
Abstract
Pretreatment with a low intracerebral dose of thrombin reduces brain edema after hemorrhagic and thromboembolic stroke. We have termed this phenomena thrombin preconditioning (TPC) or thrombin-induced brain tolerance. Red blood cell lysis and iron overload contribute to delayed edema formation after intracerebral hemorrhage. The present study examined whether TPC can attenuate the brain edema induced by lysed red blood cells or iron. It also examined whether TPC is associated with increasing hypoxia inducible factor-1α (HIF-1α) levels and alterations in two HIF-1α target genes, transferrin (Tf) and transferrin receptor (TfR), within the brain. Brain edema was measured by wet/dry weight method. HIF-1α, Tf, and TfR were measured by Western blot analysis and immunohistochemistry. We found that TPC reduces the edema induced by infusion of lysed red blood cells and iron. Thrombin increases HIF-1α levels through p44/42 mitogen activated protein kinases pathway. Thrombin also increases Tf and TfR levels in the brain. These results indicate that HIF-1α and its target genes may be involved in thrombin-induced brain tolerance.
Keywords
Intracerebral hemorrhage (ICH) is a common and often fatal stroke subtype (Kase and Caplan, 1994). Edema after ICH contributes to these outcomes causing both acute herniation-related deaths and long-term neurologic deficits (Broderick et al., 1994; Ropper and King, 1984). A number of mechanisms are involved in edema formation after intracerebral hemorrhage (Xi et al., 2002). Thrombin production, erythrocyte lysis, and hemoglobin toxicity result in perihematomal brain edema formation (Lee et al., 1996; Xi et al., 1998a,1998b). Hemoglobin-induced brain injury is, at least in part, iron-mediated (Huang et al., 2002).
Although high concentrations of thrombin are deleterious to the brain, low concentrations of thrombin are neuroprotective in vitro and in vivo. We previously found that pretreatment with a low dose of thrombin attenuates the brain edema induced by thrombin or intracerebral hemorrhage and significantly reduces infarct size and brain edema in a rat middle cerebral artery occlusion model (Masada et al., 2000; Xi et al., 1999, 2000). We have termed this phenomena thrombin preconditioning (TPC). The precise mechanisms of thrombin-induced brain tolerance to hemorrhagic and ischemic stroke are not known, although activation of thrombin receptors and stimulation of p44/42 mitogen activated protein kinases (MAPKs) phosphorylation appear to be important (Xi et al., 2003).
Hypoxia inducible factor-1α (HIF-1α) is a transcriptional factor that regulates the expression of a number of genes, including transferring (Tf), transferrin receptor (TfR), and heme oxygenase-1 (HO-1) (Lee et al., 1997b; Lok and Ponka, 1999; Rolfs et al., 1997; Semenza, 2001; Tacchini et al., 1999). Thrombin can increase brain HIF-1α levels, and HIF-1α is related to brain tolerance induced by hypoxic preconditioning (Bergeron et al., 2000; Bernaudin et al., 2002; Jiang et al., 2002b; Jones and Bergeron, 2001). After ICH, brain iron concentrations can reach very high levels. HO-1, one of the HIF-1α target genes, is a key enzyme in hemoglobin degradation. Tf and TfR play an important role in iron transport across the blood-brain barrier and neuronal membrane (Connor et al., 2001). Modification of Tf and TfR levels in the brain might change brain iron homeostasis after ICH.
The present study, therefore, examined whether TPC can attenuate the brain edema induced by lysed red blood cells (RBCs) and iron. It also examined whether TPC upregulates the brain levels of HIF-1α and its target genes, Tf and TfR. Finally, it examined the role of MAPKs in HIF-1α upregulation after TPC because there is evidence that MAPKs may regulate HIF-1α in vascular smooth muscle cells (Richard et al., 2000).
MATERIALS AND METHODS
Animal preparation
The protocols for these animal studies were approved by the University of Michigan Committee on the Use and Care of Animals. Adult male Sprague-Dawley rats (275 to 325 g, n = 85; Charles River Laboratories, Portage, MI, U.S.A.) were anesthetized with pentobarbital (40 mg/kg, intraperitoneally). Aseptic precautions were used in all procedures. A polyethylene catheter (PE-50) was then inserted into the right femoral artery to monitor arterial blood pressure and to obtain blood samples for analysis of blood gases, blood pH, hematocrit, and blood glucose concentration. Body temperature was maintained at 37.5°C by using a feedback-controlled heating pad.
Intracerebral infusion
Before intracerebral infusion, the rats were positioned in a stereotactic frame (Kopf Instrument, Tujunga, CA, U.S.A.), the scalp was incised along the sagittal midline using a sterile technique. A cranial burr hole (1 mm) was drilled near the right coronal suture 4.0 mm lateral to the midline, and a 26-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates: 0.2 mm anterior, 5.5 mm ventral, and 4.0 mm lateral to the bregma). Thrombin, saline, iron, or lysed red blood cells were withdrawn into a Hamilton syringe and were infused at 5 μL per minute into the right basal ganglia using a microinfusion pump. We infused thrombin, saline, lysed RBC, or FeCl2 into the right basal ganglia stereotaxically to ensure the same location. After infusion, the needle was removed, and the skin incisions were closed with sutures. Animals were allowed to recover.
Animal groups
There were three sets of experiments in this study. In the first set, pentobarbital anesthetized male Sprague-Dawley rats (five to six rats per group) received an intracerebral infusion of 50 μL saline or thrombin (1 U) into the right basal ganglia. They then received an infusion of 30 μL lysed RBCs 1, 3, or 7 days later. Lysed RBCs was prepared as described in our previous study (Xi et al., 1998a). Briefly, RBCs (hematocrit = 87 ± 1%) were obtained by centrifuging unclotted blood. The plasma and buffy coat were discarded. The RBCs were washed with five volumes of saline three times. The washed RBCs were then lysed by freezing in liquid nitrogen for 5 minutes followed by thawing at 37°C. All rats were killed after a further 24 hours for measurement of brain water content. In the second set of experiments, rats (six rats per group) received an intracerebral infusion of either saline or one unit of thrombin. The rats received an infusion of 20 μL FeCl2 (10 mmol/L) 3 days later, and they were killed for edema measurement after another 24 hours. In the third set, rats (three to four rats per group) had an intracerebral infusion of either saline, one unit of thrombin, or one unit thrombin plus 5 μL PD98059 (1mmol/L). The rats had no lysed RBC or FeCl2 treatment. Rats were then killed 1, 3, or 7 days later for Western blot analysis and immunohistochemistry.
Brain water content measurements
The rats (n = 5 to 6 each group) were killed by decapitation under deep pentobarbital anesthesia (60 mg/kg, i.p.). The brains were removed immediately, and a 3-mm thick coronal brain slice 4 mm from the frontal pole was cut. That slice was divided into four samples: ipsilateral basal ganglia, contralateral basal ganglia, ipsilateral cortex, and contralateral cortex. Cerebellum was obtained as a control. Tissue samples were weighed on an electronic analytical balance to obtain the wet weight (WW). The tissue was then dried in a gravity oven at 100°C for more than 24 hours to determine the dry weight (DW). Tissue water contents (%) were calculated as ((WW–DW)/WW) × 100 (Xi et al., 1998a).
Western blot analysis
The rat brains (n = 3 to 4 each group) were perfused with saline for 1 minute, and a coronal brain slice was cut as described for brain water content measurements. The ipsilateral and contralateral basal ganglia were sampled and immersed in 0.5 mL of Western blot sample buffer and then sonicated for Western blot analysis (Xi et al., 1999). The sample solution (20 μL) was taken for protein assay. Protein concentration was determined by Bio-Rad protein assay kit. Protein (50 μg) from each sample was separated by SDS-PAGE gel and transferred to a hybond-C pure nitrocellulose membrane. Membranes were probed with a 1:2000 dilution of the primary antibodies. Primary antibodies were mouse anti-human HIF-1α antibody (Novus Biologicals Inc., Littleton, CO, U.S.A.), rabbit anti-human transferrin antibody (DAKO, Glostrup, Denmark), and mouse anti-human transferrin receptor antibody (Zymed Laboratories Inc., South San Francisco, CA, U.S.A.). The relative densities of HIF-1α, Tf, and TfR protein bands were analyzed using the NIH image software. The results were repeated two times.
Immunohistochemistry
Immunohistochemical studies were performed as in our previous report (Xi et al., 1999). The rats (n = 3 to 4 each group) were reanesthetized with pentobarbital (60 mg/kg, i.p.), and the brain was perfused with 4% paraformaldehyde in 0.1 M pH 7.4 phosphate-buffered saline (PBS). Removed brain was kept in 4% paraformaldehyde for 6 hours then immersed in 25% sucrose for 3 to 4 days at 4°C. The brains were embedded in O.C.T compound (Sakura Finetek U.S.A. Inc., Torrance, CA, U.S.A.) and sectioned on a cryostat (18 μm thick). Sections were incubated according to the avidin-biotin complex technique. Primary antibodies were monoclonal mouse anti-human HIF-1α antibody (1:500 dilution; Novus Biological Inc. Littleton, CO., U.S.A.), rabbit anti-human transferrin antibody (1:500 dilution; DAKO, Glostrup, Denmark), and mouse anti-human transferrin receptor antibody (1:500 dilution; Zymed Laboratories Inc. South San Francisco, CA, U.S.A.). Hematoxylin staining was used as counterstaining. Normal rabbit IgG or normal mouse IgG was used as negative control. The results were repeated two times.
Statistical analysis
All data in this study are presented as mean ± standard deviation. Data were analyzed with ANOVA using the Scheffe F test or Student's t-test. Significance levels were measured at P < 0.05.
RESULTS
Before intracerebral infusion of lysed RBCs or iron, mean arterial blood pressure, blood pH, Pa
Intracerebral injection of lysed RBCs caused a marked increase in the water content of the ipsilateral basal ganglia (81.8 ± 0.5% versus 77.8 ± 0.3% in the saline control, P < 0.01). The time course showed that there was no protection when thrombin pretreatment was given 1 day before infusion of lysed RBCs (Fig. 1A). However, thrombin given 3 days before significantly reduced the amount of edema induced by lysed RBCs (79.4 ± 0.9% versus 81.5 ± 1.4% in the saline preconditioning group, n
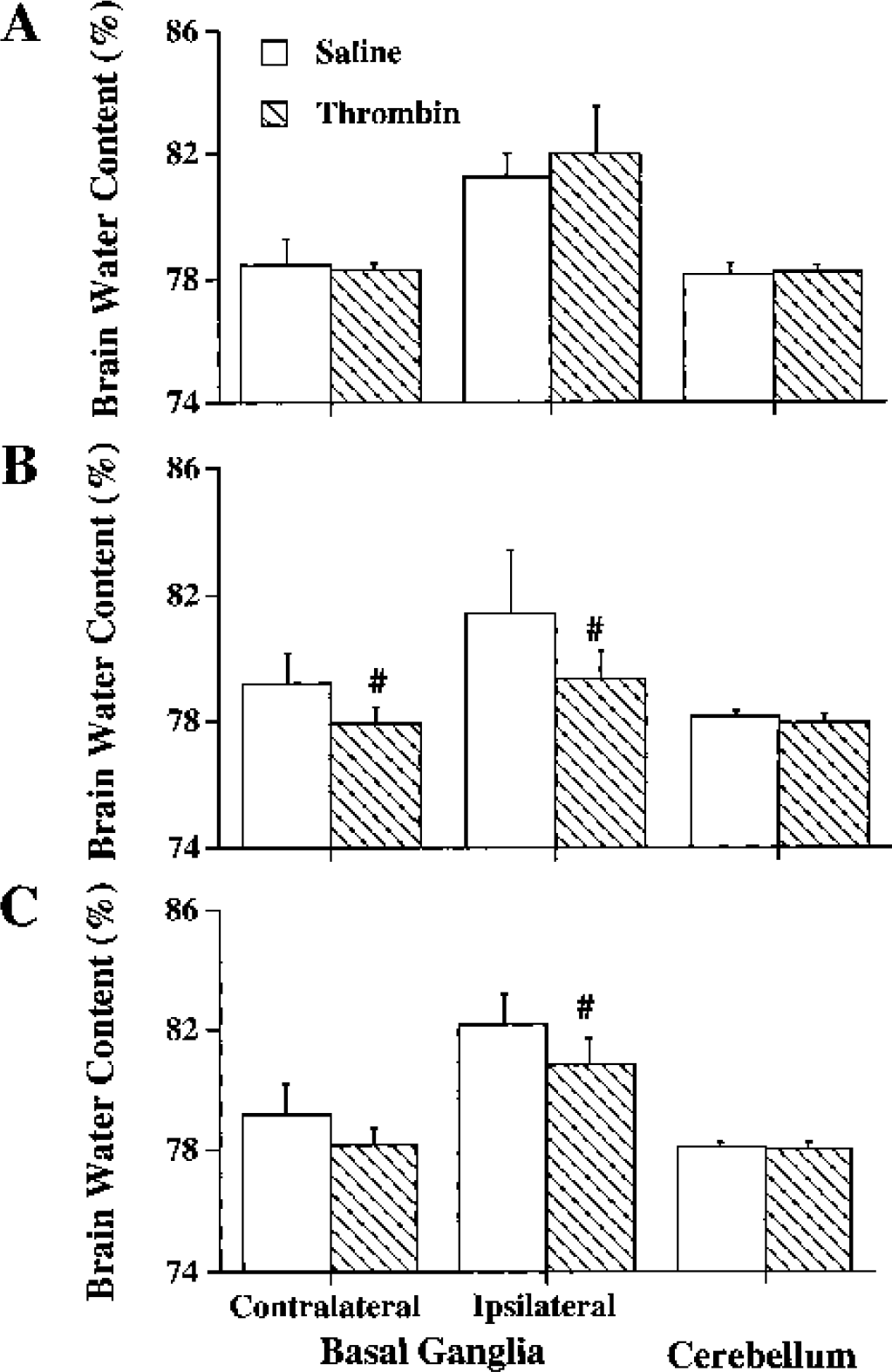
Brain water contents in the ipsilateral and contralateral basal ganglia and cerebellum at 24 hours after intracerebral infusion of 30 μL lysed RBCs. The brains had been infused with either saline or one unit thrombin at 1 (A), 3 (B), or 7 days (C) before the lysed RBCs. Values are expressed as mean ± SD, n = 5 to 6. #P < 0.05 versus saline control.
Evidence suggests that iron is a major mediator of the brain injury induced by red blood cell lysis in ICH (Huang et al., 2002). We then tested whether TPC attenuates edema induced by ferrous chloride. We chose a 3-day interval between the TPC and the ferrous chloride infusion because the lysed RBC experiments indicated maximal protection at day 3. Thrombin given 3 days before significantly reduced the amount of edema induced by iron in the ipsilateral basal ganglia (79.5 ± 1.1% versus 83.6 ± 2.0% in the saline preconditioning group, n = 6, P < 0.01) (Fig. 2) and in the ipsilateral cortex (80.0 ± 0.8% versus 82.7 ± 1.9% in the saline preconditioning group, n = 6, P < 0.01). Thrombin preconditioning had no effect on brain water contents in the contralateral cortex (79.6 ± 0.8% versus 79.0 ± 1.0% in the saline preconditioning group, n = 6, P > 0.05).
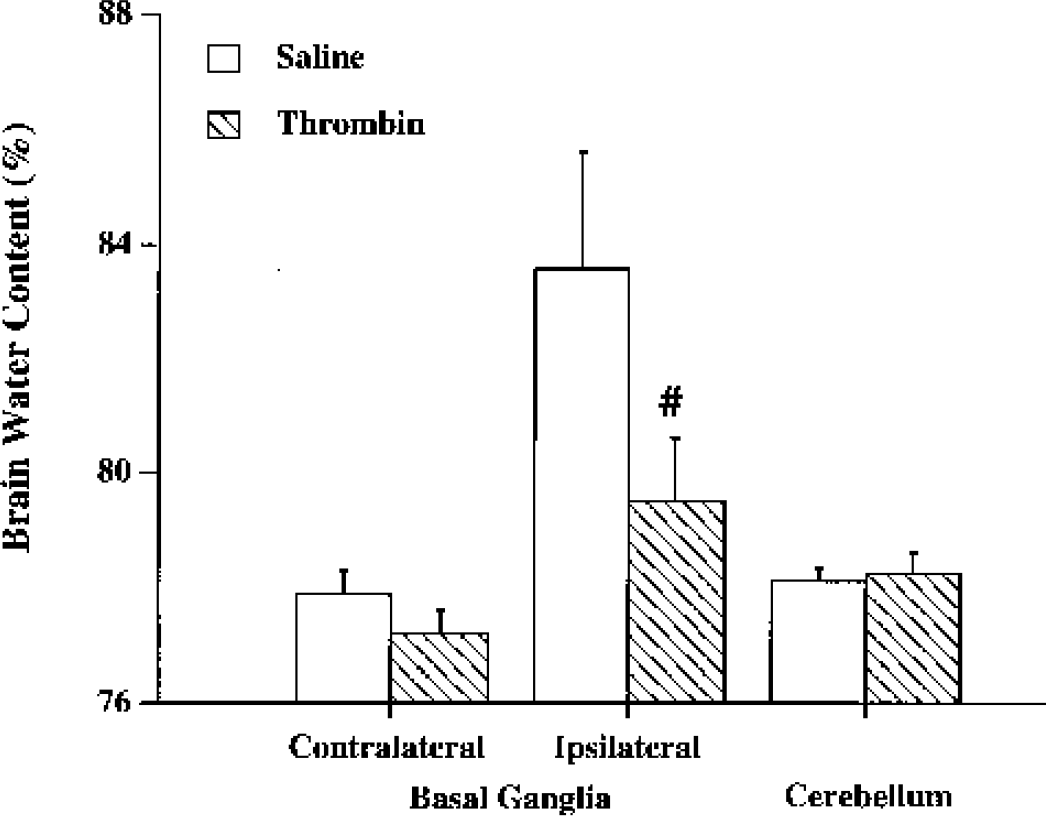
Brain water contents 24 hours after intracerebral infusion of 20 μL ferrous chloride (10 mmol/L). The brains has been infused with either saline or one unit thrombin 3 days before ferrous chloride. Values are expressed as mean ± SD, n=6. #P<0.01 versus saline control.
Hypoxia inducible factor-1α is one of the earliest molecular switches in the signaling of hypoxic tissues. Thrombin plays a key role in HIF-1α accumulation after ICH. HIF-1α protein was undetectable in the normal rat brain by Western blot analysis. However, HIF-1α protein levels in the ipsilateral basal ganglia were increased significantly at 1 and 3 days and reduced to very low levels at 7 days after intracerebral infusion of thrombin (Fig. 3). HIF-1α was not detected in the contralateral basal ganglia.
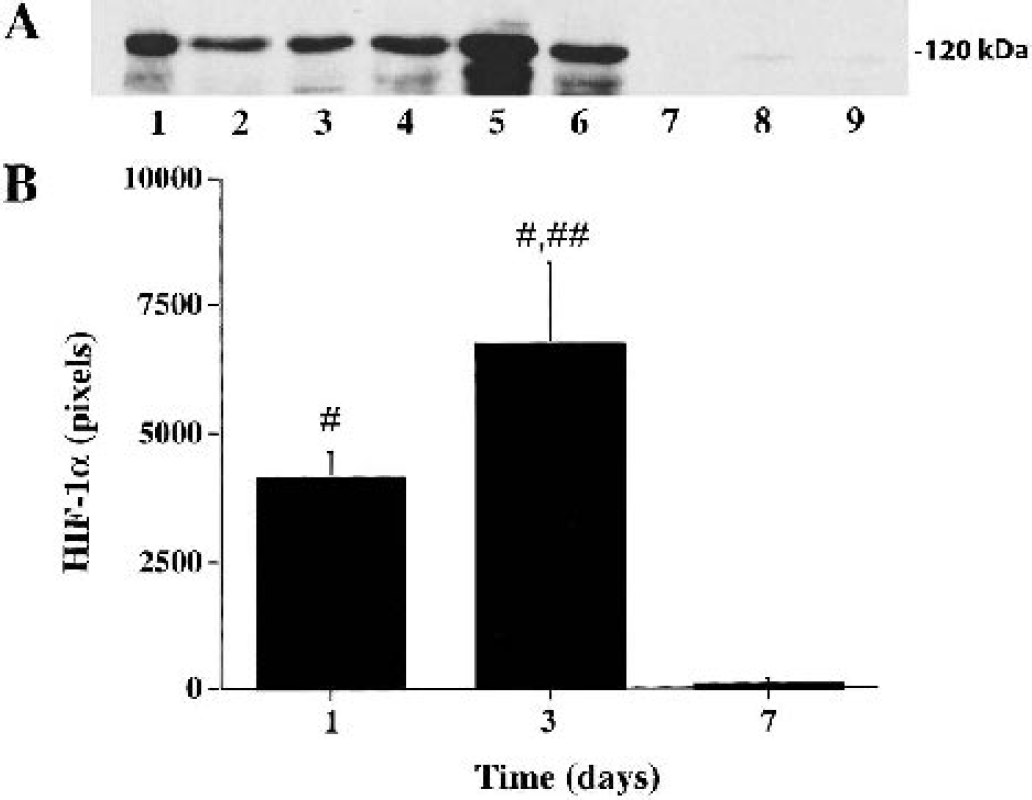
(A) Western blot analysis showing the time course of HIF-1α protein levels in the ipsilateral basal ganglia 1 (lanes 1 to 3), 3 (lanes 4 to 6), and 7 days (lanes 7 to 9) after intracerebral injection of thrombin (1 U). (B) HIF-1α protein levels were semi-quantitated by Western blot analysis. Values are mean ± SD, n=3. #P < 0.01 versus day 7; ##P < 0.05 versus day 1.
Mitogen activated protein kinases (MAPKs) are cytoplasmatic signal transducers with an important role in thrombin-induced brain tolerance. PD98059, a p44/42 MAPKs kinase inhibitor, blocks thrombin-induced p44/42 MAPKs activation and brain tolerance (Jiang et al., 2002a; Xi et al., 2001). Here, PD98059 blocked thrombin-induced HIF-1α upregulation at day 1 (Western blot analysis: 293 ± 399 pixels versus 4,004 ± 1,886 pixels in the thrombin control, P < 0.01) (Fig. 4) and day 3 (258 ± 202 pixels versus 5,085 ± 1,069 pixels in the thrombin control, P < 0.01), indicating that p44/42 MAPKs pathway is involved (Fig. 4). At day 7, HIF-1α protein levels in the ipsilateral basal ganglia were very low in both thrombin group and thrombin plus PD 98059 group (85 ± 50 pixels and 79 ± 48 pixels, respectively; P > 0.05).
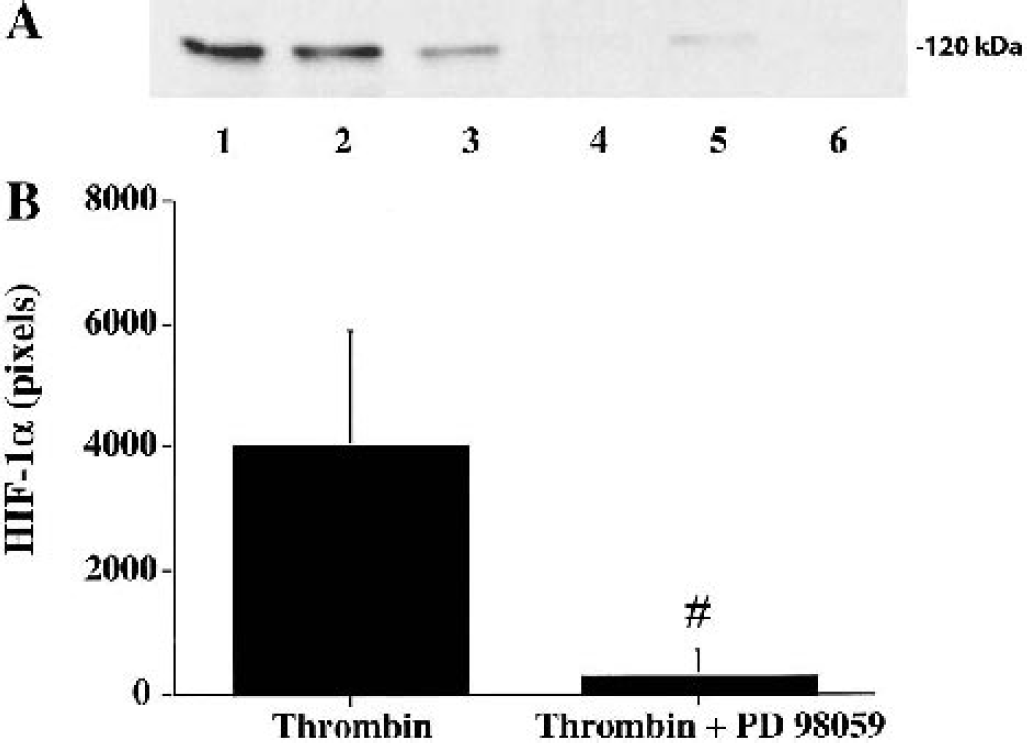
(A) Western blot showing HIF-1α protein levels in the ipsilateral basal ganglia 24 hours after intracerebral injection of either 1 U thrombin (lanes 1 to 3) or 1 U thrombin + PD98059 (lanes 4 to 6). (B) Semiquantitation of HIF-1α. Values are mean ± SD, n = 3. #P< 0.01 versus thrombin.
After intracerebral saline infusion, Tf, TfR, and HIF-1α immunoreactivities were detected in the ipsilateral basal ganglia by immunohistochemistry. However, immunohistochemistry illustrated an increase of immunoreactivities of HIF-1α, Tf, and TfR in the ipsilateral basal ganglia after one unit thrombin infusion (Fig. 5).
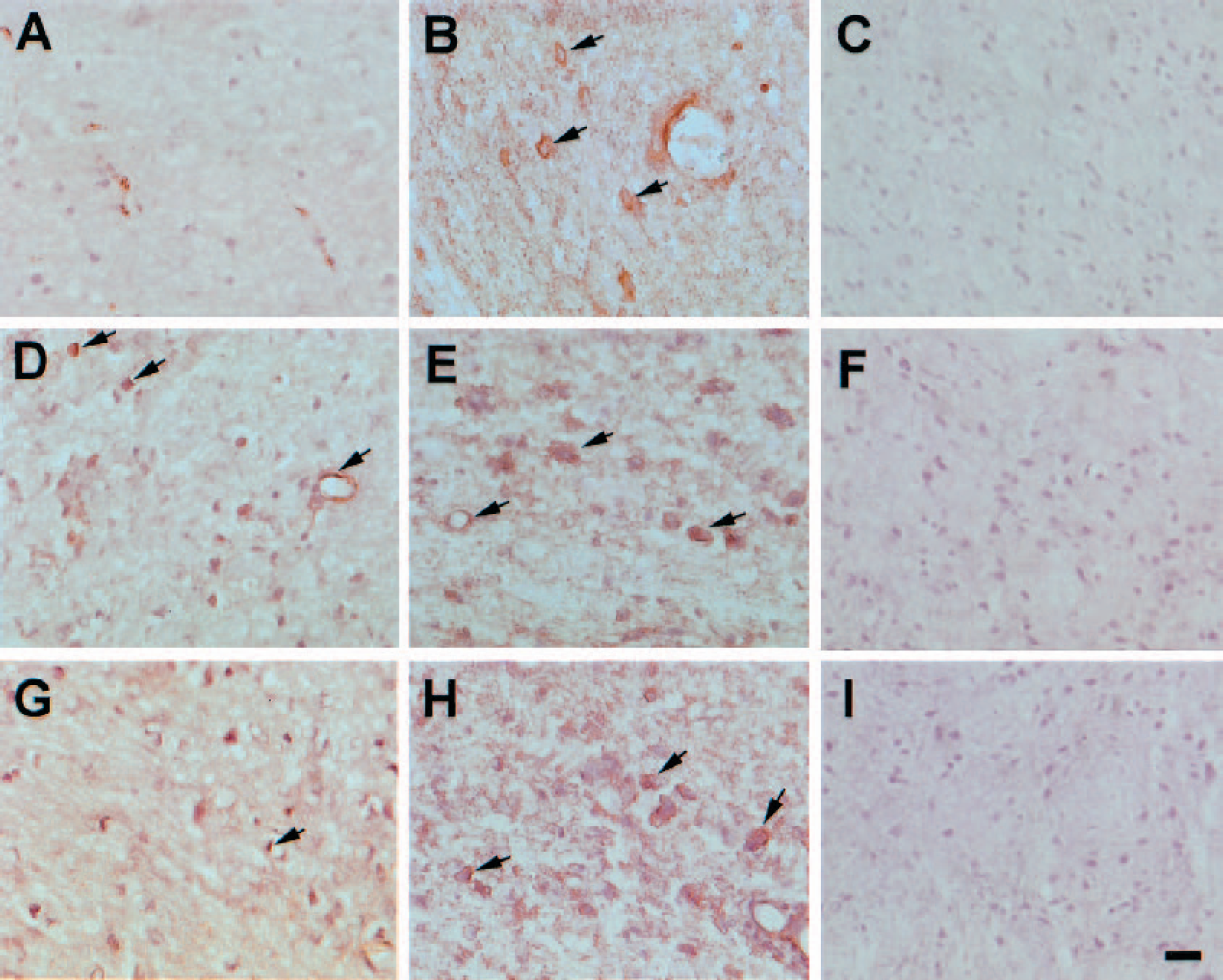
Immunoreactivities of HIF-1α, transferring, and transferrin receptor in the ipsilateral basal ganglia after intracerebral infusion of either saline (A, D, G) or one unit thrombin (B, C, E, F, H, I). Hemotoxylin staining was used as counterstaining. (A, B) HIF-1α immunoreactivities at 24 hours. (D, E) Transferrin immunoreactivities at 3 days. (G, H) Transferrin receptor immunoreactivities at 3 days. (C, F, I) Negative controls for HIF-1α, transferring, and transferrin receptor, respectively. Scale bar is 20 μm.
Brain Tf and TfR levels were significantly higher in thrombin treated group compared with those in saline control group (Figs. 6A and 7A). P44/42 MAPK inhibition with PD98059 reduced thrombin-induced upregulation of Tf (a twofold versus 100-fold increase in the thrombin control) and TfR (a twofold versus a 10-fold increase in the thrombin control). Both Tf and TfR protein levels were upregulated in the ipsilateral basal ganglia 1 day after thrombin infusion, remained at high levels at 3 days, and returned towards the baseline levels at 7 days (Figs. 6B and 7B).
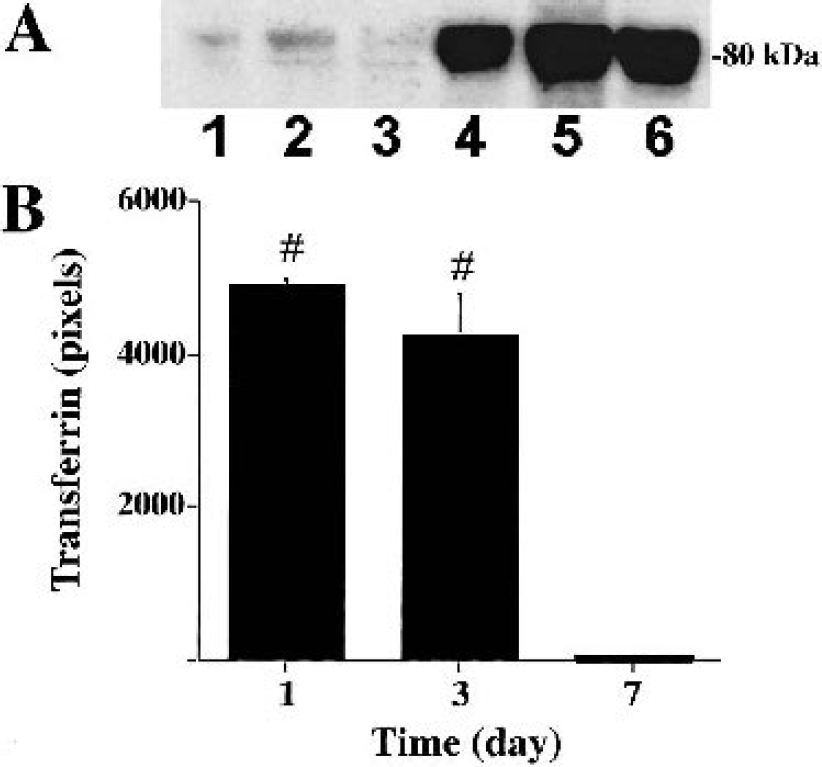
(A) Transferrin protein levels measured by Western blot analysis in the ipsilateral basal ganglia 3 days after either saline (lanes 1 to 3) or one unit thrombin infusion (lanes 4 to 6). (B) Time course of transferrin protein levels measured by Western blot analysis in the ipsilateral basal ganglia after intracerebral infusion of one unit thrombin. Values are mean ± SD, n = 3. #P < 0.01 versus day 7.
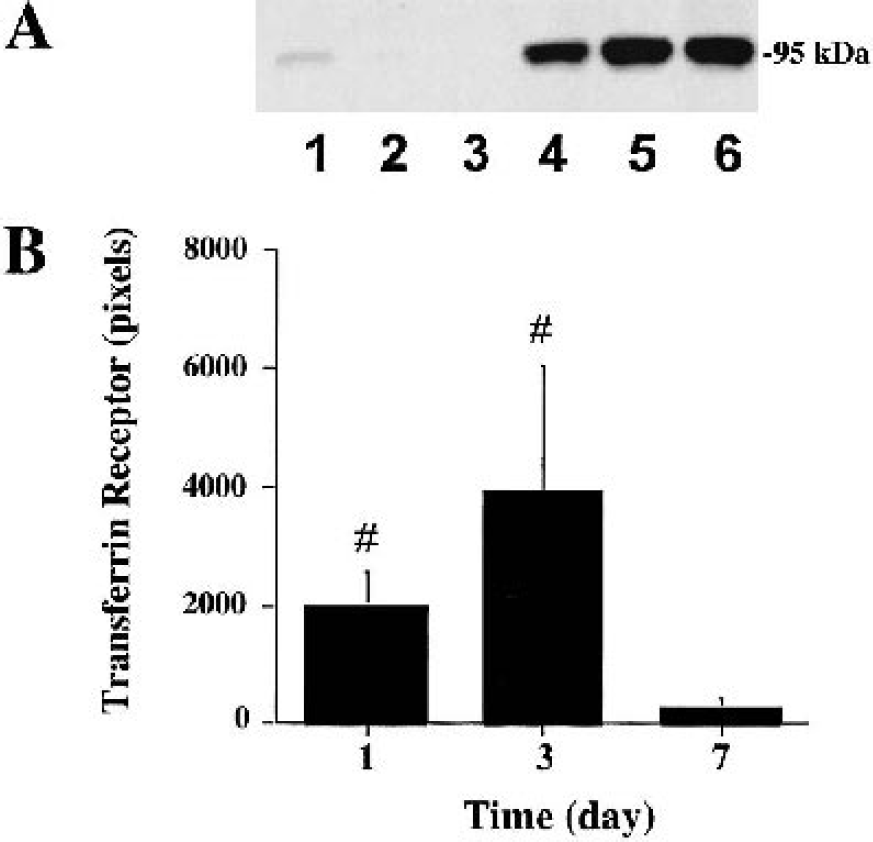
(A) Transferrin receptor protein levels measured by Western blot analysis in the ipsilateral basal ganglia 3 days after either saline (lanes 1 to 3) or one unit thrombin infusion (lanes 4 to 6). (B) Time course of transferrin receptor protein levels measured by Western blot analysis in the ipsilateral basal ganglia after intracerebral infusion of one unit thrombin. Values are mean ± SD, n = 3. #P < 0.01 versus day 7.
DISCUSSION
This study provides evidence that intracerebral thrombin preconditioning reduces brain edema induced by lysed RBCs and ferrous chloride. TPC upregulates brain HIF-1α, via a MAPK-dependent pathway, and upregulates Tf and TfR levels, two HIF-1α target genes. Together with previous data, these results suggest a scenario whereby thrombin, via its receptor(s), activates the MAPK pathway, which upregulates HIF-1α, which in turn upregulates the iron handling proteins, Tf and TfR. The change in these last two proteins may play a major role in the thrombin-induced brain tolerance to erythrocyte and iron-induced brain injury. Within the context of intracerebral hemorrhage, thrombin release during hematoma formation may initiate mechanisms that protect against iron release during clot resolution.
Thrombin at high concentrations causes brain damage and kills cultured neurons and glia, but thrombin at low concentrations is neuroprotective both in vitro and in vivo. In vitro studies have shown that thrombin protects rat astrocytes from hypoglycemia or oxidative stress-induced cell death. Thrombin also protects rat primary hippocampal neurons from cell death produced by hypoglycemia, hypoxia, or growth supplement deprivation (Striggow et al., 2000; Vaughan et al., 1995). In addition, thrombin attenuates neuronal cell death and modulates astrocyte reactivity induced by amyloid in vitro. In vivo studies have shown that TPC reduces brain injury that follows a subsequent intracerebral infusion of a high dose thrombin, an effect abolished by coinjection of a thrombin inhibitor (Xi et al., 1999). In vivo, TPC also reduced ICH-induced brain edema (Xi et al., 2000) and brain injury following focal cerebral ischemia (Masada et al., 2000). In the present study, we demonstrated that TPC can protect the brain against the brain edema formation induced by intracerebral infusion of lysed RBCs or ferrous chloride.
Iron has a key role in brain edema formation after ICH, and TPC reduces the brain edema induced by intracerebral infusion of iron. It is important to understand the mechanisms behind the thrombin-induced protective effects because they may represent naturally occurring pathways that can be manipulated pharmacologically to limit brain injury after ICH and potentially other disease states that may be associated with iron overload. Our previous experiments have indicated that lysis of erythrocytes after an ICH results in delayed brain edema to which hemoglobin and its degradation products may contribute (Huang et al., 2002; Xi et al., 1998a). After erythrocyte lysis, brain iron levels can reach very high concentrations. Iron chelation or heme oxygenase inhibition with tin-protoporphyrin to prevent heme breakdown and iron release both attenuate hemoglobin-induced edema, suggesting that iron plays an important role in edema formation after ICH (Huang et al., 2002). Although iron is essential for normal brain function, iron overload may have devastating effects (Chiueh, 2001). In addition, iron overload in the brain is associated with many neurodegenerative disorders (Thompson et al., 2001).
It is interesting that the time course of TPC-induced protection peaked at about 3 days, although there was still some protection at 7 days. In the rat, RBCs within an intracerebral hematoma start to lyse at about 3 days (Xi et al., 1998a) and brain non-heme iron starts to rise at 3 days, reaching a plateau by 7 days (Xi et al., unpublished results, 2003). Because thrombin is released during hematoma formation, it is possible that such thrombin may result in the initiation of protective mechanisms that limit brain injury when the hematoma begins to lyse.
The mechanisms of thrombin-induced brain tolerance are still not well understood, although activation of thrombin receptors and stimulation of MAPKs phosphorylation appear to be important (Xi et al., 2003). Recently, HIF-1α has been found to play an important role in hypoxia-induced ischemic tolerance (Bergeron et al., 2000). Our present data showed that HIF-1α accumulation occurs in the brain after thrombin preconditioning. TPC increases HIF-1α protein levels in neurons and endothelial cells. The thrombin effect is not likely to be mediated by hypoxia. We have previously found no reduction in cerebral blood flow after thrombin injections into the basal ganglia (Lee et al., 1997a), and other studies have found that thrombin upregulates HIF-1α levels in vascular smooth muscle cells in culture (Richard et al., 2000). It should be noted that our early study has found that HIF-1α can also be regulated by a high concentration of thrombin. This effect appears to be specific, as it was blocked by a coinfusion of hirudin. HIF-1α protein levels increased without changing HIF-1α messenger RNA levels indicating that thrombin upregulates HIF-1α by reducing HIF-1α degradation (Jiang et al., 2002b).
MAPKs are well-known cytoplasmatic signal transducers with an important role in both thrombin- and ischemia-induced brain tolerance (Irving and Bamford, 2002; Xi et al., 2001, 2003). In vivo, p44/42 MAPKs are activated in the brain after intracerebral infusion of thrombin. PD98059, a specific p44/42 MAPKs kinase inhibitor, abolished thrombin-induced activation of p44/42 MAPKs and also blocked thrombin-induced brain tolerance (Xi et al., 2001). In vitro, thrombin treatment also activated p44/42 MAPKs, and PD98059 completely blocked the cytoprotective effect of thrombin pretreatment, indicating that the p44/42 MAPKs system mediates the neuroprotective effect induced by thrombin (Jiang et al., 2002a). To test whether p44/42 MAPKs pathway is involved in thrombin-related HIF-1α upregulation, we coinjected thrombin with PD98059. Our results showed that PD98059 can also block thrombin-induced HIF-1α accumulation. This result is supported by the experiments of Richard et al. (2000) in smooth muscle cells, which indicates that HIF-1α can be phosphorylated by p44/42 MAPKs and thrombin induces HIF-1α upregulation without a change of oxygen level.
HIF-1α target genes may play a major role in the brain tolerance induced by TPC. The current study shows that brain Tf and TfR are upregulated after TPC, and these proteins may be involved in limiting iron-induced damage. HIF-1α target genes have also been linked to hypoxia-induced brain tolerance (Bergeron et al., 2000; Bernaudin et al., 2002; Jones and Bergeron, 2001). Transferrin, an 80-kDa protein, is a major iron transporter in the brain. Although many studies implicate it in the transport of iron from blood to brain across the blood-brain barrier, a recent report indicates that there also is rapid efflux of Tf from brain to blood (Zhang and Pardridge, 2001). This suggests that Tf could contribute to iron clearance when there is brain iron overload. Cellular uptake of transferrin-bound iron is achieved by binding to the TfR. In the normal brain, TfR is expressed on brain parenchymal cells at low levels. Increase of transferrin and transferrin receptor levels in the brain might change iron homeostasis after ICH. HIF-1α regulates the expression of a number of genes apart from those for transferrin and transferrin receptor (Lee et al., 1997b; Lok and Ponka, 1999; Rolfs et al., 1997; Tacchini et al., 1999). These include heme oxygenase-1 (HO-1), also known as heat shock protein 32 (HSP32). It degrades heme into iron, carbon monoxide, and biliverdin. Studies suggest that HO-1 is involved in cytoprotection from different kinds of injuries and is also upregulated after thrombin preconditioning (Sharp et al., 1999; Xi et al., 1999), although HO-1 upregulation may induce brain injury by increasing cellular iron content (Huang et al., 2002). In common with thrombin, HIF-1α and HO-1 have been implicated in both reducing and enhancing brain injury. Another HIF-1α target gene is plasminogen activator inhibitor-1. It, too, is upregulated after intracerebral thrombin injection (Hua et al., 2002) and it may regulate the rate of hematoma lysis.
In summary, our present results demonstrate that thrombin preconditioning reduces brain edema induced by both lysed RBCs and iron. Thrombin induces HIF-1α through p44/42 MAPKs pathway, and HIF-1α may be involved in thrombin-induced brain tolerance via induction of its target genes. In particular, upregulation in brain parenchymal Tf and TfR levels or enhanced iron efflux across the blood-brain barrier may limit iron-related brain damage.