
Abstract
Delayed neuroprotection against ischemic challenges is conferred by both ischemic preconditioning (IPC) and preconditioning by activation of the ε-isoform of protein kinase C (εPKC-PC). In vivo, ischemic preconditioning enhances GABA release and ameliorates glutamate release during lethal cerebral ischemia. We tested the hypothesis that IPC and εPKC-PC confer neuroprotection by GABA synapses in rat organotypic hippocampal slices. Ischemic preconditioning or εPKC-PC was induced with 15 mins oxygen-glucose deprivation (OGD) or ψεRACK, a selective εPKC activator; and test ischemia consisted of 40 mins OGD. At the time of peak neuroprotection (48 h after preconditioning), we recorded GABAA receptor-mediated miniature postsynaptic currents (GABA mPSCs) in vulnerable CA1 pyramidal neurons using whole-cell voltage clamp techniques. The frequency and amplitude of GABA mPSCs significantly increased 48 h after IPC. In contrast, εPKC-PC enhanced only the amplitude of GABA mPSCs with no effect on frequency. We next asked if neuroprotection depended on these changes in GABA synapses. Weak antagonism of the GABAA receptor with bicuculline (100 nmol/L) decreased the amplitude of GABA mPSCs by 20.9±6.1%. When applied during test ischemia, 100nmol/L bicuculline abolished neuroprotection conferred by either IPC or εPKC-PC. We conclude that neuroprotection conferred by preconditioning depends on functional modifications of GABA synapses.
Introduction
Cerebral ischemia is a devastating aspect of stroke, cardiac arrest, and surgical procedures that interrupt blood flow to the brain. CA1 hippocampal pyramidal neurons are selectively vulnerable to global cerebral ischemia (Horn and Schlote, 1992; Katz et al, 1995; Kofler et al, 2004; Petito et al, 1987). An early mild ischemic insult protects CA1 pyramidal neurons against subsequent, normally lethal ischemic challenges (Kitagawa et al, 1990). This phenomenon is referred to as ischemic preconditioning (IPC). IPC activates several endogenous signaling pathways that result in protection against ischemia. Identification of these pathways and their targets will likely contribute to the development of novel therapeutic concepts.
ε-Protein kinase C (εPKC) is one of the key signaling pathways activated by IPC in brain (Raval et al, 2003). Blockade of εPKC activation during IPC abolished neuroprotection. Using the selective peptide activator ψεRACK, preconditioning with direct activation of εPKC (here referred to as εPKC-PC) confers neuroprotection after 48 h with an efficacy similar to IPC (Raval et al, 2003). The εPKC signaling pathway was implicated in cerebral IPC via the adenosine A1 receptor (Lange-Asschenfeldt et al, 2004), adenosine-induced preconditioning in vitro (Di-Capua et al, 2003), and NMDA receptor activation (Raval et al, 2003). Triggered by diacylglycerol during IPC (Raval et al, 2003), the εPKC signaling pathway results in many downstream alterations.
One example is ERK1/2 activation (Lange-Asschenfeldt et al, 2004), which in turn increases the activity of cyclooxygenase 2 via STAT3 phosphorylation (Kim et al, 2007; Kim et al, 2008).
The GABA synapse contains many potential neuroprotective targets of preconditioning. The presynapse contains the GABA synthesizing enzymes, vesicle neurotransmitter transporters, and vesicle release machinery. The postsynapse refers to the components of the postsynaptic neuron that enable the response to GABA, particularly the GABA receptors. The potential usage of GABA synapses as a therapeutic target is emphasized by the widespread use of antiepileptic drugs targeting postsynaptic GABA receptors (Meldrum and Rogawski, 2007). Two drugs that modulate GABAA receptors failed to yield positive results in stroke clinical trials: diazepam in EGASIS (early GABAergic activation study in stroke; Lodder et al, 2005) and clomethiazole in CLASS (clomethiazole for acute stroke study; Lyden et al, 2002). Because of these failed clinical trials, enthusiasm diminished regarding the protective role of drugs that modulate GABA receptors in ischemic neurodegeneration. However, GABA receptors are only one component of the GABA synapse.
The endogenous targets of IPC may provide new pharmacologic targets at the GABA synapse in addition to GABA receptor modulators. For example, neuroprotection via IPC has been linked with changes in the subunit composition of GABAA receptors that are not easily mimicked by pharmacologic manipulations (Sommer et al, 2002, 2003). Furthermore, in vivo studies have shown that IPC greatly enhances GABA release during lethal ischemia (Dave et al, 2005). This increase in GABA release implies that presynaptic enhancements in GABA synapses may play a direct role in neuroprotection. A presynaptic change in the release of endogenous GABA cannot be mimicked by GABAA receptor agonists or modulators used in previous clinical trials.
Here, we used the technique of miniature postsynaptic current (mPSC) analysis to characterize neuroprotective changes in GABA synapses after ischemic and pharmacologic preconditioning using the in vitro organotypic hippocampal slice model. We studied GABA mPSCs in CA1 pyramidal neurons 48 h after IPC or εPKC-PC. We tested the hypothesis that preconditioning enhances GABA synapses to enable neuroprotection against subsequent lethal ischemic challenges.
Materials and methods
Slice Culture Preparation and Manipulations
The organotypic slice preparation has been described in detail (Raval et al, 2003; Xu et al, 2002). Briefly, 400 µm brain slices were obtained from rat pups of either sex between postnatal days 9 and 10. Slices were cultured for two weeks in a medium consisting of 25% heat-inactivated horse serum, 50% minimal essential medium, and 25% Hank's balanced salt solution. The final medium was supplemented with 5.5 mg/mL D-glucose and 1 mmol/L glutamine. Unless otherwise specified, all reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). This glucose supplement is 1 mg/mL less than our previous studies; however, as shown in Figure 5, the degree of neuron loss and neuroprotection is similar to our previous results. For both IPC and lethal ischemia in organotypic hippocampal slices, we used an established in vitro model consisting of combined oxygen and glucose deprivation (OGD; Raval et al, 2003; Xu et al, 2002). Oxygen-glucose deprivation duration was 15 mins for IPC and 40 mins for test ischemia. For OGD, oxygen is replaced with nitrogen and glucose with sucrose. Sham-IPC consisted of identical timing of solution changes with normal oxygen and glucose-containing medium. Pharmacological preconditioning (εPKC-PC; Raval et al, 2003) was induced using the Tat-conjugated peptide εPKC activator, ψεRACK (100 nmol/L, 15 mins; KAI Pharmaceuticals, San Francisco, CA, USA; Chen et al, 2001; Dorn et al, 1999). Sham εPKC-PC was accomplished by substituting the Tat-carrier peptide for Tat-εPKC. εPKC activity was inhibited during IPC by including the Tat-conjugated peptide inhibitor, εV1-2 (200 nmol/L; Gray et al, 1997), in the OGD medium. We conducted electrophysiology experiments and assessed cell death in parallel sets of slice cultures. All electrophysiology experiments were conducted 48 h after sham preconditioning, IPC or pharmacologic preconditioning. The electrophysiologist was blind to the experimental groups.
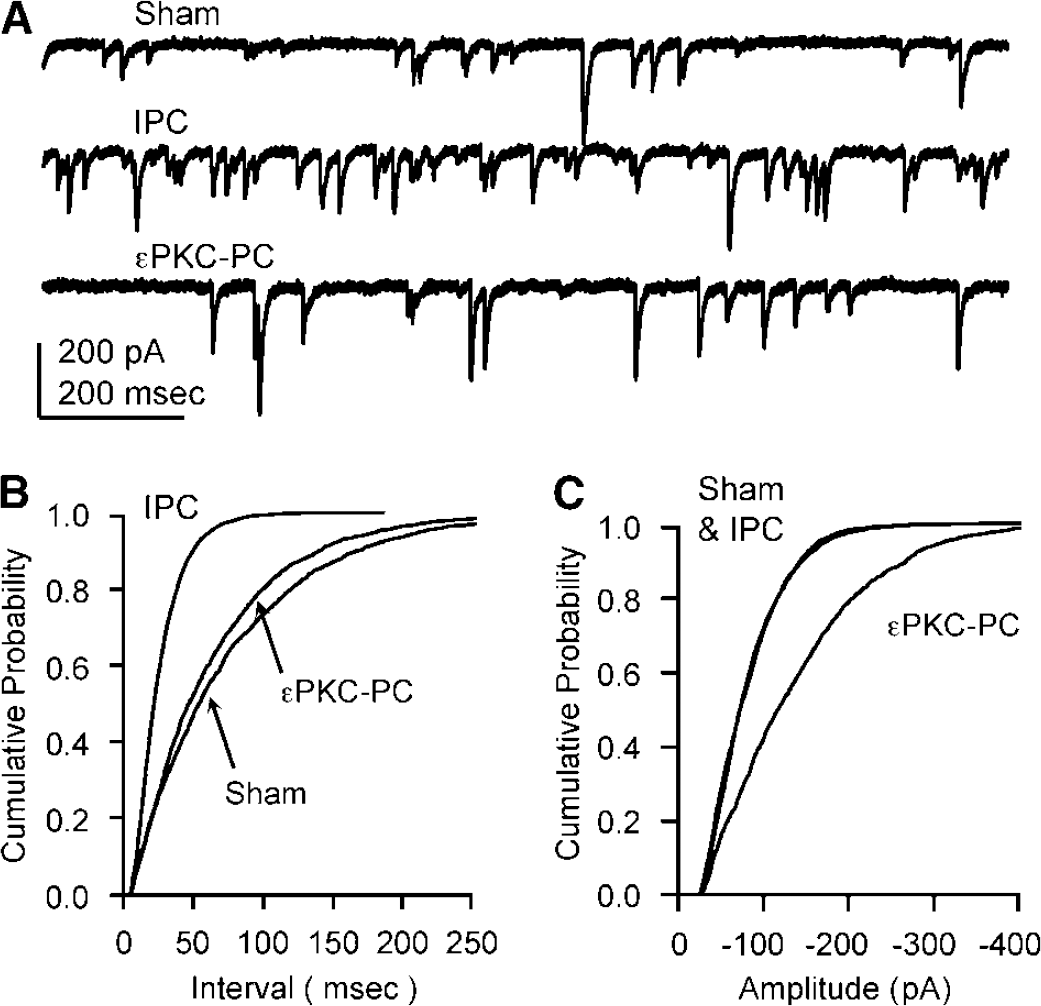
GABA miniature postsynaptic currents are enhanced after neuroprotective preconditioning. (
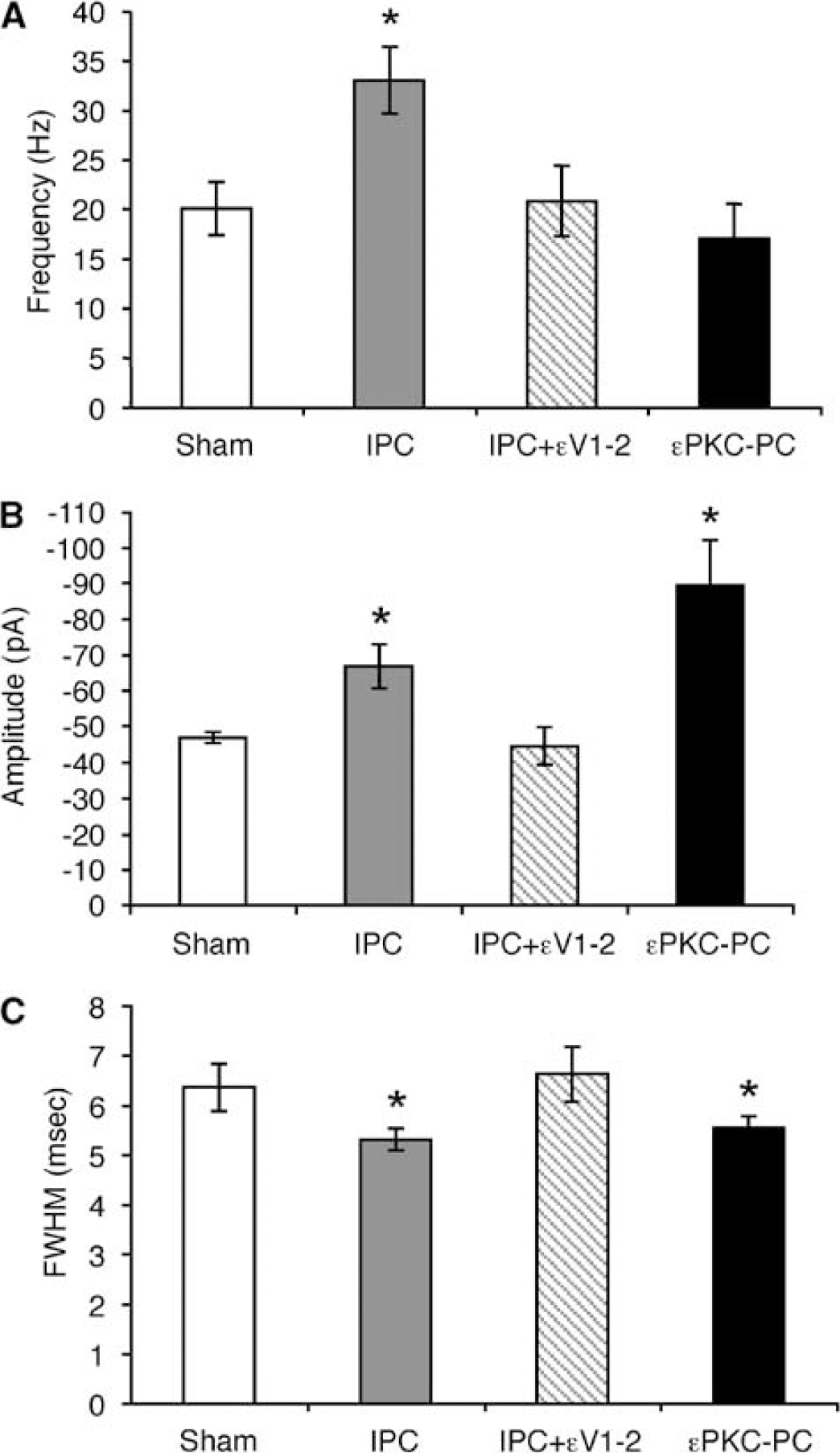
Summary of GABA mPSCs after preconditioning. (
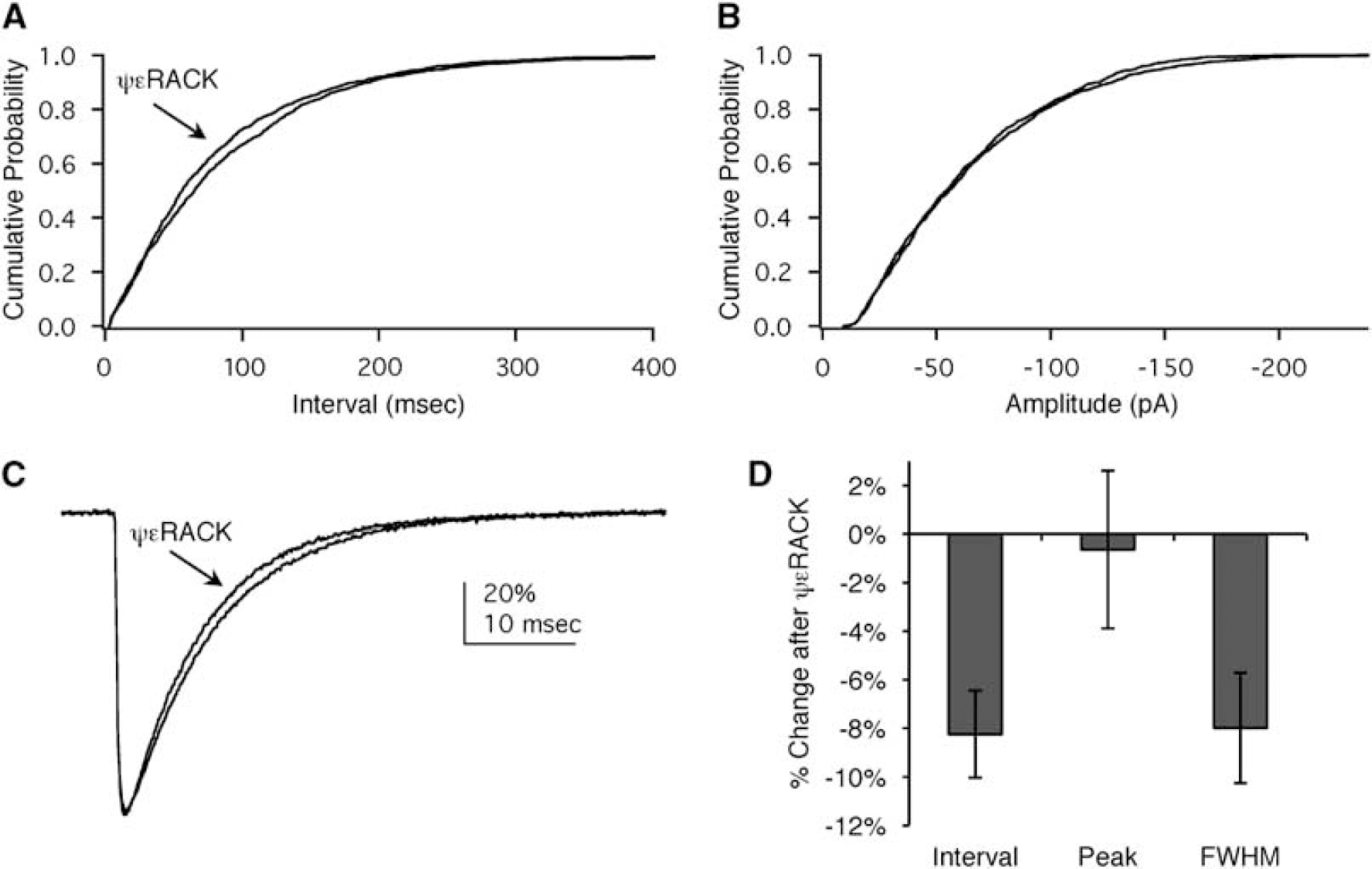
Acute activation of εPKC modifies GABA synapses. (
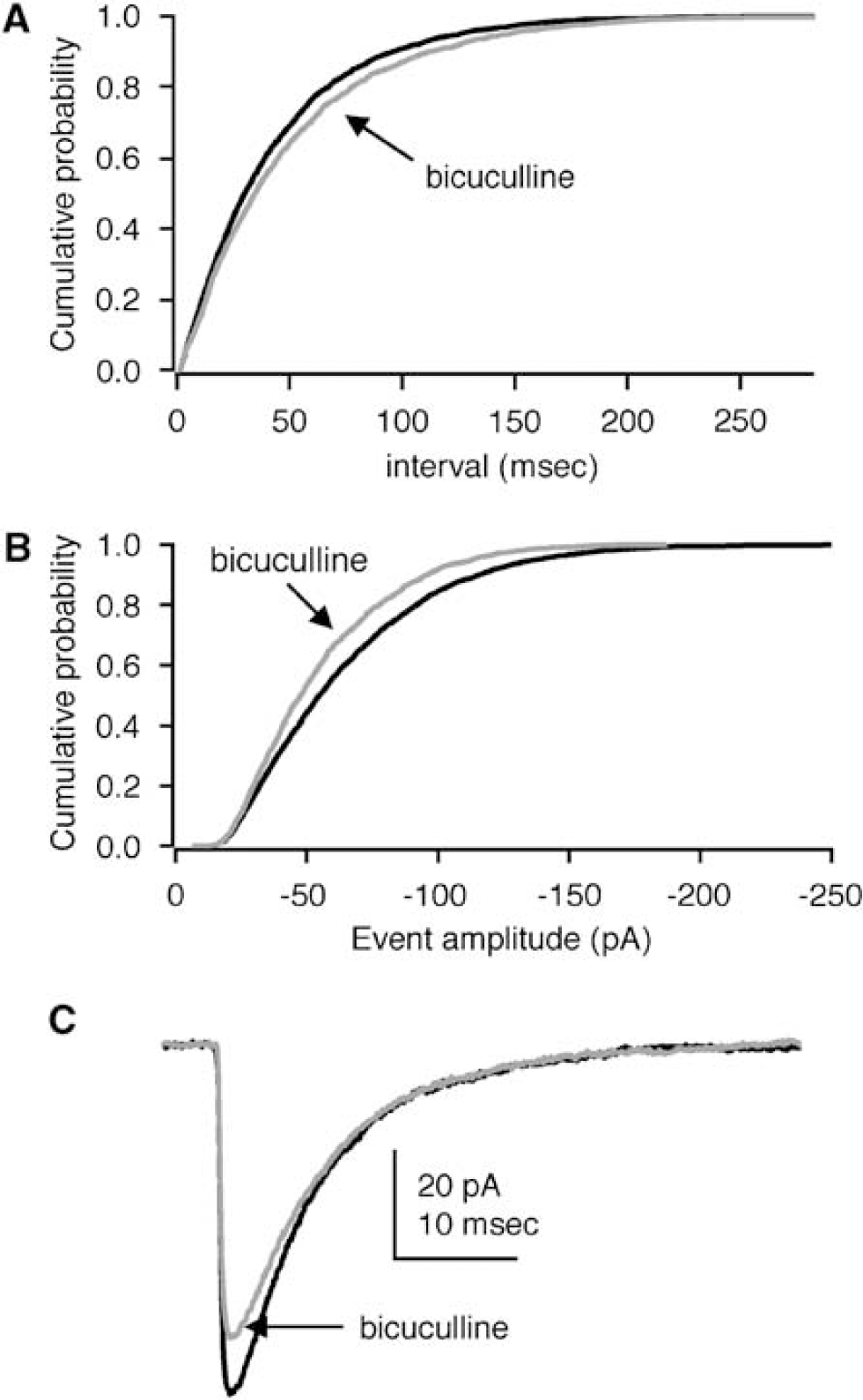
Mild antagonism of GABAA receptors decreases amplitude and frequency. (
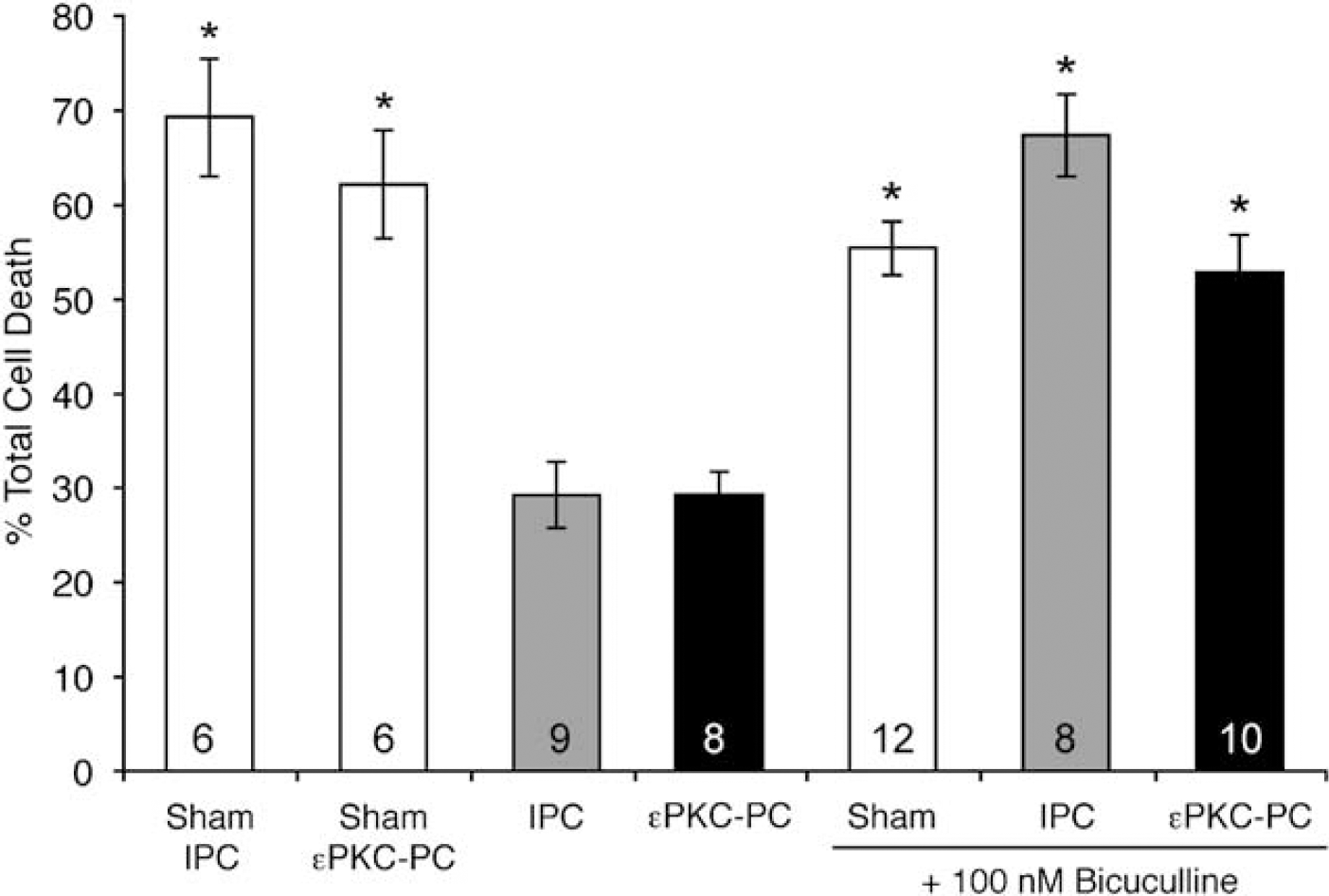
Mild antagonism of GABAA receptors abolished neuroprotection conferred by preconditioning. 48 h after preconditioning or sham treatment, slices are exposed to test ischemia (40 mins oxygen glucose deprivation, OGD). Both ischemic and εPKC-mediated pharmacologic preconditioning (IPC and εPKC-PC) significantly reduced cell death after OGD compared with sham IPC and εPKC-PC (sham IPC, sham εPKC-PC versus IPC, εPKC-PC; *P<0.05, n is indicated in each column of the graph). We tested the dependence of neuroprotection on changes in GABA synapses by weakly antagonizing the GABAA receptor during OGD. The concentration of bicuculline was selected that blocked ~20% of the synaptic current (100 nmol/L, Figure 3). In sham-preconditioned slices, bicuculline during OGD at this concentration did not significantly alter cell death compared with sham-preconditioned slices (sham +BIC versus sham IPC or sham εPKC-PC, n.s.). In ischemic or εPKC-PC slices, bicuculline during OGD abolished neuroprotection (IPC+BIC, εPKC-PC+BIC versus IPC, εPKC-PC; *P<0.05). ‘% Total cell death', as defined in the methods, reflects the ratio of propidium iodide (PI) staining 24 h after lethal ischemia (OGD) and PI staining 24 h after 100 µmol/L NMDA treatment (total cell death).
GABAA Miniature Postsynaptic Currents
To study GABA mPSCs, whole-cell voltage clamp recordings were obtained as previously described (DeFazio and Hablitz, 1998). CA1 pyramidal cells were targeted using infrared video microscopy. Slices were submerged in a circulating bath (4mL/min, 32°C). The extracellular solution contained (in mmol/L) 150 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.3 NaPO4, 25 NaHCO3, 10 glucose, 0.001 tetrodotoxin, 0.010 CNQX, 0.020 APV, and bubbled with 95%/5% O2/CO2. The pipette solution consisted of (in mmol/L) 135 CsCl, 10 HEPES, 5 EGTA, 4 MgATP, 1 NaGTP, pH 7.4 with NaOH, 320 mOsm. The small liquid junction potential was not corrected (~3 mV). Pipettes were 2 to 4MΩ. Series resistance (< 15MΩ) was not compensated because of the relatively small size of the currents (~100 pA). Recordings were rejected if the series resistance changed by more than 20% during the 2-min gap-free recordings sandwiched between tests of series resistance and membrane properties. Input resistance was > 50 and < 100MΩ; holding current, less than −500pA at −80 mV. Recordings outside this range were not included in analysis. One recording was obtained per slice.
Events were detected and analyzed with custom software written in IgorPro (Wavemetrics, Lake Oswego, OR, USA; Sullivan et al, 2003). This custom software detects events using the first derivative of the raw data. When the first derivative crossed a threshold value (10 pA/ms), the timing of this threshold crossing was used to identify the peak amplitude within 2 ms. If the peak amplitude exceeded a threshold of 5 pA, the event was accepted for further analysis. The time of the threshold crossing was also used for interevent interval calculation. All events were confirmed by eye by an investigator blind to the experimental groups. Identical detection parameters were used for all recordings.
The detection software measured the time interval between each detected event and the amplitude of each event. This analysis resulted in a list of interevent intervals and amplitudes corresponding to each event for each recording. The cumulative distribution (the values are sorted from smallest to largest values) of interevent intervals for representative cells from each group are shown in Figure 1B and for amplitudes in Figure 1C. In these cumulative probability plots, the x axis represents the interval and the y axis is the cumulative probability, i.e., at 0.5 cumulative probability, 50% of intervals are shorter and 50% are longer. A shift in the distribution to the left, as in the case of IPC in Figure 1B, indicates shorter/smaller values. We calculated frequency from the number of events occurring during the 2 mins acquisition period, i.e., frequency = number of events/120 secs. Using the average of all events detected for each cell, we measured the full-width-at-half-maximum (FWHM). This parameter reflects the duration of the GABA mPSC, which in turn is a measure of the affinity of the GABAA receptor for GABA. Longer durations indicate a higher affinity, and vice versa (see for example, DeFazio and Hablitz, 1998)).
Cell Death Assay with Propidium Iodide
Neuronal cell death was assessed by the fluorescence intensity of propidium iodide staining (a DNA stain that labels cells with compromised membranes; 2 µg/mL in medium, 1 h incubation) in the CA1 region before test ischemia (baseline), 24 h after test ischemia (OGD), and again 24 after 1 h exposure to 100 µm NMDA (total cell death; Raval et al, 2003). We previously showed that the propidium iodide staining protocol predicts neuronal cell death assessed using NeuN antibody staining of healthy neurons at 7 days after ischemia (Xu et al, 2002). Percent cell death is calculated from the average intensity of propidium iodide staining at each time point and the following ratio: (OGD—baseline)/(NMDA—baseline).
All data are summarized as mean±s.e. Unless otherwise specified, statistical significance was evaluated using ANOVA followed by the Games—Howell post hoc tests. Results were considered significant if P < 0.05.
Results
IPC Increased the Frequency of GABAA Miniature Postsynaptic Currents
We tested the hypothesis that IPC mediates neuroprotection through modifications of GABA synapses. In the CA1 region of organotypic hippocampal slices, we recorded GABA mPSCs 48 h (the time of peak neuroprotection) after sham preconditioning, IPC, or εPKC-PC (Raval et al, 2003). In the presence of ionotropic glutamate receptor antagonists to isolate GABAA receptor-mediated events and tetrodotoxin to block action potentials, prominent GABA mPSCs were detected in all CA1 hippocampal pyramidal neurons (Figure 1A). To confirm that these events are mediated by GABA acting at GABAA receptors, events were completely abolished by the GABAA receptor antagonist bicuculline (10 µmol/L, not shown).
To illustrate the impact of preconditioning in individual cells, the raw data for three representative recordings are shown in Figure 1A. Probability distributions (as described in the Materials and methods section) are used to plot the intervals and amplitudes of the individual recordings. The probability distribution of intervals is shown in Figure 1B for these three cells (group data are presented in Figure 2 and Table 1). After IPC, the increased number of events results in a decrease in the interval and a shift in the distribution toward smaller values in Figure 1B. In these representative individual recordings, εPKC-PC had no clear effect on the interval distribution.
Properties of GABA miniature postsynaptic currents
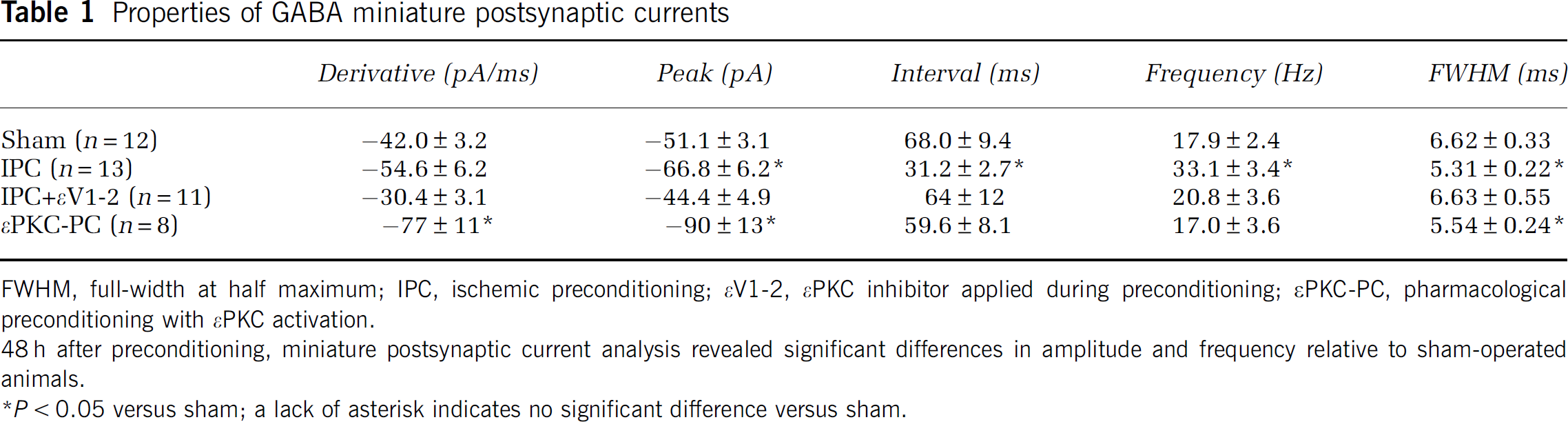
FWHM, full-width at half maximum; IPC, ischemic preconditioning; εV1-2, εPKC inhibitor applied during preconditioning; εPKC-PC, pharmacological preconditioning with εPKC activation.
48 h after preconditioning, miniature postsynaptic current analysis revealed significant differences in amplitude and frequency relative to sham-operated animals.
P<0.05 versus sham; a lack of asterisk indicates no significant difference versus sham.
Shorter intervals signify a higher frequency of GABA mPSCs. To compare data across groups, we calculated the overall frequency of events (the total number of events divided by the duration of the recording) for each cell in each treatment group. After IPC, the mean frequency of detected events was nearly doubled compared with either sham or εPKC-PC (Table 1 and Figure 2A). A significant increase in the frequency of GABA mPSCs suggested that IPC increased the number of synapses or release sites (e.g., Shao and Dudek, 2005). Note that these experiments were conducted in the presence of the sodium channel antagonist tetrodotoxin. This eliminated the influence of presynaptic excitability on the frequency of events.
We also detected a significant increase in the amplitude of GABAA mPSCs (Figure 2B). An increase in amplitude can directly affect the frequency of detected events. If the smallest events are not detected in the background signal noise, an increase in amplitude could result in an apparent increase in frequency. An increase in amplitude may allow these events to reach the detection threshold. However, an even larger change in amplitude was associated with εPKC-PC (see below, Figure 2), with no effect on the frequency.
Inhibition of εPKC Abolished Changes in GABAA mPSCs
Using cell death as the assay endpoint, εPKC is both necessary and sufficient for the induction of IPC (Raval et al, 2003). We next tested the hypothesis that εPKC activation is required for the changes in GABA synapses observed after IPC. We inhibited εPKC activity using the inhibitor peptide εV1-2 (200 nmol/L) during the induction of IPC (15 mins OGD). Inhibition of εPKC during IPC abolished the changes in frequency and amplitude of GABA synapses observed at 48 h (Figure 2; compare IPC with IPC + εV1-2). These results suggested that the impact of IPC on GABA synapses depended on downstream effectors and targets of the εPKC signaling pathway.
Preconditioning with εPKC Activation Increased the Amplitude of GABAA mPSCs
Pharmacologic preconditioning via εPKC activation (εPKC-PC) was achieved by exposing organotypic hippocampal slices to a selective peptide activator of εPKC, (ψεRACK, 100 nmol/L, 15 mins; Chen et al, 2001; Dorn et al, 1999; Raval et al, 2003). εPKC-PC conferred neuroprotection similar to that of IPC (see below; Figure 5; see also Lange-Asschenfeldt et al, 2004; Raval et al, 2003).
As with IPC, peak neuroprotection occurs 48 h after εPKC-PC. We examined the impact of εPKC preconditioning on GABA synapses at 48 h. In contrast to IPC, the frequency of GABAA mPSCs recorded from εPKC-preconditioned slices was not significantly different from sham-preconditioned or ischemic-preconditioned slices (εPKC-PC versus sham or IPC in Figure 2A and Table 1). However, the amplitude distributions were shifted toward larger values after εPKC-PC (Figure 1C). εPKC-PC significantly enhanced the mean amplitude by >40% relative to sham-treated slices (Table 1; Figure 2B). An increase in GABA mPSC amplitude suggested an increase in the postsynaptic density or affinity of GABAA receptors and/or a change in the amount of GABA released per vesicle (Vautrin and Barker, 2003).
IPC and εPKC-PC Affect the Duration of GABA mPSCs
In CA1 hippocampal pyramidal cells, postsynaptic GABAA receptors are not saturated by the release of GABA from a single vesicle (Hajos et al, 2000). Thus, a potential increase in the postsynaptic density of GABAA receptors would not result in increased amplitude of GABA mPSCs. Because the postsynaptic receptors are not saturated, pharmacologic agents known to increase GABAA receptor affinity, e.g., zolpidem, increase both the duration and amplitude of GABA mPSCs (e.g., DeFazio and Hablitz, 1998; Hajos et al, 2000; Perrais and Ropert, 1999). Because of the increase in affinity, more postsynaptic receptors are bound by GABA, which in turn increases event amplitude. We hypothesized that the increased amplitude after preconditioning is because of increased affinity of the GABAA receptors. To assess the affinity of GABAA receptors after preconditioning, we measured the duration of averaged mPSCs using FWHM (please see Materials and methods). The time course of GABA mPSCs is directly related to the binding and unbinding of GABA to the receptor (e.g., Perrais and Ropert, 1999). Based on our hypothesis, we anticipated that FWHM would be increased in conjunction with the increased amplitude. On the contrary, we found that the mean FWHM was actually decreased after both forms of preconditioning (Figure 2C; Table 1). Although these results are consistent with a subunit change in postsynaptic GABAA receptors (as in Sommer et al, 2002, 2003), our hypothesis was incorrect: an increase in affinity of the postsynaptic GABAA receptors was not responsible for the increase in GABA mPSC amplitude after IPC. Therefore, our results are consistent with an increase in the amount of GABA released from each vesicle after preconditioning.
Acute Modulation of GABA mPSCs Via Direct εPKC Activation
In light of recent reports that εPKC mediates the acute effects of ethanol and cortisol releasing factor on GABA release (Bajo et al, 2008), we tested the hypothesis that activation of εPKC acutely affects GABA mPSCs in organotypic hippocampal slices. Direct activation of εPKC with 100nmol/L ψεRACK during whole-cell recordings from control slices significantly decreased the interval and the FWHM (P < 0.05, paired two-tailed t-tests, n = 4) with no effect on amplitude (Figure 3). The decrease in interval reflected an 8.6 ± 1.6% increase in the frequency of events (P < 0.05, paired two-tailed t-test, n = 4). These small but significant changes showed that activation of εPKC rapidly affects both presynaptic release probability (increased frequency) and postsynaptic receptors (decreased duration/affinity).
Neuroprotection After Preconditioning was Abolished by Bicuculline
As both IPC and εPKC-PC significantly modified GABA synapses in electrophysiology assays at the time of peak neuroprotection, we tested the hypothesis that these functional changes conferred neuroprotection during test ischemia. First, we identified an agent that would decrease the amplitude of GABA mPSCs without completely abolishing GABAergic transmission. Bicuculline is a competitive antagonist of the GABAA receptor with an IC50 of ~2 to 3µmol/L (see e.g., McCartney et al, 2007). Full blockade of GABA mPSCs was accomplished with 10 µmol/L bicuculline (not shown).
We tested the impact of mild antagonism of GABAA receptors on GABA mPSCs. The acute effect of 100 nmol/L bicuculline is illustrated in Figure 4. An increase in the interval distribution is illustrated in Figure 4A. The shift to larger intervals in bicuculline likely reflects the smallest events falling below the detection threshold. As expected, bicuculline decreased the amplitude of GABA mPSCs, i.e., shifted the amplitude distribution to the left (Figure 4B). This reflects the impact of the low concentration of the competitive antagonist on GABA binding to the GABAA receptor. Averaged events indicated the overall effect of bicuculline: a small reduction in amplitude (Figure 4C). At 100nmol/L bicuculline, our population data show that the GABA mPSC mean amplitude was reduced by an average of 21.2 ± 6.0% whereas the mean frequency was reduced by 21.6 ± 7.3% (n = 4). Thus, mild antagonism of GABAA receptors with 100nmol/L bicuculline diminished the amplitude of GABA mPSCs.
Armed with these results, we next tested our hypotheses regarding the role of GABA synapses in neuroprotection. As previously reported, robust neuroprotection against test ischemia (40 mins OGD) was conferred 48 h after either IPC or εPKC-PC (Raval et al, 2003). Both IPC and εPKC-PC significantly decreased cell death after test ischemia (Figure 5).
Next, we tested the effect of mild blockade of GABA synapses on neuroprotection conferred by preconditioning. In sham-preconditioned slices, weak blockade of GABAA receptors with 100 nmol/L bicuculline during test ischemia (40 mins OGD) did not affect neuronal cell death (Figure 5; white bar sham + BIC). In our paradigm, test ischemia yields 60% to 70% of the total cell death observed after NMDA treatment. Thus, this parameter can decrease (as with preconditioning) or potentially increase if a treatment (e.g., bicuculline) exacerbates cell death. Mild antagonism of GABAA receptors with 100 nmol/L bicuculline did not exacerbate cell death.
Application of 100 nmol/L bicuculline during OGD abolished the neuroprotection conferred by either IPC or εPKC-PC (IPC + BIC and εPKC-PC +BIC; Figure 5). Combined with our electrophysiology data, the abolition of neuroprotection by mildly antagonizing GABAA receptors supports an important neuroprotective role for GABA synapses in both ischemic and εPKC-mediated preconditioning in organotypic hippocampal slices.
Discussion
Our results suggest that endogenous mechanisms already in place in the brain have the capacity to confer dramatic neuroprotection by presynaptic and/or postsynaptic modification of GABA synapses. 48 h after preconditioning, IPC significantly enhanced the frequency of GABAA miniature postsynaptic currents in CA1 pyramidal neurons. This is consistent with an increase in the number of presynaptic GABA release sites or an increase in release probability. Both IPC and εPKC-PC increased the amplitude of GABA mPSCs. Our study is the first to show enhancement of hippocampal GABA synapses 48 h after direct and transient activation of εPKC. In the context of neuroprotection, mild antagonism of GABAA receptors abolished neuroprotection conferred by preconditioning—consistent with the hypothesis that these preconditioning-induced changes in the GABA synapse play a necessary role.
To examine the role of GABA synapses in neuroprotection, we used miniature postsynaptic current analysis to study changes in GABA synapses at the time of peak neuroprotection (48 h after preconditioning). After pharmacologic blockade of glutamate receptors and in the presence of tetrodotoxin to block action potentials, GABAergic synaptic currents are referred to as miniature postsynaptic currents (GABA mPSCs or ‘minis’ (Bekkers and Stevens, 1989; Edwards et al, 1990)). The frequency of GABA mPSCs reflects the number of synapses and the release probability (see for example, Shao and Dudek, 2005). The amplitude of events is determined by presynaptic factors (e.g., the amount of GABA in each vesicle, for a recent discussion see (Vautrin and Barker, 2003)) and postsynaptic factors including the number and subunit composition of postsynaptic receptors (e.g., DeFazio and Hablitz, 1998; Del Castillo and Katz, 1954; Wan et al, 1999).
Previous in vitro studies have shown that activation of postsynaptic GABAA receptors can protect neurons during lethal ischemia. Activation of GABAA receptors during lethal ischemia has been proposed as a mechanism of acute isoflurane neuroprotection (Bickler et al, 2003). Antagonism of GABAA receptors during lethal ischemia abolished isoflurane neuroprotection. Muscimol (a GABAA receptor agonist) mimicked the effect of isoflurane (Bickler et al, 2003). Thus, activation of GABAA receptors during lethal ischemia can confer neuroprotection. It is important to point out that this approach represents an acute treatment during lethal ischemia that directly activates postsynaptic GABAA receptors. In this context, our data suggest that IPC induces presynaptic modifications in GABA synapses via endogenous signaling pathways. In contrast to the acute neuroprotection afforded by GABA agonists and modulators, IPC confers neuroprotection in a time frame of 24 to 72 h after the treatment.
IPC and εPKC-PC Converge at the GABA Synapse
To identify functional neuroprotective alterations in GABA synapses at the time of peak neuroprotection, we targeted CA1 hippocampal pyramidal cells at 48 h after preconditioning. We showed that at the time of peak neuroprotection, IPC doubled the frequency of GABA mPSCs in these vulnerable neurons. The increased frequency of GABAA mPSCs observed 48 h after IPC suggests an increase in the number of release sites; however, we cannot rule out the possibility of other presynaptic enhancements of GABA synapses that result in increased probability of vesicle release. Increased release of GABA (via more GABA per vesicle, more release sites or enhanced probability of release) correlates well with in vivo microdialysis experiments that showed a tremendous increase in GABA release during lethal ischemia after IPC (Dave et al, 2005). This previous result was associated with a trend toward increased activity of the rate-limiting enzyme in the GABA synthesis pathway, glutamatic acid decarboxylase.
Both forms of preconditioning studied here evoked changes at the GABA synapse. εPKC-PC with the peptide ψεRACK nearly doubled the amplitude of GABA mPSCs with no effect on frequency. εPKC is associated with both GABA and glutamate synapses (Bajo et al, 2008; Olive and Hodge, 2000; Saitoh et al, 2001). The increased amplitude of GABAA mPSCs after preconditioning with εPKC is consistent with a postsynaptic increase in affinity of GABAA receptors (Hajos et al, 2000), or a presynaptic change such as increased GABA concentration in vesicles (Rahamimoff and Fernandez, 1997). Instead of increased affinity, we observed a decrease in the affinity of postsynaptic GABAA receptors as indicated by the decrease in the FWHM of GABA mPSCs. Therefore, our data suggest that εPKC-PC increased the amount of GABA in each vesicle.
Functional Divergence
With cell survival as an endpoint, it is difficult to distinguish IPC from εPKC-PC because survival can occur via different pathways and targets with the same apparent outcome. In this context, εPKC activation is both necessary and sufficient for the induction of neuroprotection during IPC (Raval et al, 2003). At the level of GABA mPSCs, there was a clear distinction between the two forms of preconditioning: IPC increased the frequency and amplitude, whereas εPKC-PC increased only the amplitude. In addition to the εPKC signaling pathway, IPC activates other signaling pathways that contribute to the changes in GABA synapses. It is likely that these other pathways also contribute to the neuroprotective changes in GABA synapses. If the number of GABA synapses can be increased in combination with the profound increase in amplitude observed with εPKC-PC, even greater neuroprotection might be conferred. Thus, dissection of the multiple signaling pathways evoked by IPC and their impact on GABA synapses is an important target for future studies.
Acute Modulation via εPKC
Acute εPKC modulation of GABAergic synaptic activity been reported previously (Bajo et al, 2008; Proctor et al, 2003). In knockout mice lacking εPKC expression, GABA postsynaptic currents were rapidly potentiated by ethanol in the hippocampus (Proctor et al, 2003). This effect was absent in the wild-type control group. In the wild-type group, εPKC apparently occludes the impact of ethanol on GABAergic transmission. However, this effect of εPKC varies with brain region. For example, in the central amygdala a presynaptic role for εPKC was identified (Bajo et al, 2008). εPKC knockout abolished the increase in the frequency of GABA mPSCs induced by ethanol or cortisol releasing factor. These results indicated that εPKC mediates ethanol and cortisol releasing factor modulation of GABA synapses via presynaptic effectors.
In the hippocampal organotypic slice, our results showed acute modulation of GABA mPSCs after direct activation of εPKC with ψεRACK (Figure 3). Activation of εPKC affected both presynaptic and postsynaptic components of the GABA synapse. Both the interval and the FWHM were decreased by ~8% with no effect on amplitude (Figure 3D). The effect on interval suggested a presynaptic target: an increase in the probability of release. The decrease in the FWHM indicated a decrement in affinity of the GABAA receptor for GABA, i.e., a postsynaptic effect. However, none of the recordings showed an increase in amplitude similar to that observed 48 h after IPC or εPKC-PC. Also, the acute impact of εPKC on the frequency of events (~9%) was much smaller than the >50% increase observed after IPC (Figure 2A). In the context of preconditioning, these results suggest that εPKC alters GABA synapses during the 48 h after preconditioning via activation of downstream signaling pathways.
Consequences for Neuroprotective Therapeutics
Weak blockade of GABAA receptors abolished neuroprotection conferred by either IPC or εPKC-mediated preconditioning (Figure 5). This result showed that preconditioning activates endogenous adaptive mechanisms to confer powerful neuroprotection via changes in GABA synapses. Although systemic modulation of GABAA receptors is effective for anxiety and many forms of epilepsy, clinical trials using systemic GABAA receptor modulators as neuroprotective agents failed (Lodder et al, 2005; Lyden et al, 2002). Our data suggest that targeting therapeutics to enhance endogenous release of GABA at vulnerable CA1 hippocampal pyramidal cells may provide a superior outcome. For example, clinical trials are currently evaluating gene therapy to express glutamic acid decarboxylase (the primary GABA synthesis enzyme) in the glutamatergic subthalamic nucleus—a primary excitatory input to the substantia nigra. In animal models and patients with Parkinson's disease, hijacking the excitatory inputs to the substantia nigra with the inhibitory GABA synthesis enzyme enhances GABA release, and showed both tolerability and improved outcome (Kaplitt et al, 2007; Luo et al, 2002).
In addition to viral hijacking of glutamate synapses, specific signaling pathways could be targeted to replicate the preconditioning-induced enhancement of GABA synapses in vivo. In contrast to global manipulation of GABA release or GABA receptor activation, it may be necessary to target specific synapses or receptor subtypes to obtain neuroprotection without adverse effects. Thus, greater understanding of the structure and function of GABA synapses in disease and neuroprotective states will lead to novel therapies. Therapeutics targeting the presynaptic component of GABA synapses on vulnerable CA1 hippocampal pyramidal cells may significantly improve neurologic outcome after stroke, cardiac arrest, or surgical treatments that result in cerebral ischemia.
Footnotes
Acknowledgements
The authors thank Dr Tatjana Abaffy and Dr Suzanne M Moenter for critical reading of early versions of the article.
Disclosure/conflict of interest
Each of the authors has declared no conflict of interest.