
Abstract
The authors previously found that pretreatment with a low dose of thrombin attenuates the brain edema induced by a large dose of thrombin or an intracerebral hemorrhage, and reduces infarct volume after focal cerebral ischemia (i.e., thrombin preconditioning). This study investigated whether thrombin preconditioning is caused by activation of the thrombin receptor, also called protease-activated receptor. In the in vivo studies, thrombin-induced brain tolerance was eliminated by RPPGF (Arg-Pro-Pro-Gly-Phe), a thrombin-receptor antagonist. Pretreatment with a thrombin-receptor agonist reduced the amount of edema induced by a large dose of thrombin infused into the ipsilateral basal ganglia 7 days later (81.3 ± 0.7% vs. 82.6 ± 0.8% in the control, P < 0.05). In the in vitro study, low doses of thrombin (1 or 2 U/mL) did not induce cell death. However, doses greater than 5 U/mL resulted in dose-dependent lactate dehydrogenase release (P < 0.01). Thrombin and thrombin receptor-activating peptide preconditioning reduced lactate dehydrogenase release induced by a high dose of thrombin (10 and 20 U/mL), whereas RPPGF blocked the effect of thrombin preconditioning in vitro. Western blots indicated that p44/42 mitogen-activated protein kinases were activated after thrombin preconditioning. Finally, inhibition of p44/42 mitogen-activated protein kinases activation by PD98059 abolished the thrombin-preconditioning effect. Results indicate that thrombin-induced brain tolerance is in part achieved through activation of the thrombin receptor. Activation of the thrombin receptor in the brain may be neuroprotective. The protective effect of thrombin preconditioning is achieved through the p44/42 mitogen-activated protein kinase signal-transduction pathway.
Thrombin is a serine protease and an essential component of the coagulation cascade. It is produced immediately in the brain after intracerebral hemorrhage, brain trauma, or blood–brain barrier breakdown after many kinds of brain injury (Gingrich and Traynelis, 2000). Direct infusion of large doses of thrombin into brain causes inflammatory cell infiltration, mesenchymal cell proliferation, brain edema formation, and seizures (Lee et al., 1995, 1996a,b, 1997; Nishino et al., 1993; Xi et al., 1998a,b). High concentrations of thrombin kill neurons and astrocytes in vitro (Striggow et al., 2000; Vaughan et al., 1995). Our laboratory has shown that thrombin is responsible for early brain edema formation after intra-cerebral hemorrhage (Lee et al., 1996b).
Although a large dose of thrombin causes brain injury and cell death, low concentrations of thrombin are neuroprotective. We previously found that pretreatment with a low dose of thrombin attenuated the brain edema induced by thrombin or hemorrhage, and significantly reduced the infarct size in a rat middle cerebral artery occlusion model (Masada et al., 2000; Xi et al., 2000, 1999). We have termed this phenomena thrombin preconditioning (TPC), or thrombin-induced brain tolerance, by analogy to ischemic preconditioning.
The physiologic actions of thrombin either are non-receptor mediated (e.g., the cleavage of fibrinogen to fibrin) or are receptor mediated. A family of protease-activated receptors (PAR) has been identified (Coughlin, 2000), of which PAR-1, PAR-3, and PAR-4 are activated by thrombin. The relative role of the receptor- and non-receptor-mediated effects of thrombin in brain injury and brain protection (i.e., TPC) has been studied incompletely, though our recent in vivo study found that thrombin-induced brain tolerance is derived through p44/42 mitogen-activated protein kinases (MAPK), suggesting a receptor-mediated pathway (Xi et al., 2001). The current study seeks to address this issue both in vivo and in vitro.
MATERIALS AND METHODS
Studies in vivo
Studies in vitro
RESULTS
Studies in vivo
Physiologic parameters including blood pH, blood gases, mean arterial blood pressure, hematocrit, and blood glucose were measured immediately before an intracerebral infusion, and all the values were within the normal range (data not shown).
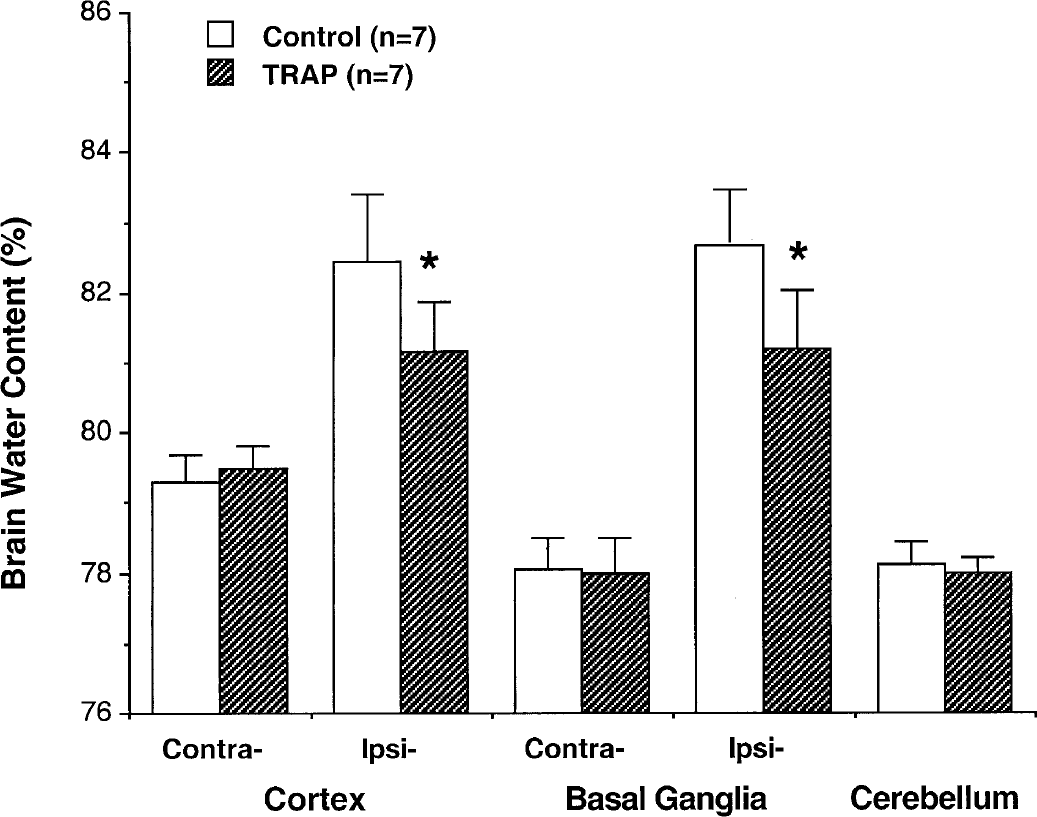
Thrombin-receptor agonist and brain tolerance. Brain water content 24 hours after intracerebral infusion of 5 U thrombin into the right basal ganglia is shown. The brains had been infused with either saline or thrombin-receptor agonist (5-nmol/L TRAP) 7 days before the large dose of thrombin. Values are expressed as mean ± SD, n = 7. *P < 0.05 versus control.
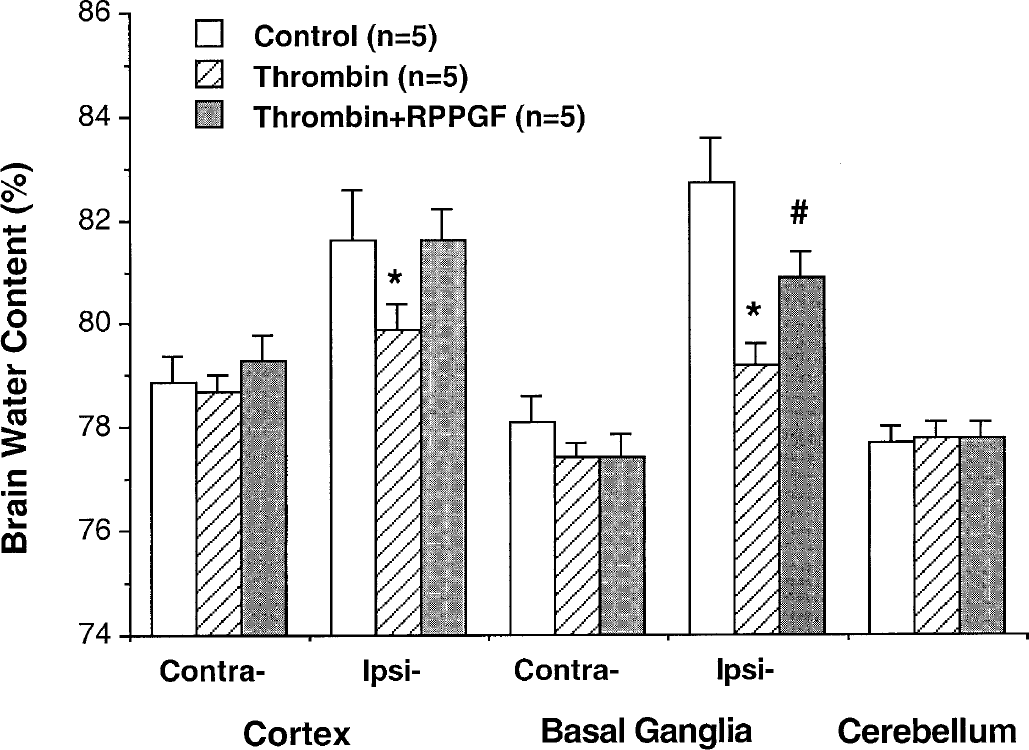
Thrombin-receptor antagonist and thrombin-induced brain tolerance. Brain water content 24 hours after intracerebral infusion of 5 U thrombin is shown. The brains had been infused with saline, 1 U thrombin, or 1 U thrombin and 5-nmol/L thrombin-receptor antagonist RPPGF 7 days before the large dose of thrombin was administered. Values are expressed as mean ± SD, n = 5. *P < 0.01 versus the other groups. #P < 0.05 versus thrombin.
Studies in vitro
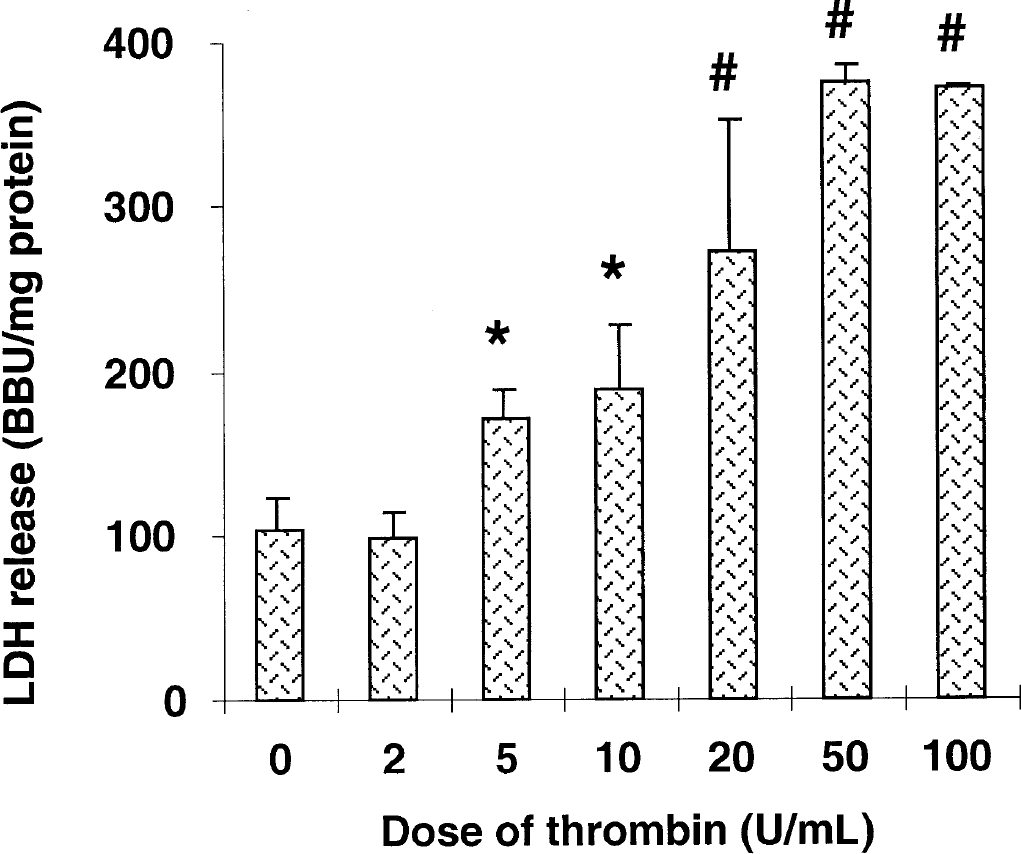
Dose effect of thrombin on cell viability in a neuron/astrocyte culture. Cells were treated with different doses of thrombin, and LDH release in the medium was measured 24 hours later. Thrombin caused a dose-dependent LDH release. Values are mean ± SD. *P < 0.05 versus dose 0 and 2. #P < 0.01 versus dose 0, 2, 5, and 10. BBU, Berger-Broida unit.
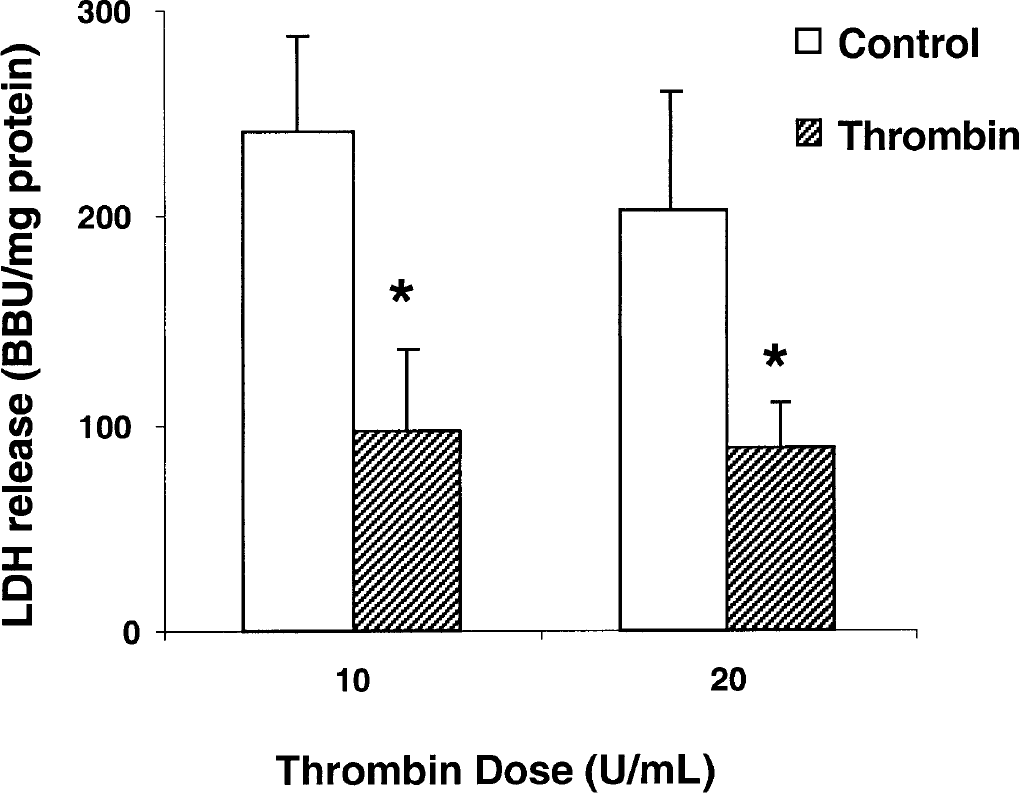
The effect of thrombin preconditioning on thrombin-induced cell death. Cells were pretreated with vehicle (control) or 1 U/mL thrombin (Thrombin). Forty-eight hours later, the cells were incubated with large doses of thrombin (10 U or 20 U/mL). Lactate dehydrogenase levels in the medium were measured at 24 hours. Values are mean ± SD. *P< 0.05 versus vehicle group. BBU, Berger-Broida unit.
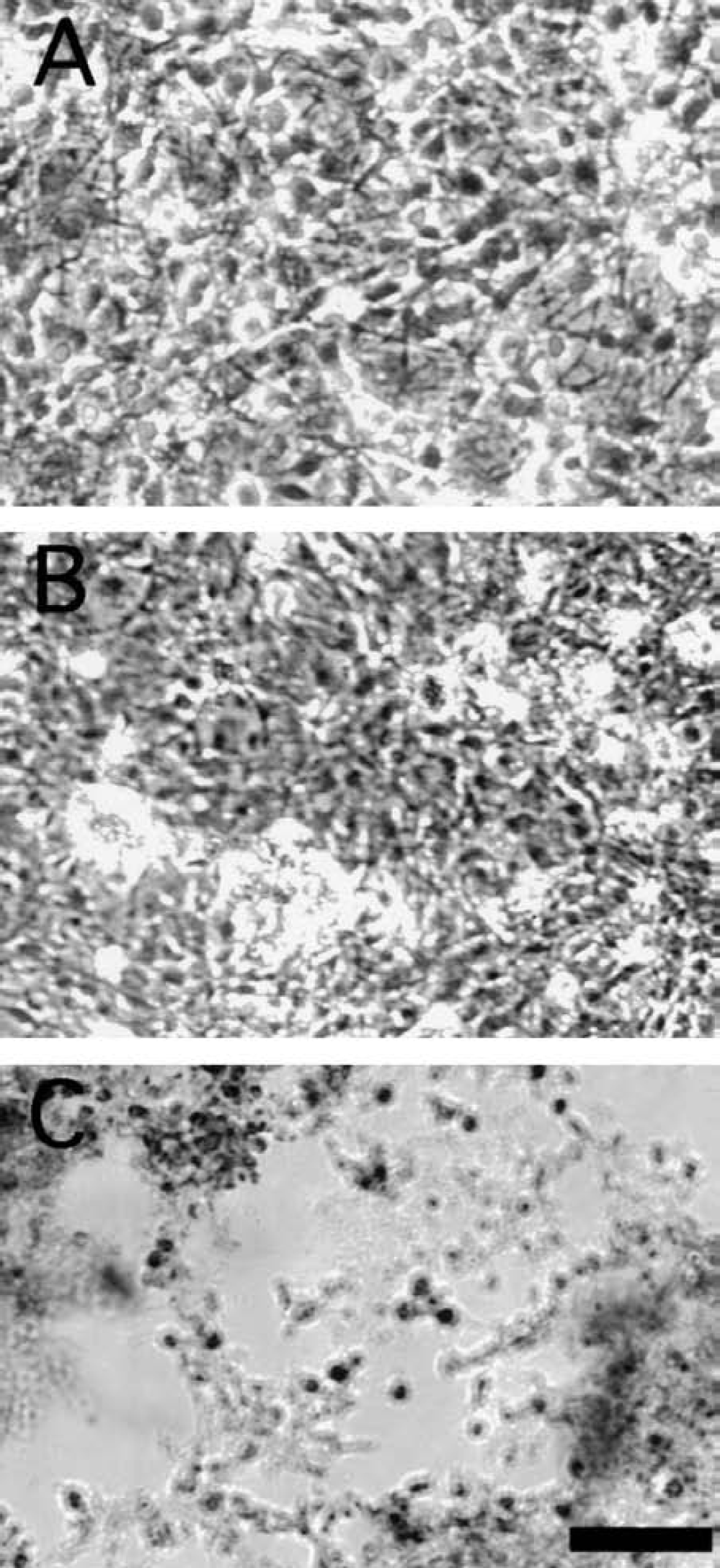
Thrombin preconditioning and cell death caused by thrombin. Cells were pretreated with vehicle or 1 U/mL thrombin (TPC). Forty-eight hours later, the cells were incubated with large doses of thrombin (10 U /mL) and observed under microscopy.
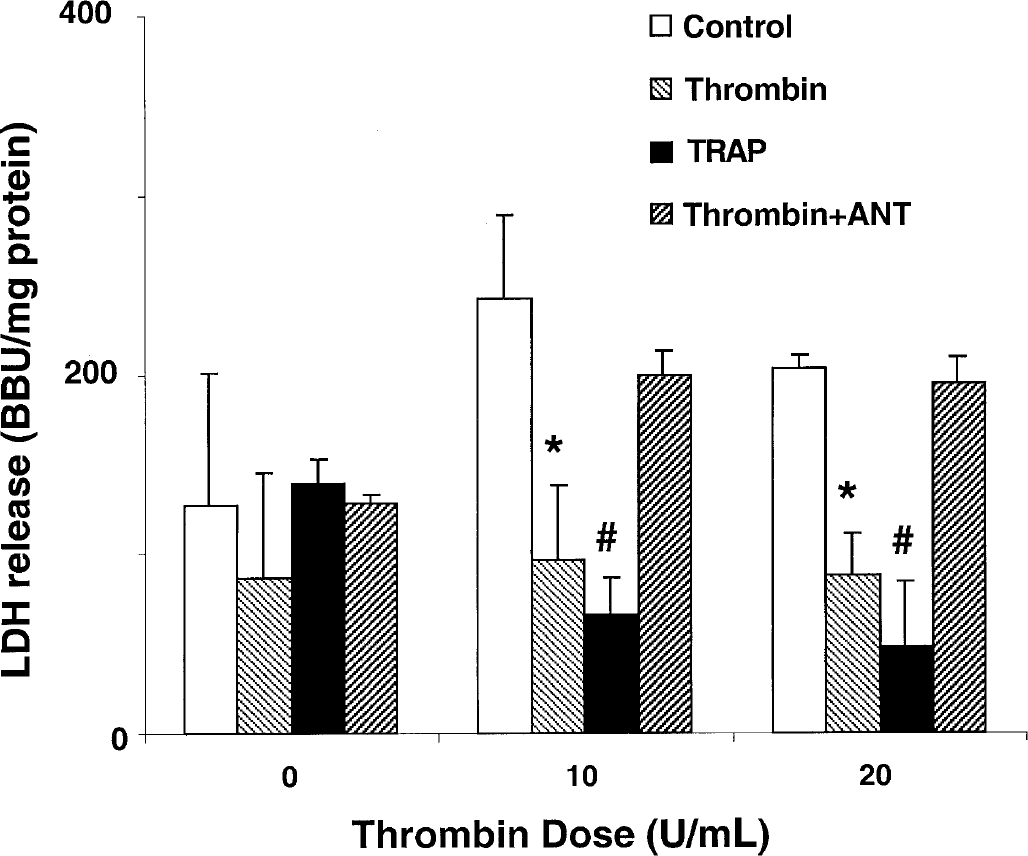
Thrombin receptor and thrombin preconditioning. Cells were pretreated with vehicle (Control), 1U thrombin (Thrombin), 5-μmol/L TRAP (TRAP), or 1 U thrombin plus 500-nmol/L thrombin-receptor antagonist (Thrombin + ANT). After 48 hours, the cells were treated with 10 U/mL thrombin and LDH levels were measured 24 hours later. Values are mean ± SD. *,#P < 0.05 versus control or thrombin plus thrombin-receptor antagonist. BBU, Berger-Broida unit.
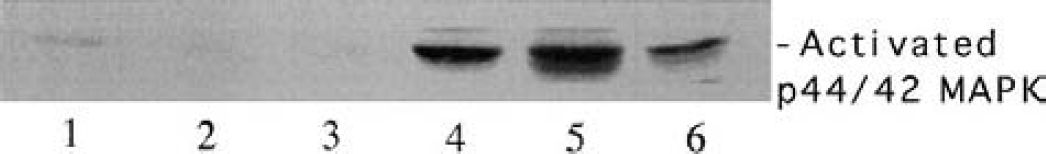
Thrombin preconditioning and p44/42 mitogen-activated protein kinase (MAPK) activation. Activated p44/42 MAPK in the cell 48 hours after vehicle (lanes 1–3, control) or 1 U thrombin (lanes 4–6, TPC) treatment.
Lactate dehydrogenase release 24 hours after 10 U/mL thrombin treatment

Values are mean ± SD.
LDH, lactate dehydrogenase; TPC, thrombin preconditioning; BBU, Berger-Broida unit.
P < 0.01 vs. the other groups.
DISCUSSION
Our present study found that thrombin-receptor agonist can mimic the effect of thrombin preconditioning, and a thrombin-receptor antagonist blocks thrombin-induced brain tolerance. These studies also showed that thrombin caused dose-dependent death of neurons and astrocytes in vitro. Pretreatment with a low dose of thrombin or thrombin receptor–activating peptides reduced thrombin-induced cellular death, whereas a thrombin-receptor antagonist blocked the protective effect of thrombin. In addition, PD98059 abolished the protective effect of thrombin. These results suggest that thrombin-induced tolerance results from activation of the thrombin receptor and the p44/42 MAPK pathway.
There is increasing evidence that, in addition to its role in homeostasis and thrombosis, thrombin plays an important role in developmental, physiologic, and pathological functions in the nervous system (Coughlin, 1999; Gingrich and Traynelis, 2000; Turgeon et al., 2000). Thrombin has been found in astrocytes, neuroblastomas, and motor neuron cultures. It is also known that exogenous thrombin alters cell morphology and differentiation, including neurite retraction, reversal of astrocyte astellation, glial proliferation, and axonal growth cone collapse (Beecher et al., 1994; Brewer, 1995a; Grabham and Cunningham, 1995; Nelson and Siman, 1990). At high concentrations, thrombin induces apoptotic cell death (Donovan et al., 1997; Smirnova et al., 1998). The present data also show that in a neuron–astrocyte mixed culture, thrombin decreased viability in a dose-dependent fashion, which is consistent with findings a previous report (Smith-Swintosky et al., 1995). An intracerebral injection of thrombin induces a variety of inflammatory changes and increases brain water content (Lee et al., 1995, 1996a,b). Some of these effects can be suppressed by thrombin inhibitors, such as hirudin. Thrombin is produced immediately at sites of cerebrovascular injury such as stroke and trauma, and persists for several days after injury. These findings indicate that thrombin is involved in the pathophysiologic processes of several nervous system diseases.
Recent reports show that thrombin, at a low dose, can protect neuronal cells from toxic insults such as hypoxia and β-amyloid peptides (Debeir et al., 1996; Pike et al., 1996; Vaughan et al., 1995). Our previous studies also found that preconditioning with a low dose of thrombin attenuates the brain edema induced by an intracerebral injection of a high dose of thrombin or autologous blood, and reduces the infarct volume in a rat model of permanent middle artery occlusion (Masada et al., 2000; Xi et al., 2000, 1999). In the current study, we have confirmed that TPC prevents cell damage induced by a large dose of thrombin.
Because of thrombin can cause cell injury at high concentrations and be protective at low concentrations, it is important to know what concentrations of thrombin may occur in healthy and injured brain. Direct thrombin measurements are complicated because of questions regarding assay specificity, binding of thrombin to endogenous inhibitors within the brain, interactions with fibrin and, in the case of intracerebral hemorrhage, interference by hemoglobin with the assay. However, the concentration of prothrombin in the plasma is high enough (1–5 μmol/L) to produce a substantial amount of thrombin in the brain parenchyma after a hemorrhage. In the rat, the brain edema produced by a 50-μL intracerebral hematoma can be significantly inhibited by thrombin inhibitors, and an intracerebral infusion of 5 U thrombin causes a similar degree of edema (Lee et al., 1996b; Xi et al., 1998a). Because 1 mL whole blood can produce approximately 260 to 360 U thrombin, a 50-μL clot could be expected to produce up to 15 U thrombin. Thus, very high concentrations of thrombin appear to occur near an intracerebral hemorrhage, though lower concentrations may occur distant from the clot. Thrombin concentrations may also be elevated after brain injury where there is no hemorrhage because of blood–brain barrier breakdown or prothrombin synthesis within the brain. After 24 hours of permanent middle cerebral artery occlusion in the rat, we have measured a thrombinlike protease activity of 0.7 U/g in the ipsilateral hemisphere compared with 0.3 U/g in the contralateral hemisphere (Xi et al., unpublished results, 2001). Thrombin has also been detected in chronic neurodegenerative diseases such as Alzheimer disease and cerebral vascular dementia. Recently, zymogen precursors of thrombin, thrombinlike proteases, and their receptors have been localized in several distinct regions of the developing and adult brain (Dihanich et al., 1991).
The mechanism of TPC cytoprotection is not clear. Nevertheless, the majority of cellular functions influenced by thrombin are mediated by the thrombin receptor under normal conditions (Coughlin, 2000). The present data show that TPC protection results from activation of thrombin receptor because the effect is mimicked by a thrombin-receptor agonist and suppressed by a thrombin-receptor antagonist. Thrombin receptor is a proteolytically activated, seven-transmembrane domain, G-protein–coupled receptor (Gerszten et al., 1994; Vouret-Craviari et al., 1992). Striggow et al. (2000) also found that a low concentration of thrombin (50 pmol/L) or TRAP reduces ischemic neuronal injury in vitro.
Previous studies have implicated several different signaling cascades induced by thrombin-receptor activation, including phospholipase activation, adenyl cyclase, phosphoinositide hydrolysis, intracellular calcium release, tyrosine phosphorylation and, possibly, RhoA activation (Donovan and Cunningham, 1998; Grand et al., 1996; Van Obberghen-Schilling and Pouyssegur, 1993). Mitogen-activated protein kinases (including p38, p44/42, and JNK) are well-known cytoplasmic signal transducers with an important role in cell responses to external stimuli (Davis, 1993; Fukunaga and Miyamoto, 1998). Gonzalez-Zulueta et al. (2000) reported that activation of the p44/42 MAPK pathway is required for preconditioning induced by oxygen-glucose deprivation in primary cortical cell culture. It is also activated after ischemic preconditioning and cerebral ischemia (Gu et al., 2000; Shamloo et al., 1999). In a recent in vivo study, showed that p44/42 MAPK is activated after TPC (Xi et al., 2001). The current study confirms that activated p44/42 MAPK is upregulated during TPC in vitro. In addition, PD98059, a specific inhibitor of p44/42 MAPK, completely blocked the TPC cytoprotective effect, indicating that the p44/42 MAPK system mediates the neuroprotective effect of TPC.
How p44/42 MAPK pathway activation results in brain tolerance to injury is not clear. An earlier study showed that activation of p44/42 MAPK is associated with upregulation of heat shock protein 27 expression (Xi et al., 2001). Heat shock protein 27 exerts its cytoprotective effects by stabilizing actin filaments, by its role as a molecular chaperone, and by increasing glutathione levels. The antiapoptotic effect may also contribute to thrombin-induced brain tolerance. In Drosophila, p44/42 MAPK directly inactivates the head involution defective (HID) gene that is proapoptotic (Kurada and White, 1998). Recently, Anderson and Tolkovsky (1999) reported that MAPK also protects sympathic neurons from apoptosis induced by cytosine arabinoside. In addition, translocation of the activated p44/42 MAPK into the nucleus can initiate transcription factors that lead to trophic responses (Brunet et al., 1999; Robinson et al., 1998). Whether TPC can regulate thrombin-receptor expression is still to be determined.
We conclude that thrombin preconditioning prevents thrombin-induced edema in vivo and astrocyte/neuron death induced by a high dose of thrombin in vitro. The protective effects come through activation of thrombin receptor and the p44/42 MAPK signal transduction pathway. Further study of the molecular pathways by which activation of thrombin receptor mediates cytoprotective effects should allow us to understand and manipulate cellular mechanisms of viability and death related to thrombin and other stimuli.