
Abstract
Aquaporin 4 (AQP4) is a water channel involved in water movements across the cell membrane and is spatially organized on the cell surface in orthogonal array particles (OAPs). Its role in edema formation or resolution after stroke onset has been studied mainly at late time points. We have shown recently that its expression is rapidly induced after ischemia coinciding in time with an early swelling of the ischemic hemisphere. There are two isoforms of AQP4: AQP4-M1 and AQP4-M23. The ratio of these isoforms influences the size of the OAPs but the functional impact is not known. The role of the early induction of AQP4 is not yet known. Thrombin preconditioning in mice provides a useful model to study endogenous protective mechanisms. Using this model, we provide evidence for the first time that the early induction of AQP4 may contribute to limit the formation of edema and that the AQP4-M1 isoform is predominantly induced in the ischemic tissue at this time point. Although it prevents edema formation, the early induction of the AQP4 expression does not prevent the blood—brain barrier disruption, suggesting an effect limited to the prevention of edema formation possibly by removing of water from the tissue.
Introduction
Brain edema is a serious complication of several brain diseases such as stroke and significantly affects the long-term prognosis (Klatzo, 1985). The discovery of specific water channel molecules (aquaporins, AQPs) in the brain provided new insights into water routes through the plasma membrane (Verkman et al, 2006) and opened avenues into the research of novel pharmacologic tools to treat edema formation. On the basis of experiments conducted with AQP4-knockout (KO) mice, it was concluded that the role of the AQP4 is dual in edema formation and resolution depending on the pathologic model studied (Badaut et al, 2007b; Tait et al, 2008). In a model of transient ischemia, the level of expression of AQP4 is significantly increased on astrocyte endfeet in the ischemic hemisphere, 1 h after stroke onset coinciding with an early cerebral swelling (de Castro Ribeiro et al, 2006b). The question remains whether this increased expression contributes to edema formation, or is aimed at reducing it. Moreover, several studies have shown a close association between the formation of the blood—brain barrier (BBB) and an increase in AQP4 expression during development (Badaut et al, 2002; Nicchia et al, 2004). Orthogonal array particles (OAPs) are visualized by freeze-fracture electron microscopy at the astrocyte endfeet in contact with the vessels (Wolburg, 1995). These OAPs are constituted by AQP4 proteins (Rash et al, 2004) and their appearance during development is associated with the polarization of astrocytes and the development of the BBB (Wolburg, 1995). In disorders where the BBB is disrupted such as in global cerebral ischemia (Suzuki et al, 1984) as well as in tumors (Warth et al, 2004) the density of the OAPs is reduced.
Two splice variants of AQP4 have been described corresponding to two isoforms, AQP4-M1 and AQP4-M23, both of which are present in OAPs (Furman et al, 2003). Little is known about the physiological role of each isoform in astrocytes. Interestingly, the ratio of the two isoforms in M1 and M23 expressing CHO cells affects the size of these OAPs in that an increase in the level of AQP4-M1 reduces the size of the OAPs (Furman et al, 2003). Recently, it was shown in cultured astrocytes that the formation of OAPs and the ratio of each isoform are affected by one of the isoforms of agrin, a protein expressed in the basal lamina (Noell et al, 2007). To date, the expression of AQP4 isoforms in transient cerebral ischemia has never been evaluated. Modifications of the level of expression of AQP4-M1 and AQP4-M23 may affect water movements through astrocytes or the properties of the BBB.
The serine protease thrombin has been shown to be involved in brain injury such as ischemia (de Castro Ribeiro et al, 2006a), intracerebral hemorrhage, and traumatic brain injury (Xi et al, 2003). High doses of the serine protease thrombin are known to induce swelling and death of cells in vitro and thrombin is a key player in cerebral edema formation after intracerebral hemorrhage (Xi et al, 2003). In contrast, a low dose of thrombin injected into the brain tissue, 24 h or 7 days before a severe injury, such as a stroke or subarachnoid hemorrhage, induces a protection of the nervous tissue known as thrombin preconditioning (TPC; Granziera et al, 2007; Henrich-Noack et al, 2006; Xi et al, 1999). Therefore, as thrombin can induce edema formation and as low doses of thrombin are able to prevent edema formation, TPC is an appropriate model to study the role of AQP4 in early edema formation after stroke.
A model of TPC by injection of a low dose of thrombin intracerebroventricularly (i.c.v.), 24 h before a transient middle cerebral artery occlusion (MCAo) in mice, has been previously established in our lab (Granziera et al, 2007). Using this model, we provide evidence for the first time that the early induction of AQP4 may contribute to limit edema formation but does not prevent early BBB disruption. We also show for the first time that AQP4 isoforms are differentially regulated as the AQP4-M1 isoform is more abundant in the ischemic tissue of both TPC and control mice at this early time point.
Materials and methods
Animal Procedures for TPC and MCAo
All animal experiments were conducted in accordance with the guidelines of the cantonal veterinary service. The animals were maintained in standard laboratory conditions with a 12/12 h light—dark cycle (lights on at 0700 hours). Food and water were available ad libitum. TPC and MCAo were performed as previously described (Granziera et al, 2007). Briefly, male ICR-CD1 mice (weight 26.2 ± 2g, n = 24) were anesthetized with halothane (2.5% for induction and 1.5% during surgery) and 2 µL of 0.9% NaCl solution containing 0.01U of thrombin (Sigma, Switzerland) or vehicle solution were injected i.c.v. (0.9 mm laterally, 0.1 mm posteriorly, 3.1 mm deep from the bregma) using a Hamilton syringe 24 h before stroke (Granziera et al, 2007). MCAo was induced by introducing a silicone-coated 8-0 filament from the common carotid artery into the internal carotid artery and advancing it (regional cerebral blood flow 14%±1% of baseline). The filament was removed after 30 mins to allow reperfusion (regional cerebral blood flow 98%±8%) measured by laser-Doppler flowmetry with a flexible probe fixed on the skull (1 mm posteriorly and 6 mm laterally from the bregma). Rectal temperature was controlled and maintained at 36.5°C with a temperature control unit and a heating pad during the anesthesia period (36.6°C±0.1°C). Sham animals have received i.c.v 0.01U of thrombin 24 h before being killed.
Some of the animals were killed 1 h (n = 15), 24 h (n = 12), and 48 h (n = 12) after MCAo for immunohistochemistry and histology. Mice were deeply anesthetized with an overdose of pentobarbital and perfused transcardially with 100mL of paraformaldehyde 4% in phosphate-buffered saline (PBS)0.1mol/L, pH 7.4 at 4°C. Fixed brains were removed from the skull and immersed overnight in fixative solution. Brains were then stored in PBS containing 30% sucrose for 48 h at 4°C for cryoprotection. Serial coronal sections (40 µm) were then cut on a freezing microtome (Leica, Glattbrugg, Switzerland).
The other animals (n = 27) were killed by an overdose of pentobarbital for Western blot analysis. Some of the mouse brains were rapidly removed from the skull and frozen in liquid nitrogen vapor. Others were freshly dissected for Western blot studies.
Histology and Brain Swelling Measurement
Brain swelling was quantitated on adjacent 40-µm cresyl violet-stained coronal sections at the level of the anterior commissure. Histologic sections were digitalized using optical microscopy coupled with a CCD camera (Wild-Leica, Glattbrugg, Switzerland) and analyzed with Morpho-Expert (Explora-Nova, France) for sham (n = 3), 1 h (n = 6), 48 h (n = 6) for each group, the TPC-treated and nontreated mice. The cerebral hemisphere swelling was analyzed by measuring the percentage of hemispheric enlargement ((ischemic hemisphere area)–(contralateral hemisphere area)/(contralateral hemisphere area)×100) (de Castro Ribeiro et al, 2006b).
The lesion area (outlined in Figure 1A) characterized by reduced labeling on cresyl violet-stained sections was measured with Morpho-Expert (Explora-Nova, La Rochelle, France) in the TPC-treated (n = 6) and vehicle-treated mice (n = 6) at 48. Lesion area was expressed as a percentage of the ipsilateral hemisphere surface: (lesion area)/(ipsilateral hemisphere) ×100.
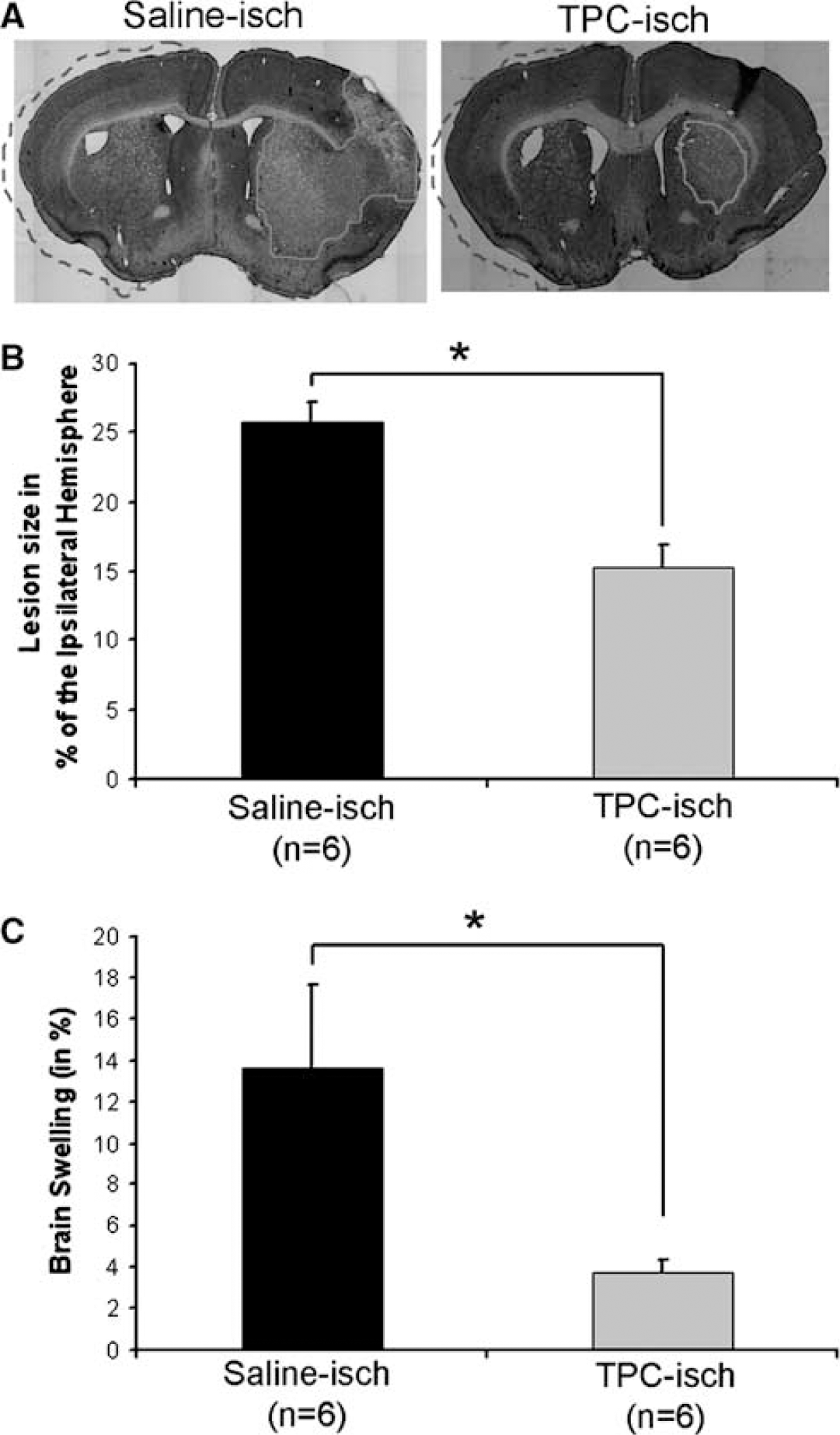
Infarct size and brain swelling. (
Water Content, Wet Weight/Dry Weight Ratio
The water content of the brain was measured to evaluate the formation of edema after ischemia in saline- and TPC-treated mice. At 1 h after stroke onset, animals were killed by an overdose of pentobarbital. The brain was rapidly removed from the skull and the olfactory bulbs and brain stem with the cerebellum were eliminated before the wet weight (Weightwet) measurements, which were performed less than 2 mins after the decapitation. The brain was split down the midline into ischemic and contralateral hemispheres, and the wet weight of each of them was measured. The tissue was then dried at 100°C for 24 h, and its dry weight (Weightdry) was measured. Brain water content (as a percentage) was determined by the following calculation:
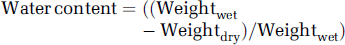
Water content was also determined on sham animals (with vehicle or thrombin injection) where there was no difference between hemispheres (data not shown).
IgG staining for BBB evaluation: Free-floating sections were mounted on slides for immunoglobulin G (IgG) immunostaining. The sections were incubated with biotin-conjugated affinity-purified donkey anti-mouse IgG coupled with infrared dye 800nm (Molecular Probes, Eugene, OR, USA) diluted (1:200) in PBS containing 0.1% Triton X-100 and 1% bovine serum albumin for 4 h at room temperature. After washing, sections were scanned on an infrared scanner (Odyssey System; LI-COR Biosciences, Germany) to quantify fluorescence in two boxes per region of interest on two adjacent slices. The IR was quantified and fluorescence was converted into averaged integrated intensities from the ipsilateral and contralateral hemispheres (Wang et al, 2007).
AQP Immunohistochemistry
Commercially available, affinity-purified rabbit polyclonal antibodies were used for AQP4 immunolabeling (Chemicon International, Temecula, CA, USA) (de Castro Ribeiro et al, 2006b). Immunostaining was performed in PBS containing 0.1% Triton X-100 and 0.3% bovine serum albumin. After each incubation, sections were rinsed in PBS three times for 10 mins. For immunolabeling, the sections were first incubated overnight at 4°C with anti-AQP4 (1:100). After washing, floating sections were incubated for 2 h at room temperature with an Alexa Fluor 568 nm-coupled secondary rabbit antibody (1:500; Molecular Probes-Invitrogen, Switzerland). Sections were mounted and coverslipped with antifading medium Vectashield containing 46-diamidino-2-phenyl indole (Vector; Vector Laboratories, Burlingame, CA, USA). Controls were performed by omitting primary antibody. All controls gave negative results with no detectable labeling. Depletion of the antibody by an excess of the specific peptide (from Chemicon International) was also performed and gave negative results as previously observed (Badaut et al, 2004; de Castro Ribeiro et al, 2006b).
Immunohistochemistry Imaging and Quantification of the Immunolabeling
Immunofluorescent preparations were examined using confocal laser scanning microscopy (Zeiss, Feldbach, Switzerland) and epifluorescence microscopy (Olympus BX41; Olympus, Switzerland). The image from the fluorescent staining under the confocal laser scanning microscope was processed using 568nm (Alexa Fluor) using the same parameter of acquisition to perform after an analysis of fluorescence.
The AQP4 immunoreactivity was quantified in three different fields (422 ×338 µm) obtained in the ischemic and contralateral hemispheres in saline and TPC mice. Optical density was measured using Morpho-Expert (image analysis software from Explora-Nova). The analysis was performed on 1 µm thick confocal laser scanning microscope images at three different levels in the slices and in three different areas inside the ipsilateral and contralateral striata, which was not different from AQP4 staining in sham animals. The optical density was measured by a masked experimenter (JB) on nontreated images acquired using the confocal laser scanning microscope (Zeiss). Then, the ratio of the mean optical density between the ischemic and contralateral hemispheres was calculated.
RT-PCR Experiments
Frozen sections were cut using a cryostat (Leica) at −20°C and the ipsilateral and contralateral striata were dissected and collected in separate Eppendorf tubes. Total RNA from ipsilateral and controlateral striata of eight cryosections was extracted with Trizol (Invitrogen, Switzerland) and 1 µg total RNA was used for first-strand synthesis using MulVRTM0253S (New England Biolabs Inc., Ipswich, MA, USA) as described by the supplier. Classical direct AQP4 and actin PCRs were performed with a HotStarTaq Polymerase Kit (Qiagen GmbH, Switzerland) with 5pmol of primers per reaction (mouse AQP4 forward: 5′-CATCATTGCACAGTGCCTGGG-3′; and reverse: 5′-CTTCTGCCTTCAGTGCTGTCC-3′, amplicon size: 586 bp; mouse β-actin forward: 5′-GG GCTGTATTCCCCTCCATCGTGG-3′ and reverse: 5′-C AGTGGCCATCTCCTGCTCGAAG-3′). A seminested PCR was used to distinguish between the three isoforms. Each isoform was first amplified with its specific forward primer: AQP4-M23, 5′-GGAGACTTTAGAAGCAGTC-3′; AQP4-M1, 5′-GAGGGAAGGATGAGTGACG-3′; and common reverse 5′-CTTCTGCCTTCAGTGCTGTCC-3′ primers (see Supplementary Figures 1 and 2 for the specificity of the primers). Then, the seminested PCR was performed with the same couple of primers described above for the amplification of AQP4. Each reaction was performed separately with the HotStarTaq mix (Qiagen GmbH, Switzerland) as described by the supplier. All PCRs were performed in the Personal Thermocycler (Biometra GmbH, Germany) with one first activation step (15 mins, 95°C), followed by 35 cycles of denaturation (94°C, 30 secs), annealing (56°C, 45 secs) and elongation (72°C, 90 secs), and a final extension step (72°C, 90 secs). The infrared fluorescent nucleic acid dye Syto60 (Molecular Probes, Invitrogen, Switzerland) was incorporated in the loading buffer (1:6,000) to detect the amplicons with the Odyssey (LI-COR Biosciences, Germany) infrared imaging system after migration on a 0.8% agarose gel.
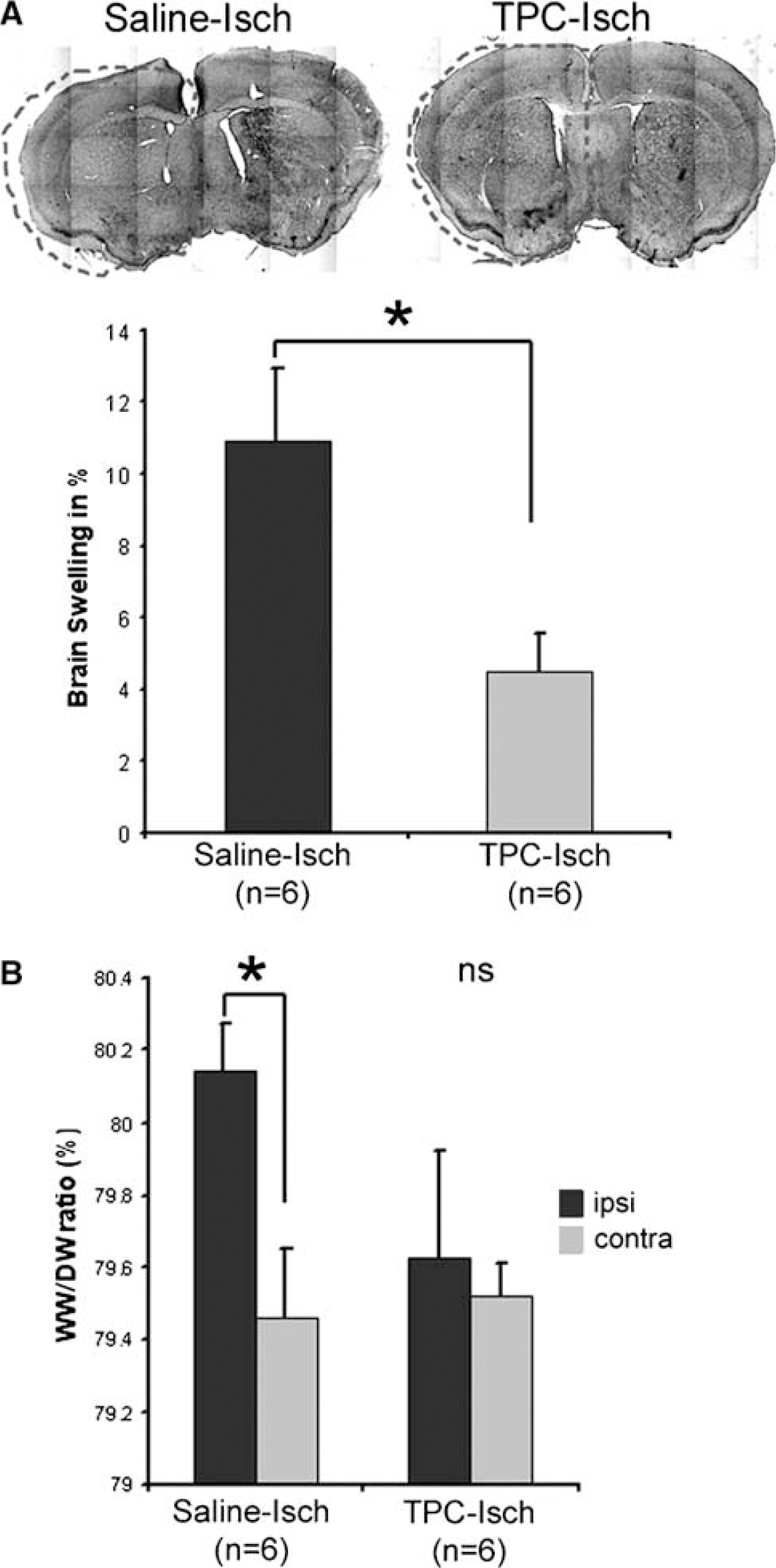
Analysis of the brain swelling and the water content 1 h after stroke onset in TPC and control saline mice. (
Western Blot
Immunoblotting was used to determine the changes in AQP4 expression with two protocols for tissue preparation.
For quantification of total AQP4, 1 µg of protein was prepared from the ischemic-ipsilateral (ipsi) and the contralateral hemispheres (ctl) as described above for RT-PCR experiments. Dissected slices are prepared in the buffer described below.
For quantification of each isoform of AQP4, proteins were directly prepared from freshly dissected ipsilateral and contralateral hemispheres in a buffer containing 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.6), 42 mmol/L KCL, 5 mmol/L MgCl2, 1% SDS, 1 mmol/L phenylmethylsulflonyfluoride, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1.5 mmol/L pepstatin, 2 mmol/L leupeptin, 0.7 mmol/L aprotinin, and sonicated for 30 secs.
Then, proteins were subjected to SDS—polyacrylamide gel electrophoresis on a 12% gel (Nupage; Invitrogen, Switzerland) for the quantification of total AQP4. In the study of each isoform of AQP4, samples were subjected to SDS—polyacrylamide gel electrophoresis electrophoresis with a gradient gel 4% to 12% (Nupage; Invitrogen, Switzerland). In both experiments, proteins were then transferred to a polyvinylidene fluoride membrane (PerkinElmer, Germany). The blot was incubated with a polyclonal antibody against AQP4 (1:1,000; Chemicon International) and a monoclonal antibody against actin (1:25,000; Sigma) in Odyssey blocking buffer (LI-COR Biosciences, Germany) overnight at 4°C. After washing, the filter was incubated with two fluorescence-coupled secondary antibodies (1:10,000, anti-rabbit Alexa Fluor 680 nm; Molecular Probes and anti-mouse IR-Dye 800-nm; Roche, Germany) for 2 h at room temperature. After washing, fluorescence was scanned with an infrared scanner (Odyssey System; LI-COR Biosciences, Germany). The advantage of the infrared detection system for quantification is the linearity, and the number of photons emitted by the blot was recorded and used for quantification (Badaut et al, 2007a). Quantification was blindly done by two experimenters (JB and BT) on total AQP4 (both isoforms). In the second series of experiments, the level of expression of each AQP4 isoform was quantified in the same way. The peak of each band was used to quantify the level of expression of AQP4. Isoforms were selected as a function of their relative molecular weights 34 kDa for AQP4-M1 and 30 kDa for AQP4-M23.
Statistical Analysis
All the data were presented as mean±s.e.m. and the statistical analysis was performed using GraphPad InStat version 3.05 (GraphPad Software, San Diego, CA, USA). The Kolmogorov—Smirnov test was first performed to assess the Gaussian distribution of the data. Data that passed the test were then analyzed with an unpaired t-test (lesion size, brain swelling, AQP4-IR) or a paired t-test (water content, IgG staining, AQP4 isoform comparisons between hemispheres). For the other data nonparametric Wilcoxon and Kruskal—Wallis tests (Western blot analysis) were used.
Results
Validation of the TPC Model
We have recently shown that TPC 24 h before 30 mins MCAo reduces the infarct volume after ischemia and improves the behavioral outcome at 72 h (Granziera et al, 2007). To confirm theses findings, we tested whether TPC reduced the lesion size 48 h after ischemia. Injection of a low dose of thrombin (0.01 U) i.c.v 24 h before stroke triggered endogenous mechanisms of protection leading to a decrease in lesion extent evaluated at 48 h (Figure 1A and 1B) thereby confirming our previous observation at 72 h that TPC induces a powerful neuroprotection (Granziera et al, 2007). We also show that TPC attenuates swelling of the ischemic hemisphere 48h after stroke (Figure 1C). We then went on to study the effect of TPC on changes occurring early after ischemia.
Effect of TPC on Edema Formation and the BBB 1 h After Stroke Onset
We have shown previously that AQP4 expression on astrocyte endfeet is increased 1 h after ischemia onset in parallel with an early swelling of the ischemic hemisphere (de Castro Ribeiro et al, 2006b). Our next step was to evaluate the effect of TPC on edema development 1 h after stroke onset. Here, we show that TPC before transient MCAo in mice strongly reduces brain swelling 1 h after stroke onset (4.5%±2%), compared to saline-treated mice (11%±5%) (Figure 2A). Brain swelling is an indicator of brain edema formation (de Castro Ribeiro et al, 2006b; Gerriets et al, 2004). This result was confirmed by measuring the water content using the wet weight/dry weight ratio method, which showed a significant increase in the ratio between the ischemic hemisphere of the saline-treated mice and the contralateral hemisphere, but not in TPC-treated mice at 1 h after stroke onset (Figure 2B). The prevention of edema formation correlates with a significant decrease of the lesion size by 42% 48 h after stroke onset (Figure 1A).
Cerebral edema after ischemia is partly cytotoxic and partly vasogenic (Gartshore et al, 1997; Loubinoux et al, 1997). Cytotoxic edema is characterized by an intracellular water accumulation without BBB disruption. In contrast, vasogenic edema appears after physical disruption of the BBB leading to a diffusion of proteins from the blood to the tissue followed by water (for review, see Unterberg et al, 2004). To analyze the mechanisms underlying early edema formation we decided to study the permeability of the BBB by examining the extravasation of circulating IgGs into the brain parenchyma. In our ischemia model, a significant increase in the IgG staining was observed in the ischemic hemisphere (88% increase compared to the contralateral hemisphere; Figure 3) in saline-treated animals 1 h after ischemia onset, showing an early disruption of the BBB in our mild model of ischemia, 30 mins after reperfusion in accordance with the recent observation in the rat (Strbian et al, 2008). An increase in IgG staining was observed also in TPC-treated animals (with 41% increase compared to the contralateral hemisphere; Figure 3), showing that TPC does not prevent BBB disruption. The molecular mechanisms underlying the decrease in edema in the TPC model are not yet understood and are possibly complex. In the following part, we have focused on AQP4, a molecule likely to be involved in this process.
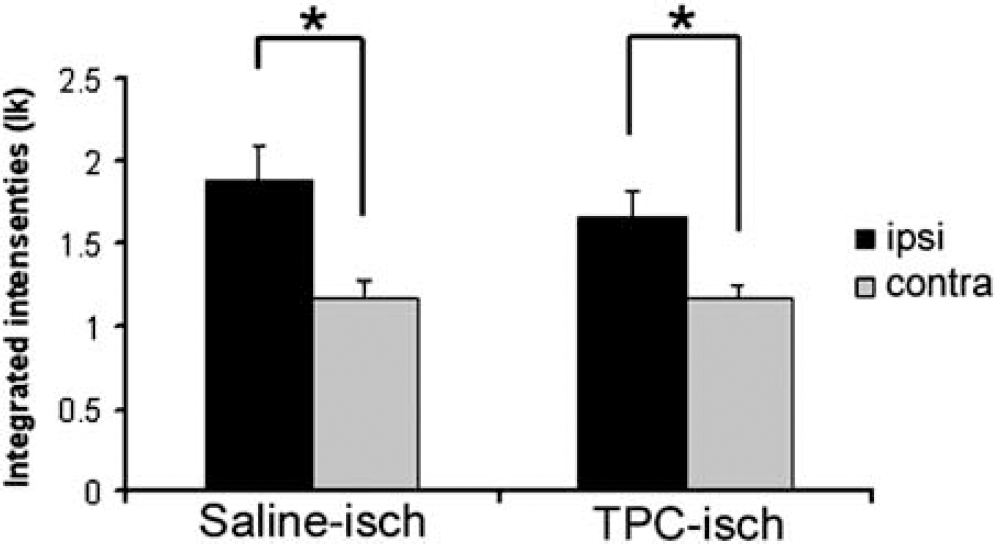
IgG staining for assessment of BBB disruption. In saline- and TPC-treated mice, IgG staining is significantly increased in the ischemic hemisphere (paired t-test, P<0.05) 1 h after stroke onset. This difference in staining between the ischemic (ipsi) and control (contra) hemispheres shows that there is a disruption of the BBB.
Effects of TPC on AQP4 Expression 1 h After Stroke Onset
AQP4 is a water channel and during development its expression correlates with the maturation of the BBB.
We have shown previously that AQP4 expression on astrocyte endfeet is increased 1 h after ischemia onset, in parallel to an early swelling of the ischemic hemisphere (de Castro Ribeiro et al, 2006b). We have now shown that TPC prevents early edema without preventing the opening of the BBB. We studied the effect of TPC on AQP4 expression 1 h after stroke by comparing the level of expression of AQP4 1 h after ischemia onset in saline-treated and thrombin-preconditioned mice.
In the group of mice used to measure the brain swelling, the level of AQP4 expression was measured by immunohistochemistry (Figure 4). We confirm that the expression of AQP4 is significantly increased on astrocyte endfeet in contact with the blood vessels in the ischemic territory 1 h after stroke in saline-treated mice (Figure 4A; de Castro Ribeiro et al, 2006b).
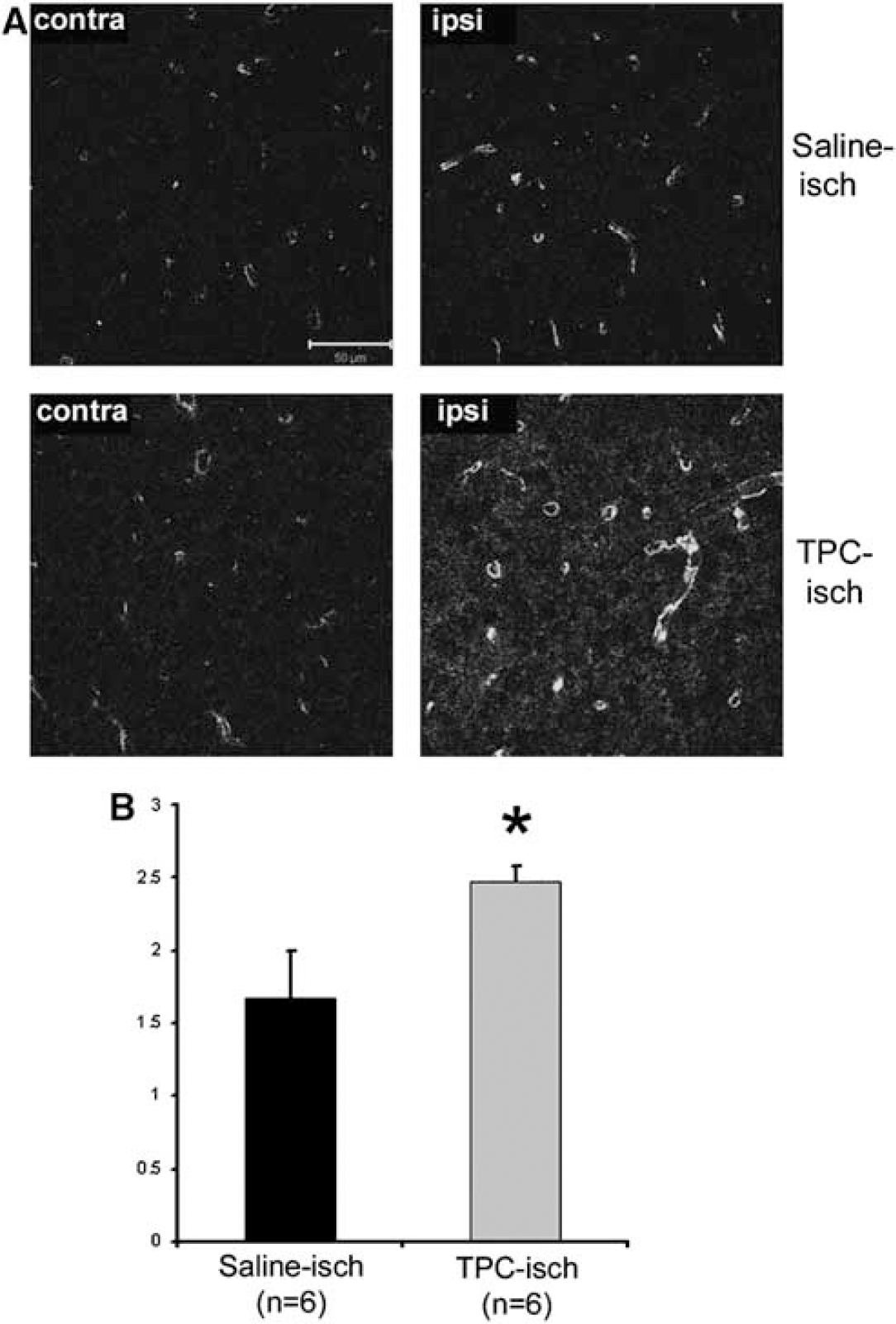
Illustration of the AQP4-ir in the contralateral hemisphere (contra) and in the future ischemic lesion (ipsi) at 1 h after stroke onset in saline- (Saline-isch) and TPC-treated mice (TPC-isch). (
Interestingly, the increase in AQP4 immunoreactivity (AQP4-ir) on astrocytes is significantly increased in TPC-treated mice (47%) compared to control mice (Figure 4B). The level of AQP4 was also investigated on Western blot (Figure 5A). The increased AQP4-ir in the ischemic hemisphere is confirmed by Western blot of control mice 1 h after stroke onset. The level of expression of AQP4 is significantly increased (37%) in the ischemic hemisphere compared to the control hemisphere. This result shows that the increased AQP4-ir is due to increased AQP4 protein levels. TPC also induced a significant increase in the level of AQP4 in the ischemic hemisphere (52% compared to the control hemisphere; Figure 5). The level of expression of AQP4 is higher in the ischemic hemisphere of TPC mice (0.81 ± 0.14 AU, n = 7) than in the ischemic hemisphere of saline-treated mice (0.47 ± 0.01 AU, n = 8, P = 0.048). Therefore, TPC induces a strong increase in the level of AQP4 in the ischemic hemisphere 1 h after stroke onset. No significant effect of thrombin injection on AQP4 expression is observed without ischemia in sham animals (data not shown).
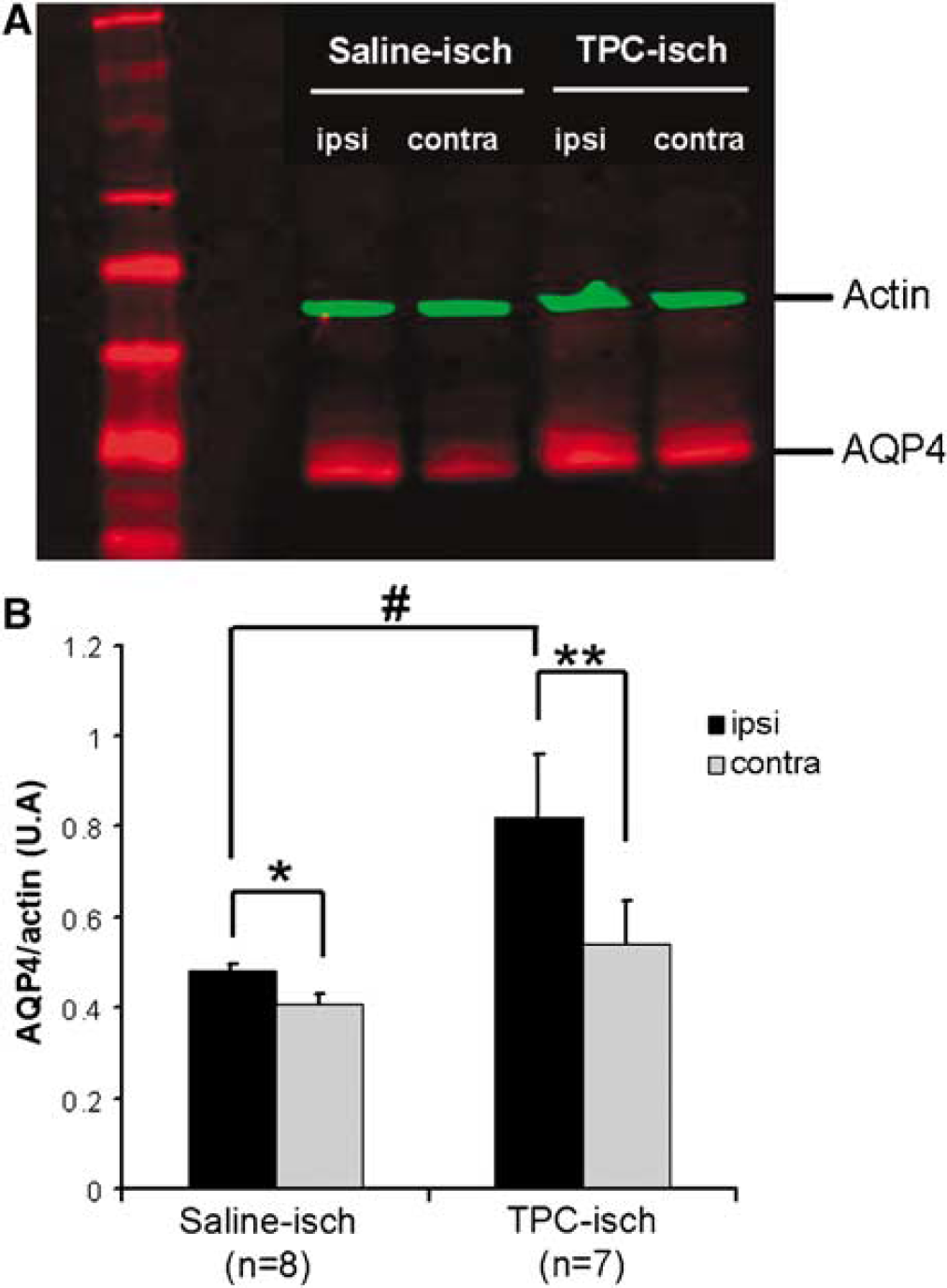
AQP4 expression quantified from Western blot experiments in saline- and TPC-treated mice before ischemia. (
As TPC prevents early edema formation in our model, it is clear that the significant increase in expression of the AQP4 water channel does not result in more water in the ischemic tissue. On the contrary, enhanced AQP4 expression prevents edema and the higher expression (in TPC) contributes to remove the water. The water can diffuse through this channel depending on the osmotic gradient. To date, all the mechanisms underlying this decrease in early edema formation are not known.
As mentioned in the introduction, two AQP4 isoforms have been described in astrocyte endfeet: AQP4-M1 and AQP4-M23. Here we provide a first study of the level of expression of each AQP4 isoform after ischemia. We show by Western blot that the commercial AQP4 antibody (Chemicon International) detects a double band of 30 and 34 kDa, corresponding in size to the M1 (34 kDa) and M23 (30 kDa) isoforms. By band intensity quantification (using specific Odyssey software; LI-COR Biosciences), we confirm an increased expression of AQP4 in the ischemic compared to the contralateral hemisphere (Figure 6).
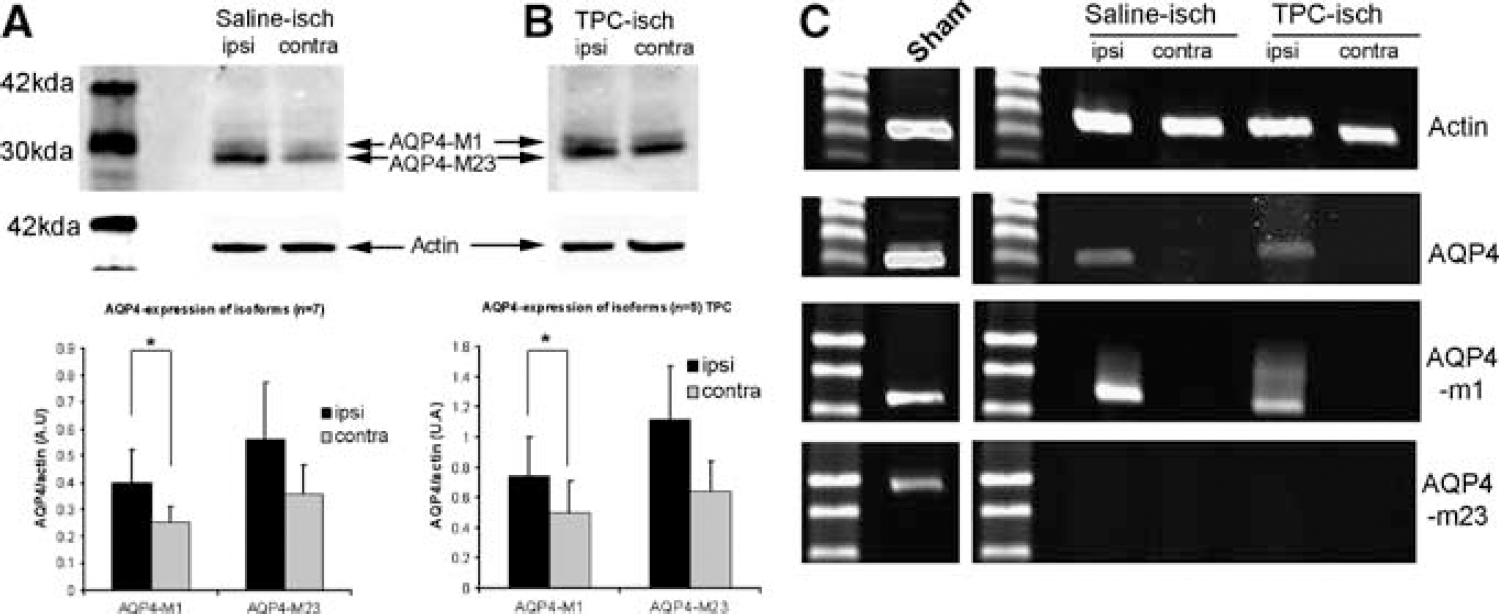
Expression of the AQP4 isoforms after stroke onset in saline- and TPC-treated mice. (
By comparing the relative intensity of the upper and lower bands, we show that AQP4-M1 is significantly increased in ischemic extracts compared to contralateral hemisphere extracts (Figure 6B). AQP4-M23 is also increased but without reaching statistical significance (Figure 6A). This larger amount of AQP4-M1 isoform could induce a small change in the ratio between AQP4-M23 and AQP4-M1 in the ischemic tissue. In TPC mice, the overall expression of AQP4 is higher than in saline-treated animals, with a significant increase of AQP4-M1 and again a nonsignificant increase of AQP4-M23 (Figure 6B). The modification of the ratio AQP4-M1 and AQP4-M23 after ischemia is similar in saline- and TPC-treated mice.
In a second set of experiments on adjacent slices, the level of expression of the mRNA was investigated using RT-PCR with three sets of specific primers (Supplementary Figures 1 and 2), recognizing either all AQP4 mRNAs, the M1 isoform or the M23 isoform (Figure 6A). In the brain of sham mice, AQP4 mRNA, the M1 isoform, and the M23 isoform are all detected (Figure 6C). After ischemia, AQP4 mRNA and the M1 isoform mRNA can be detected whereas the mRNA of the M23 isoform cannot be detected in the ischemic hemisphere, both in saline- and thrombin-treated mice (Figure 6C). The detection of M1 mRNA and not of M23 mRNA coincides with the higher level of AQP4-M1 protein seen on Western blots and indicates a potential change in the isoform ratio. Surprisingly, AQP4-mRNAs are not detected in the contralateral hemisphere. This difference between hemispheres for AQP-mRNAs is well correlated with Western blot results showing an induction of the AQP4 protein in the ischemic hemisphere 1 h after stroke onset.
Discussion
We have recently shown that AQP4 is induced on astrocyte endfeet (perivascular compartment) very early after cerebral ischemia, coinciding with an early swelling of the ischemic hemisphere (de Castro Ribeiro et al, 2006b), suggesting a role for AQP4 in edema development or in water clearance. In young rats, AQP4 expression was increased 24 h after stroke at the edge of the lesion where there was no significant modification of water content measured by magnetic resonance imaging (Badaut et al, 2007a), suggesting that increased AQP4 expression could limit or prevent the formation and extension of edema. The role of AQP4 in cerebral edema is dual and depends of the model used (Badaut et al, 2007b; Verkman et al, 2006). Indeed, the absence of AQP4 was shown to prevent the formation of edema in a permanent ischemia model in AQP4-KO mice (Manley et al, 2000). Similarly, edema formation is prevented in α-syntrophin-KO mice at 24 h after stroke (Amiry-Moghaddam et al, 2003). This decrease of brain swelling was correlated with the loss of the perivascular AQP4 domain in α-syntrophin-KO mice (Amiry-Moghaddam et al, 2003). These results suggest that perivascular AQP4 is important in edema formation. However, the absence of AQP4 in AQP4-KO mice also prevents water clearance in an experiment of intrastriatal infusion of a saline solution, showing that AQP4 is critical for water removal from tissue (Papadopoulos et al, 2004).
On the basis of our previous time course study (de Castro Ribeiro et al, 2006b), we have focused on the functional consequences of an early induction of the AQP4 expression. Using a preconditioning paradigm, we have studied the consequences of an early induction of the AQP4 expression, with the hypothesis that early upregulation of AQP4 expression is aimed at reducing edema formation. We and others have shown previously that exposure to a low dose of thrombin can protect the brain parenchyma against subsequent ischemia (Granziera et al, 2007; Masada et al, 2003; Xi et al, 1999). We chose to explore the effect of TPC both on early edema formation and AQP4 expression. We now show, both by immunohistochemistry and Western blotting, that in TPC mice the induction of AQP4 expression is stronger than in saline-treated mice, and correlates with a reduced edema formation 1 h after stroke onset (Figure 2). Intriguingly, these results suggest that the early induction of AQP4 on astrocyte endfeet prevents the development of edema. Furthermore, we also show that the upregulation of AQP4 does not prevent the early opening of the BBB, suggesting that BBB disruption is not the main mechanism of early edema formation or that TPC does not prevent edema formation but rather promotes water clearance from the tissue. However, TPC decreases the IgG labeling in the ischemic hemisphere at 48 h after stroke onset (see Supplementary Figures 1 and 2). Therefore, TPC has an effect on BBB permeability at this later time point.
Our results suggest that AQP4 upregulation on astrocyte endfeet facilitates water movement in the tissue and favors the removal of water from the ischemic hemisphere to reduce the edema formation. This hypothesis is supported by a recent publication showing an increase of AQP4 in ependymal cells in the border of the ventricles in a traumatic brain injury model (Guo et al, 2006). To date, the exact mechanism causing decreased edema formation is not yet fully understood. Osmotic gradients can have an important function and recently high AQP4 expression was observed in hypersaline treatment after stroke correlating with decreased edema formation at 48 h (Chen et al, 2007). This role for AQP4 in water removal from brain tissue is also supported by previous observations obtained during the resolution of the vasogenic edema in a model of freeze injury (Papadopoulos et al, 2004). In our stroke model, this early induction in the absence of TPC may be insufficient to sustain water removal required after stroke onset and later this induction could even become deleterious for the tissue as suggested by the study performed in AQP4-KO mice showing that the presence of AQP4 contributes to the cytotoxic edema formation at 24 h (Manley et al, 2000). Because we looked at the expression of AQP4 at an early time point (1 h after stroke onset), we believe that the higher early expression of perivascular AQP4 contributes to prevent edema formation by helping water clearance. We also show a reduced swelling of the ischemic hemisphere after 48 h, but this is partly because of a reduced infarct volume and we cannot conclude that preconditioning has a selective effect on edema development at later time points such as 48 h.
Another surprising result is that the increase of AQP4-ir on perivascular astrocyte endfeet is the result of an increased level of protein (Figure 4) with modifications also at the mRNA level (Figure 5), showing that AQP4 expression can be rapidly regulated. It was shown previously that AQP4-ir increased in the rat brain after systemic hyponatremia (Vajda et al, 2000). Because there was no increase in protein levels in Western blots, the authors proposed that the phosphorylation of AQP4 facilitated the detection of the protein in the cell membrane by immunohistochemistry (Vajda et al, 2000). Several putative PKA, PKC, CKII, and CaMKII phosphorylation sites have been described which could allow a rapid regulation of AQP4 (Gunnarson et al, 2004). Here we show that the level of AQP4 protein is increased at early times after reperfusion (30 mins). It was recently shown that the thyroid transcription factor-1 is induced in response to hypertonicity and activates the expression of AQP1 (Kim et al, 2007). Such a mechanism could also trigger the increase in AQP4 expression at an early time point after stroke. MicroRNAs directed against AQP4 have been described (GenBank) and would be a possible mechanism of rapid mRNA and protein regulation (Filipowicz et al, 2008).
An exciting issue is the cellular localization of AQP4, its organization into OAPs, in which both the two AQP4 isoforms have a role. AQP4-M23 and AQP4-M1 contribute to the formation of OAPs observed in astrocyte endfeet in contact with the basal lamina of brain vessels (Rash et al, 2004). Experiments in oocytes show that AQP4-M1 appears to disorganize the OAPs (Furman et al, 2003). The ratio AQP4-M1 and AQP4-M23 was not yet investigated under pathologic conditions in vivo. In this study, we show that AQP4 mRNA is present both in control animals and after ischemia. After ischemia, only the AQP4-M1 mRNA and total AQP4 mRNA can be detected, whereas M23 mRNA is not detected. Intriguingly, none of these mRNAs are detected in the contralateral hemisphere, whereas the house-keeping gene actin is unchanged. The presence of only AQP-M1 mRNA early after ischemia could favor a shift in the balance between M1 and M23 toward M1, which is known to favor small-sized OAPs. Our Western blots show a double band at approximately 30 to 34 kDa, which we quantitated individually with the number of photons emitted by infrared fluorescence (Odyssey System; LI-COR Biosciences), using the peak intensity of each band. Both isoforms are increased after ischemia, but the increase is only statistically significant in the case of M1, which also supports a trend toward a shift in favor of M1. To date, the role of each AQP4 isoform has not been studied in normal and pathologic brains. We assume that a predominance of AQP4-M1 in the ischemic hemisphere could contribute to the disorganization of the OAPs as is the case in oocytes. Early disorganization of OAPs on the astrocyte endfeet has been previously observed in freeze fracture after global cerebral ischemia and was shown to precede astrocyte swelling (Suzuki et al, 1984). Recently, a new role has been proposed for AQP4 in cell adhesion (Hiroaki et al, 2006). AQP0 has been well described in the epithelial cells of the lens, is present in OAPs, and participates in the linkage between epithelial cells but does not facilitate water flux (Gonen et al, 2004). In this respect, the presence of AQP4 in the endfoot membrane depends on the presence of proteins in the basal lamina such as agrin, α-dystroglycan and laminin (Guadagno and Moukhles, 2004; Warth et al, 2004) and on syntrophin and dystrophin protein complexes (Amiry-Moghaddam et al, 2003; Neely et al, 2001). The connection of AQP4 to proteins in the basal lamina may explain the ability of astrocytes to maintain the integrity of the BBB, a possible role for AQP4. Modification of the ratio of AQP4-M23 to AQP4-M1 could affect the connection with the basal lamina and possibly contribute to the early BBB disruption. This hypothesis is supported by the fact that in TPC animals BBB disruption is not protected and the modification of the ratio AQP4-M23 and AQP4-M1 is similarly affected in TPC and control mice (Figure 6B). Functional experiments in oocytes show no significant difference in water conductance between isoforms in vitro (Furman et al, 2003). This result is in contrast with recent papers showing that the increase in AQP4-M23 is correlated with an increase in the permeability of water in astrocytes and the LLC-PK1 cell line (Noell et al, 2007; Silberstein et al, 2004). A direct effect of the modification of the ratio AQP4-M1 and AQP4-M23 on water permeability is not yet directly investigated in vivo. In the TPC model, the strong increase in AQP4 expression is correlated with a reduction in edema development and less water in the tissue, suggesting an increased water diffusibility to remove excess liquid in brain tissue. The exact mechanism is not yet fully understood as the direction of water through the channel depends on the osmotic pressure (Figure 7).
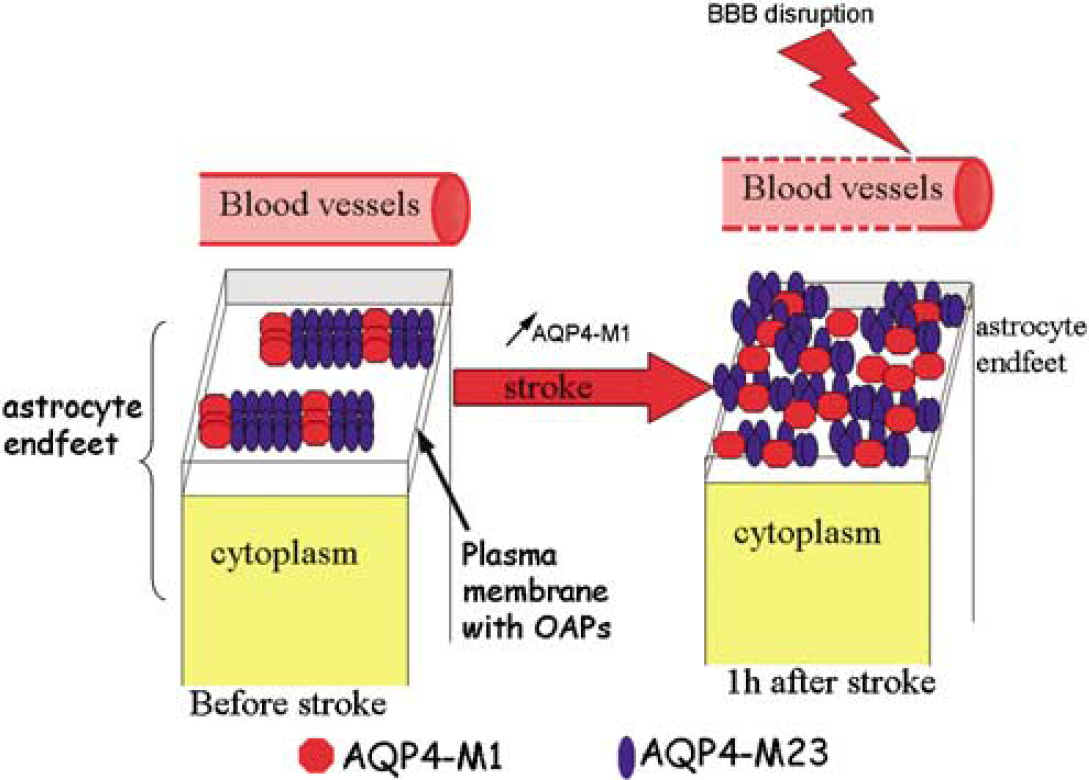
Level of expression is increased at early time point (1 h) after stroke onset in the ischemic hemisphere. The isoform AQP4-M1 (red circles) is highly induced than AQP4-M23 (blue circles) suggesting that a disorganization of the OAPs in astrocyte endfeet in contact with vessels. In association with modification of the AQP4 isoforms ratio, BBB disruption was observed at this time point. Experiments with a model of preconditioning show that the early induction of AQP4 is a protective mechanism against edema formation.
To summarize, we have observed a significant early induction of the AQP4 protein correlating with an absence of edema formation 1 h after ischemia onset with TPC. Increased AQP4 expression has been shown to increase water permeability in oocytes, and it is very likely that increased AQP4 expression on astrocyte endfeet contributes to attenuate cytotoxic edema after cerebral ischemia. Our data suggest a shift toward the M1 isoform, known to contribute to OAP disorganization and modify cellular adhesion properties of astrocyte endfeet (Figure 7). This intriguing result requires further investigation of the AQP4 isoforms toward an understanding of the specific roles of AQP4.
Footnotes
Acknowledgements
We are grateful to Dr Karine Wiegler and Dr Delphine Michel for their help with the surgery, and to Professor Luca Regli (Neurosurgery, Department, UMC, Utrecht, Netherland) for his support.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.