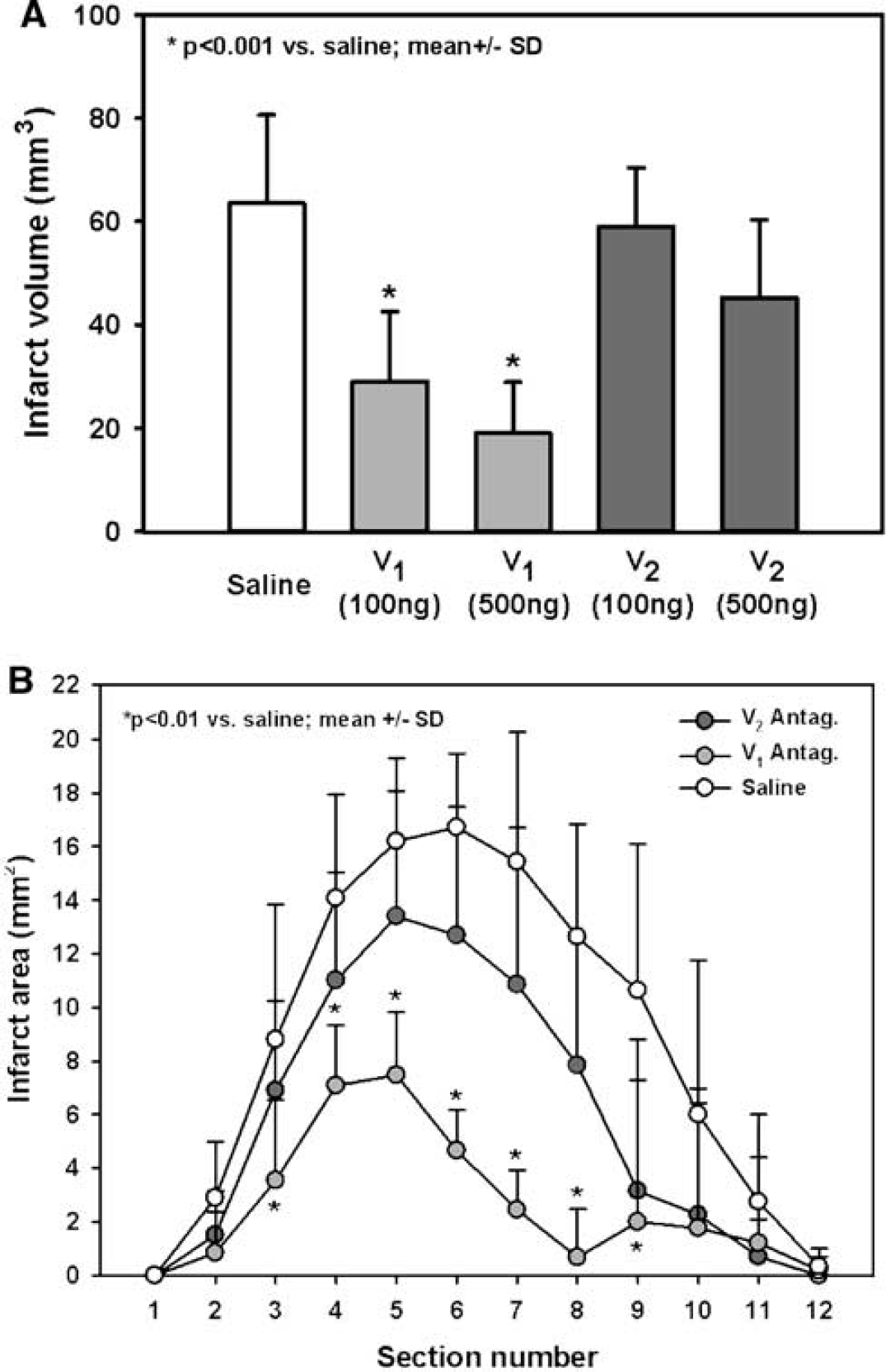
(
Effect of V1 and V2 Receptor Antagonization on Brain Water Content
Hemispheric brain water content of sham-operated mice (anesthesia, 2 μL saline intracerebroventricularly) 24 h after surgery was 79.2%±0.2% and 79.1%±0.7%. Focal cerebral ischemia significantly increased brain water content of the ischemic hemisphere by more than 3.5% to 82.7%±1.2% (P<0.001), while the brain water content of the contralateral hemisphere did not change significantly (79.8%±0.4%) (Figure 4). Inhibition of AVP V1 receptors reduced the postischemic increase of brain water content by 67% (from 82.7%±1.2% in control animals to 80.4%±1.0%; P<0.001). Inhibition of V2 receptors had no significant effect on brain water content of the ischemic hemisphere (82.8±0.9 versus 82.7±1.2).
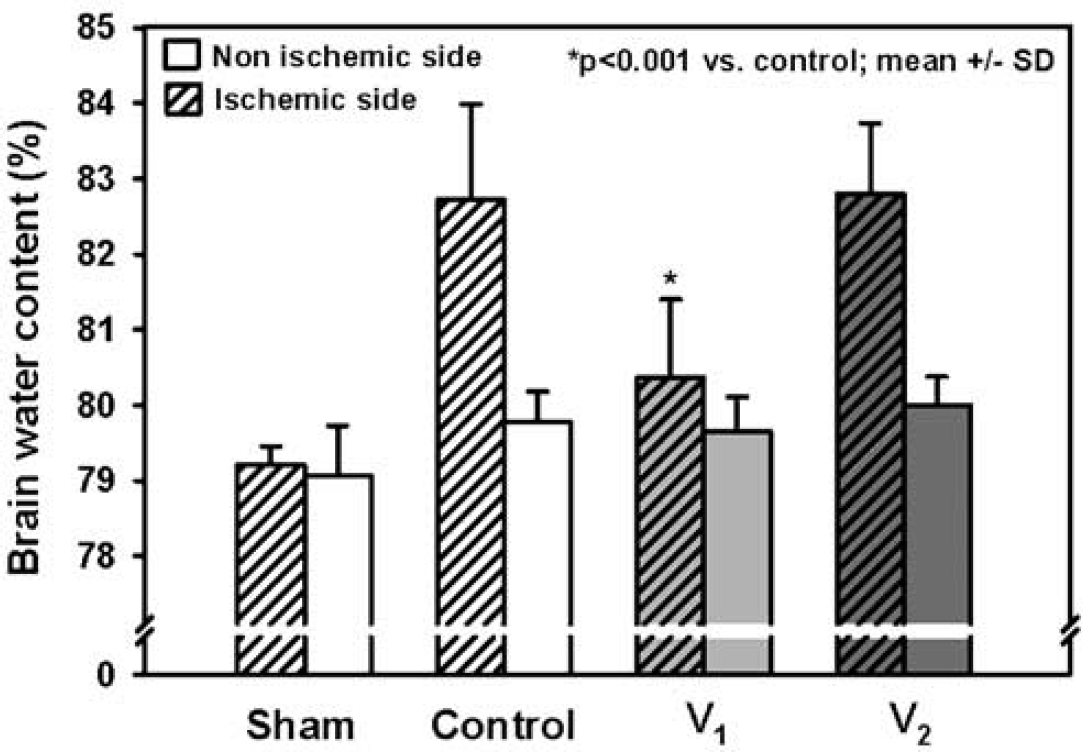
Brain water content 23 h after 60 mins MCAO in normal mice or animals treated with saline, AVP V1, or AVP V2 receptor antagonist (500 ng) (n = 6 to 7 each) 5 mins after initiation of MCAO.
Role of V1 and V2 Receptors on Blood-Brain Barrier Permeability
To investigate if AVP receptor-mediated brain edema formation was associated with opening of the BBB, we quantified EB extravasation 24 h after MCAO. Cerebral ischemia resulted in a tissue EB content of 87±33 μg/g tissue, while tissue EB in the nonischemic hemisphere (44±11 μg/g tissue) was not different from values previously obtained in non-operated animals (data not shown). Treatment with an AVP V1 receptor antagonist (500 ng) resulted in 75% less EB extravasation into ischemic brain (55±8 versus 87±33 μg/g tissue; P<0.001), while BBB integrity of the contralateral hemisphere was not affected (Figure 5).
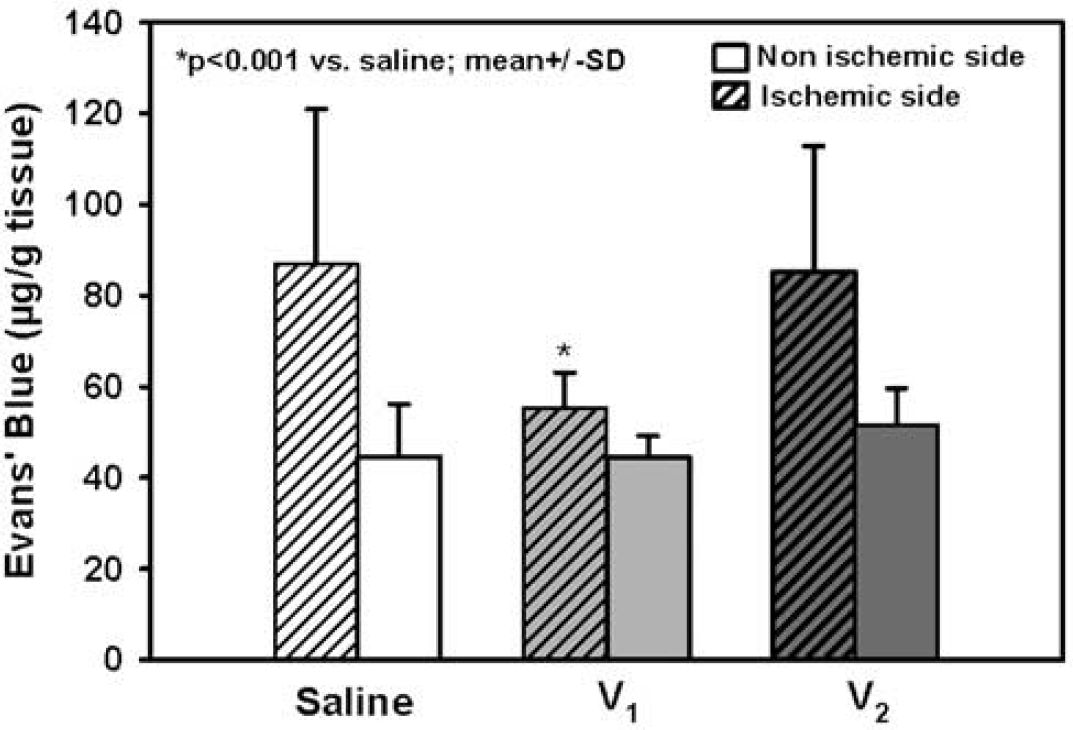
Blood-brain barrier permeability assessed by EB extravasation 23 h after 60 mins MCAO in mice treated with saline, AVP V1, or AVP V2 receptor antagonist (500 ng) (n = 7 each).
In animals receiving an AVP V2 receptor antagonist (500 ng), EB extravasation was not different from saline-treated animals (Figure 5).
Effect of V1 and V2 Receptor Blockade on Functional Outcome
In the saline-treated control group, the neurologic deficit score was 1.9±0.4 24 h after MCAO (Figure 6). Inhibition of AVP V1 receptors 5 mins after MCAO resulted in a significantly improved neurologic deficit score 24 h after initiation of cerebral ischemia (1.1±0.4 versus 1.9±0.4; P<0.05). All other treatment groups did not show an improvement of the neurologic function as compared with control animals (Figure 6).
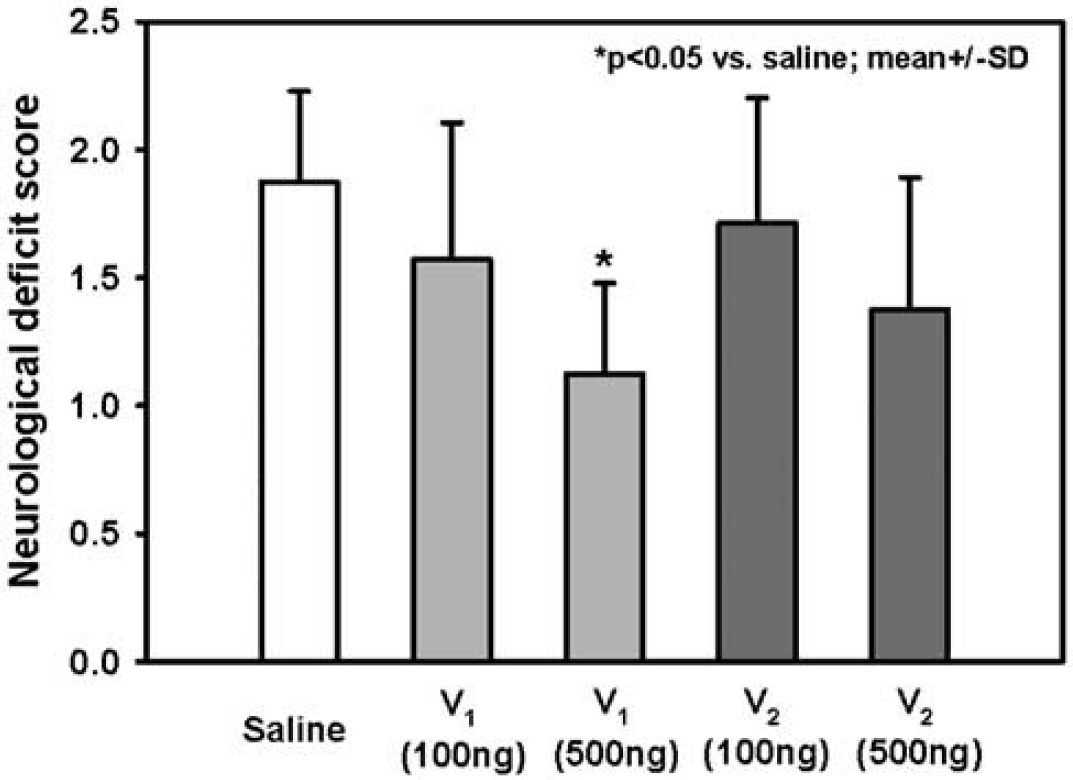
Effect of treatment with saline, AVP V1, or AVP V2 receptor antagonist (100 or 500 ng) (n = 7 each) on functional outcome 60 mins and 23 h after MCAO.
DISCUSSION
Our findings show, to the best of our knowledge for the first time, that inhibition of vasopressin V1 receptors reduces brain edema formation, BBB disruption, infarct volume, and improves functional outcome in an animal model of stroke followed by reperfusion, while inhibition of V2 receptors had no effect.
Model
Transient focal cerebral ischemia of the MCA was induced in mice by an intraluminal filament, a reliable and highly reproducible method routinely used in our laboratory (Plesnila et al, 2004; Kataoka et al, 2004). Successful and sufficient MCAO (<20% of baseline cerebral blood flow (CBF)) was demonstrated by laser-Doppler fluxmetry in all experimental animals and blood pressure, blood gases, and body temperature were maintained within the physiologic range during all experimental procedures.
Our results show that centrally applied AVP receptor antagonists do not have an effect on blood pressure, CBF, and body temperature as also observed by others (Rosenberg et al, 1992). Hence, hypothermia or any other disturbances of animal physiology can be ruled out as a possible nonspecific mechanism of the neuroprotection observed in the current study.
AVP during Pathologic Conditions of the Brain
Our results are well in agreement with other experimental studies showing that AVP is involved in the pathophysiology of acute brain damage, for example, after subarachnoid or intracerebral hemorrhage (Doczi et al, 1984; Rosenberg et al, 1992), cortical focal lesions (Reeder et al, 1986; Bemana and Nagao, 1999), traumatic brain injury (Armstead, 2001), global cerebral ischemia (Liu et al, 1991, Liu et al, 1996; Ikeda et al, 1997), and permanent focal cerebral ischemia (Dickinson and Betz, 1992; Shuaib et al, 2002). The amount of AVP mRNA and of AVP protein increase after experimental cerebral ischemia (Liu, 1992; Liu et al, 2000) and levels of AVP are elevated in the CSF of stroke patients (Barreca et al, 2001).
Role of AVP for the Formation of Brain Edema
Although intracerebroventricular infusion of AVP does not alter brain water content in normal brain, it does significantly increase brain edema formation and cerebral sodium uptake during hypernatremia (Vajda et al, 2001), after cold injury-induced brain edema (Reeder et al, 1986), and after postischemic brain edema (Liu et al, 1996). In the Brattleboro rat, which does not express AVP, brain sodium uptake was reduced during hypernatremia and after cerebral ischemia by 61% and 36%, respectively, and brain edema formation was reduced by 1/3 (DePasquale et al, 1989; Dickinson and Betz, 1992) while all changes were reversible when AVP was administered intracerebroventricularly (Dickinson and Betz, 1992). Most interestingly, AVP seems to influence brain edema formation after both cold injury and global cerebral ischemia, two conditions known to be associated almost exclusively with the development of vasogenic or cytotoxic brain edema, respectively (Reeder et al, 1986; Liu et al, 1996). These findings indicate that AVP may influence brain water and brain volume homeostasis by multiple pathways, for example, by influencing the permeability of the BBB and by directly modulating the cell volume of neurons and astrocytes. This hypothesis is supported by our current findings and by other studies showing reduced BBB permeability after inhibition of AVP V1 receptors (Figure 4; Bemana and Nagao, 1999; Shuaib et al, 2002), and by experiments showing that AVP leads to swelling of cultured astrocytes and this response can be inhibited by AVP V1 receptor antagonists and by the Na+−K+−Cl− cotransport inhibitor bumetanide (Latzkovits et al, 1993), a transporter involved in pathologic cell swelling of astrocytes (Ringel et al, 2000).
Furthermore, these data clearly indicate that brain edema formation is mainly mediated by the activation of AVP V1 receptors. AVP V2 receptors, which have been claimed to exist in the brain (Kozniewska and Szczepanska-Sadowska, 1990; Gouzenes et al, 1999), had no effect on BBB permeability and brain edema formation after transient focal cerebral ischemia.
Alternative Mechanisms of AVP-Mediated Brain Damage
Although our data strongly suggest that AVP V1 receptor-mediated neuroprotection is due to the reduction of postischemic brain edema, AVP V1 receptors have also additional functions that may suggest alternative mechanisms of neuroprotection by AVP receptor inhibition. AVP V1 receptors were, for example, identified on cerebrovascular smooth muscle cells and capillary endothelium (Hess et al, 1991; Fernandez et al, 2001) and have been implicated in the regulation of CBF (Fernandez et al, 2001). Because brain AVP content increases after cerebral ischemia in experimental animals and in stroke patients (Liu, 1992; Liu et al, 2000; Barreca et al, 2001) and AVP mediates vasoconstriction through V1 receptors (Fernandez et al, 2001) it might be speculated that AVP induces vasoconstriction after cerebral ischemia, thereby leading to reduced collateral blood flow to ischemic tissue and cell death. This effect, if present, seems to be restricted to penumbral tissue, because inhibition of AVP V1 receptors does not affect CBF in the infarct core (Figure 1). Another explanation for AVP V1 receptor-mediated neuroprotection might be that AVP V1 receptors are widely expressed in the brain parenchyma (Brinton et al, 1984; Pearlmutter et al, 1988; Phillips et al, 1988; Young et al, 1999; Tribollet et al, 1999; Fernandez et al, 2001) where they have been implicated in intracellular calcium mobilization, activation of NMDA receptors, and superoxide anion generation (Urban and Killian, 1990; Hess et al, 1991; Gouzenes et al, 1999; Armstead, 2001), all conditions well known to be associated with neuronal cell death after cerebral ischemia (Lo et al, 2003). Accordingly, the positive effect AVP V1 receptor inhibition on cell death might also be explained by a reduction of glutamate- and superoxide anion-mediated toxicity, as also discussed previously (Tanaka et al, 1994).
Conclusion
Taken together, our data show that AVP mediates brain tissue damage after cerebral ischemia by activation of V1 receptors; however, it remains to be clarified if AVP has a direct effect on cell survival or if it adds damage to the postischemic brain by initiating brain edema formation.
Regardless of the underlying mechanisms, V1 receptor antagonists might be potent drugs for reducting brain damage after cerebral ischemia followed by reperfusion, thereby reducing the deleterious sequels of reperfusion injury, for example, brain edema formation in stroke patients receiving thrombolysis.