
Abstract
Stress proteins are induced after a variety of neuronal injuries. The inducible 72-kDa heat shock protein (hsp70) is a stress protein that protects neurons from glutamate toxicity in vitro. Hsp70 has also been proposed to underlie the phenomenon of ischemic tolerance whereby brief sublethal intervals of global ischemia protect the hippocampus from subsequent lethal prolonged ischemia. To determine if the phenomenon of tolerance occurs in cortex after focal ischemia, the rat middle cerebral artery (MCA) was occluded by the suture method. Three 10-min intervals of transient ischemia (3 × 10-isc) separated by 45-min periods of reperfusion made up the most effective paradigm of preconditioning ischemia studied, and substantially reduced the volume of infarction 72 h after subsequent 100-min MCA occlusion. This approach induced protection if the interval between the 3 × 10-isc and the 100-min ischemia was 2, 3, or 5 days but not 1 or 7 days. Three 10-min intervals of transient ischemia alone produced minimal histological changes in the cortex at 72 h. Moreover, there were no significant changes in regional cerebral blood flow in the tolerant regions at 72 h after 3 × 10-isc before or during MCA occlusion. To explore the role of stress proteins in the induction of tolerance, expression of hsp70 and the glucose-regulated proteins grp75 and grp78 were studied. Samples from tolerant regions of the brain that had undergone preconditioning ischemia were evaluated at 1, 2, 3, 5, 7, and 14 days after 3 × 10-isc by Western blot analysis. The time course of hsp70 expression most closely correlated with tolerance. Hsp70 protein expression increased during times when tolerance was present (at 2–5 days) but did not increase thereafter (at 7 and 14 days). However, hsp70 was also increased before tolerance was present (at 1 day). Immunocytochemistry showed that hsp70 protein was expressed in neurons in the tolerant regions 24 h after 3 × 10-isc and was expressed in both neurons and glia after 72 h. Although immunocytochemistry suggested that there was increased neuronal expression of grp75 and grp78, no significant differences were found in protein expression as determined by Western blot before (at 1 day), during (at 2–5 days), and after (at 7 days and thereafter) tolerance. Thus, the time course of grp75 and grp78 expression did not correlate with that of tolerance. This model of ischemic tolerance is a useful method by which mechanisms of endogenous neuroprotection may be explored.
Ischemic tolerance is the phenomenon whereby a preconditioning ischemia of brief duration, inadequate to infarct the brain, protects it from subsequent severe ischemia. Global ischemia was the first model used to describe ischemic tolerance, which protected hippocampal neurons from subsequent ischemic necrosis (Kato et al., 1991; Kitagawa et al., 1991). These initial observations of ischemic tolerance in the brain were found in global ischemia models, where protection is limited to the hippocampus. We have shown previously that brief intervals of global ischemia decrease the size of cortical infarcts after middle cerebral artery (MCA) occlusion in the rat and that this protection is associated with the induction of heat shock protein hsp70 (Simon et al., 1993). These results suggest that ischemic tolerance is also relevant to cortical neuronal injury in focal ischemia.
Heat shock proteins such as hsp70 have attracted major interest as potential neuroprotectants because their prior induction, by a variety of stimuli, protects cultured cells against subsequent injury. This protective effect requires the synthesis of new protein and RNA (Rordorf et al., 1991). Furthermore, preinduction of hsp70 protects neurons from glutamate toxicity (Lowenstein et al., 1991). Since ischemic tolerance is accompanied by hsp70 protein expression, it has been proposed that hsp70 expression may be responsible for tolerance (Kato et al., 1991; Kitagawa et al., 1991; Nakata et al., 1992; Aoki et al., 1993; Liu et al., 1993; Nishi et al., 1993; Parsell and Lindquist, 1993).
To determine if temporary focal ischemia, like global ischemia, induces ischemic tolerance in the cortex, brief periods of ischemia were induced by advancing a suture into the internal carotid artery (ICA). At various intervals later, the MCA was occluded for 100 min, producing near-complete infarction of the MCA territory in naive rats. The duration of brief ischemia that produces optimal reduction in infarct size and the effect of this optimal preconditioning ischemia on neuropathology and regional cerebral blood flow were determined. The time course of expression of the stress protein hsp70 and the glucose-regulated proteins grp75 and grp78 were characterized after the preconditioning ischemia.
METHODS
Temporary focal ischemia
Focal cerebral ischemia was induced in rats by reversible occlusion of the MCA, using the suture method previously described (Longa et al., 1989), with modifications. A total of 260 male Sprague–Dawley rats (280–300 g) were anesthetized with chloral hydrate (400 mg/kg i.p.). Rectal temperature was monitored and kept at 37 ± 0.5°C with a heating pad, and left temporalis muscle temperature was kept at 37.5 ± 0.2°C with a heating lamp. One femoral artery was cannulated to monitor blood pressure, arterial blood gases, and blood glucose. Under the operating microscope, the bifurcation of the common carotid artery was exposed, the external carotid artery was coagulated and dissected distally, and the ICA was isolated and separated from the vagus nerve. The extracranial branch of the ICA was then ligated close to its origin with a 5–0 silk suture. To block the origin of the MCA, a 3–0 monofilament nylon suture, its tip rounded by heating, was introduced into the ICA lumen through the stump of the external carotid artery and gently advanced into the ICA 20–22 mm past the common carotid artery bifurcation. After completion of the proximal MCA occlusion, the suture was gently withdrawn to permit reperfusion. The external carotid stump was ligated and the wound closed, and the rats were maintained under temperature control until they were returned to animal cages ∼1 h later.
At a later time, a second MCA occlusion was performed. The rats were anesthetized as already described. The wound was reopened, a microvascular clip was applied to the proximal origin of the external carotid artery, and the stump ligation was undone. The monofilament nylon suture was reinserted into the ICA through the stump for 100 min, a duration of ischemia that produces almost complete infarction of the MCA territory in naive rats (Chen et al., 1991). Sham-operated rats underwent identical surgery but did not have the suture inserted or the ICA manipulated.
Infarct volume measurement
Animals were killed at either 24 or 72 h after MCA occlusion by an overdose of chloral hydrate injected i.p.; brains were rapidly removed and sectioned coronally at 2-mm intervals. Sections were immersed in 2% 2,3,5-triphenyltetrazolim hydrochloride (TTC) in buffered Ringer's solution at pH 7.4 (37.5°C) for 20 min and then transferred to 4% formaldehyde buffer for 15 min before photography (Bederson, et al., 1986). Photographs of the six sections were then analyzed for infarct size by a blinded observer using an image-analysis system (MCID, St. Catherine's, Ontario, Canada). The hemispheric infarct area in each section was calculated by subtracting the area of normal, TTC-staining brain in the ipsilateral ischemic hemisphere from the contralateral nonischemic area. This technique minimizes the effect of edema on measurement of infarct size (Swanson et al., 1990). Infarct volume was then calculated by summing the infarct areas over all sections and multiplying by the slice thickness.
For confirmation of the TCC determinations of infarct volume, some experiments were repeated and formal histology was obtained. Animals were killed with chloral hydrate, and their brains were removed, frozen in 2-methyl-butane at −30°C, and transferred to powdered dry ice. Brains were sectioned coronally into 20-μm-thick slices on a cryostat every 0.5 mm from the anterior limit of the caudate to the posterior hippocampus (20 sections per brain). Sections were stained with cresyl violet. The infarct volume was determined using the method already described.
Histopathology
For assessment of microscopic cytopathology, rats were anesthetized with chloral hydrate and perfused via the ascending aorta with 100 ml of heparinized Ringer's solution followed by 500 ml of formalin in 0.1 M phosphate buffer solution (pH 7.4). The brains were removed and postfixed in 10% buffered formalin for 2 weeks. They were then embedded in paraffin, and 6-μm-thick coronal sections were taken at three coronal levels: +2.2, −0.3, and −2.8 mm from bregma. Sections were stained with cresyl violet. Brain injury was assessed and graded using a 0–4 scale (Pulsinelli et al., 1982): 0, normal brain; 1, scattered neurons damaged; 2, moderate number of neurons damaged; 3, many neurons damaged; and 4, majority of neurons damaged.
Cerebral blood flow measurement
Quantitative [14C]iodoantipyrine autoradiography was used to determine changes in regional cerebral blood flow (rCBF) after a period of focal ischemia sufficient to result in tolerance. Twenty-four rats underwent either a sham operation or three 10-min intervals of brief ischemia. The rCBF was determined 72 h later, either just before or 30 min after the onset of the secondary MCA occlusion (six rats per group). Surgery was performed in all animals under the same anesthetic conditions already described. Ventilation was used to ensure uniform normoxia and normocapnia. A femoral artery and a femoral vein were cannulated, and 20 μCi of [14C]iodoantipyrine (Du Pont, Boston, MA, U.S.A.), dried and diluted in 0.5 ml saline, was infused i.v. for 30 s by an infusion pump. During the infusion, seven 50-μl samples of free-flowing arterial blood from the femoral artery catheter were collected in heparin-coated sample tubes. The [14C]iodoantipyrine concentration in the blood samples was determined by liquid scintillation counting after 24 h of decolorization in a mixture with 1 ml of tissue and gel solubilizer. The rats were decapitated 30 s after the start of infusion. The brains were quickly removed and frozen in −30°C 2-methyl-butane and then in powdered dry ice. Each brain was serially sectioned into 20-μm-thick slices every 0.5 mm on a cryostat at −20°C; the sections were air-dried before exposure to x-ray film (Kodak NMC-1, Rochester, NY, U.S.A.) with an autoradiographic 14C standard microscale (Amersham, San Diego, CA, U.S.A.) in x-ray cassettes for 1 week. Quantitative rCBF for each brain was determined by means of a computer-based image analysis system (MCID).
Western analysis
Brain tissue, dissected from cortical regions (Fig. 5A) where tolerance was induced following three 10-min intervals of MCA occlusion and a variety of durations of reperfusion, was homogenized and lysed in 0.1 M NaCl, 0.01 M Tris–HCl (pH 7.6), 0.001 M ethylenediaminetetracetate (EDTA) (pH 8.0), 1 μg/ml aprotinin, and 100 μg/ml phenylmethylsulfonyl fluoride (PMSF). Lysates were cleared by centrifugation at 14,000 g for 30 min at 4°C and boiled at 100°C in sodium dodecyl sulfate (SDS) gel-loading buffer (100 mM Tris–HCl, 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue, and 20% glycerol) for 6 min before being run on a 12% SDS-polyacrylamide gel. Western blots were performed as previously described (Sambrook et al., 1989). The transferred polyvi-nylidene difluoride (PVDF) membrane was incubated in the primary antibodies (monoclonal antibodies against hsp70, grp75, or grp78) in a dilution range of 1:500 to 1:1,000 at 4°C overnight. This step was followed by three washes in washing buffer (0.1% Tween-20, 0.5% bovine serum albumin, and 1% nonfat dry milk in phosphate-buffered saline buffer) and then incubated in the alkaline-phosphatase (AP)-conjugated goat antimouse secondary antibodies at room temperature for 60 min. Chemiluminescent substrate (CSPD, 25 mM) was applied to the side of the membrane containing the blotted proteins. The blot was wrapped in plastic wrap and exposed to a Kodak X-OMAT film. The film was developed and the band optical density was measured by the MCID image system.
Effect of altering time variables of pretreatment with brief focal ischemia on infarct size after subsequent 100-min MCA occlusion.
Effect of three 10-min periods of ischemia pretreatment on infarct size (determined 72 h after 100-min ischemia) in serial frozen sections.
Representative photomicrographs from brains removed either 24 h (
Representative [14C]iodoantipyrine autoradiographs at +2.2 mm (
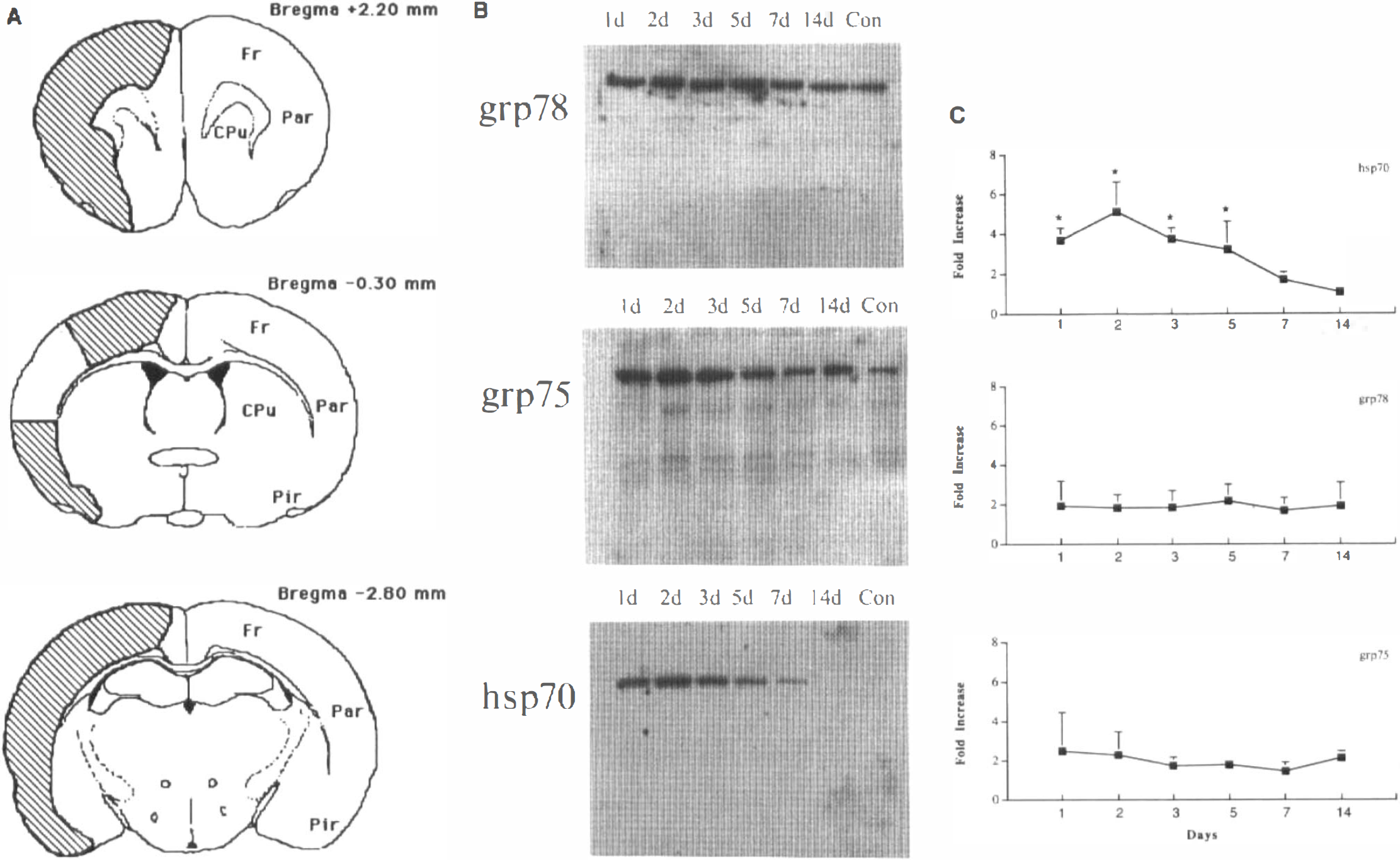
Expression of stress proteins after 3 × 10-isc in regions of brain where tolerance is induced.
Immunocytochemistry
Animals were killed 24 or 72 h after three 10-min intervals of ischemia and perfused via the ascending aorta with normal saline followed by paraformaldehyde in phosphate buffer (pH 7.4). The brains were removed, postfixed for 24 h, sectioned (100 μm) on a vibratome (Vibratome, Series 1000, Ted Pella, Inc., CA), and reacted with a monoclonal antibody to hsp70 (Amersham) and the avidin-biotin-peroxidase method previously described (Sharp et al., 1991). Alternative sections were immunoreacted with monoclonal antibody to grp75 or grp78 (Stressgen, Victoria, Canada) or without primary antibody as a control.
Statistics
The statistical significance between groups was determined with analysis of variance (ANOVA). Posthoc testing used the Bonferroni t test, and p < 0.05 was accepted as statistically significant. Differences in physiological parameters and rCBF were analyzed using repeated-measures ANOVA. All values are expressed as means ± SD.
RESULTS
Infarct volume
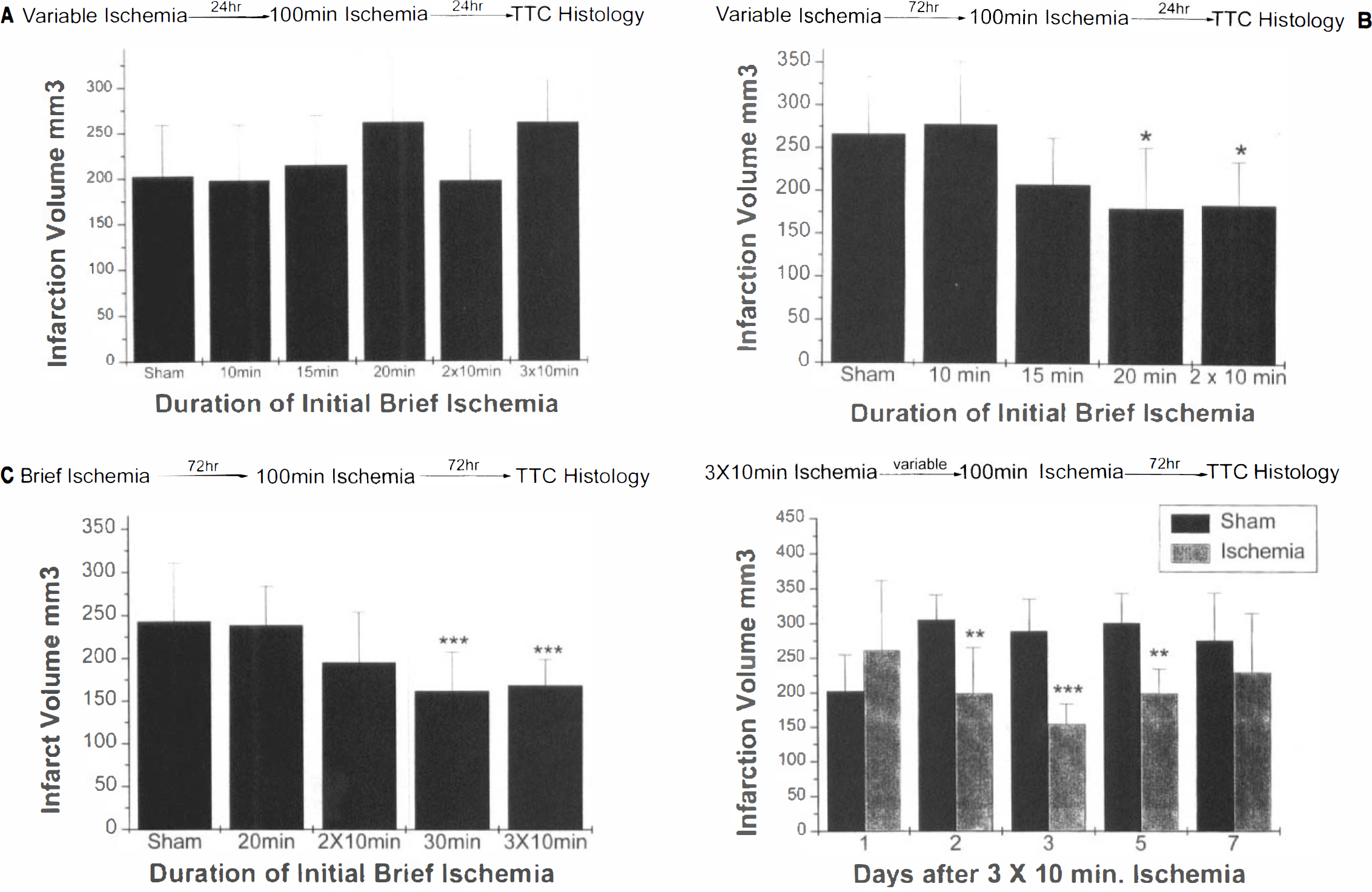
Previous experiments in global ischemia suggested that a 24-h interval between pretreatment conditioning ischemia and the prolonged ischemic insult was necessary for the induction of tolerance. These studies also suggested that multiple brief pulses of ischemia induced a greater degree of protection than did the same total duration of ischemia delivered as a single event (Kitagawa et al., 1990; Kirino et al., 1991). Rats were randomly divided into six groups (n = 6 per group). On day 1 the animals received either a sham operation, MCA occlusion for 10, 15, or 20 min, or two 10-min intervals of ischemia (2 × 10-isc) or three 10-min intervals of ischemia (3 × 10-isc) separated by 45 min. Twenty-four hours later, all animals underwent MCA occlusion for 100 min, a duration of ischemia that causes reproducible infarction in the MCA territory. Brains were removed 24 h after the 100-min ischemia to determine infarct volume using the TTC method. Figure 1A charts the results of this study. There was no significant difference in the infarct size in any of the groups studied.
To determine if a longer interval between preconditioning ischemia and 100 min of ischemia was required for the induction of tolerance, 33 rats were randomized into five groups. On day 1 the rats received either a sham operation (n = 9), transient MCA occlusion for 10, 15, or 20 min, or two intervals of 10-min transient MCA occlusion (n = 6 per group). Seventy-two hours later, all animals received 100 min of MCA occlusion; after an additional 24 h, the infarct volume was determined by the TTC method. Figure IB illustrates the results of these experiments. There was a significant reduction in infarct size in both the 20-min ischemia and the 2 × 10-isc groups compared with the animals without pretreatment.
We then studied the effect of a 72-h rather than 24-h interval between the 100-min occlusion and death for histological changes. We also investigated whether a longer period of prior ischemia (30 min, either uninterrupted or as separate 10-min intervals) was more efficient in inducing tolerance. Figure 1C illustrates the results of these experiments. Contrary to the protection observed after a 24-h interval between prolonged ischemia and death, no significant reduction in infarct size between the 20-min pretreatment groups and the sham-operated controls was noted after 72 h. These results suggest that shortened periods of ischemia might delay rather than prevent infarction. However, 30-min ischemia and 3 × 10-isc afforded significant protection even after the longer interval to death.
We also investigated the optimal interval between preconditioning ischemia and prolonged ischemia. Fifty-four rats received either sham operations or 3 × 10-isc, a duration demonstrated to produce maximal protection. Infarct size was determined by the TTC method 72 h after the 100 min of ischemia. No significant protection was seen if the interval between the pretreatment and the 100-min ischemia was 24 h (Fig. ID). However, with 48-, 72-, and 120-h (5 days) intervals between pretreatment and prolonged ischemia, the subsequent infarct size was significantly smaller; the difference between pretreatment and sham controls was most significant with a 72-h interval. If the interval between the pretreatment and the prolonged ischemia was 168 h (7 days), the protective effect was no longer seen.
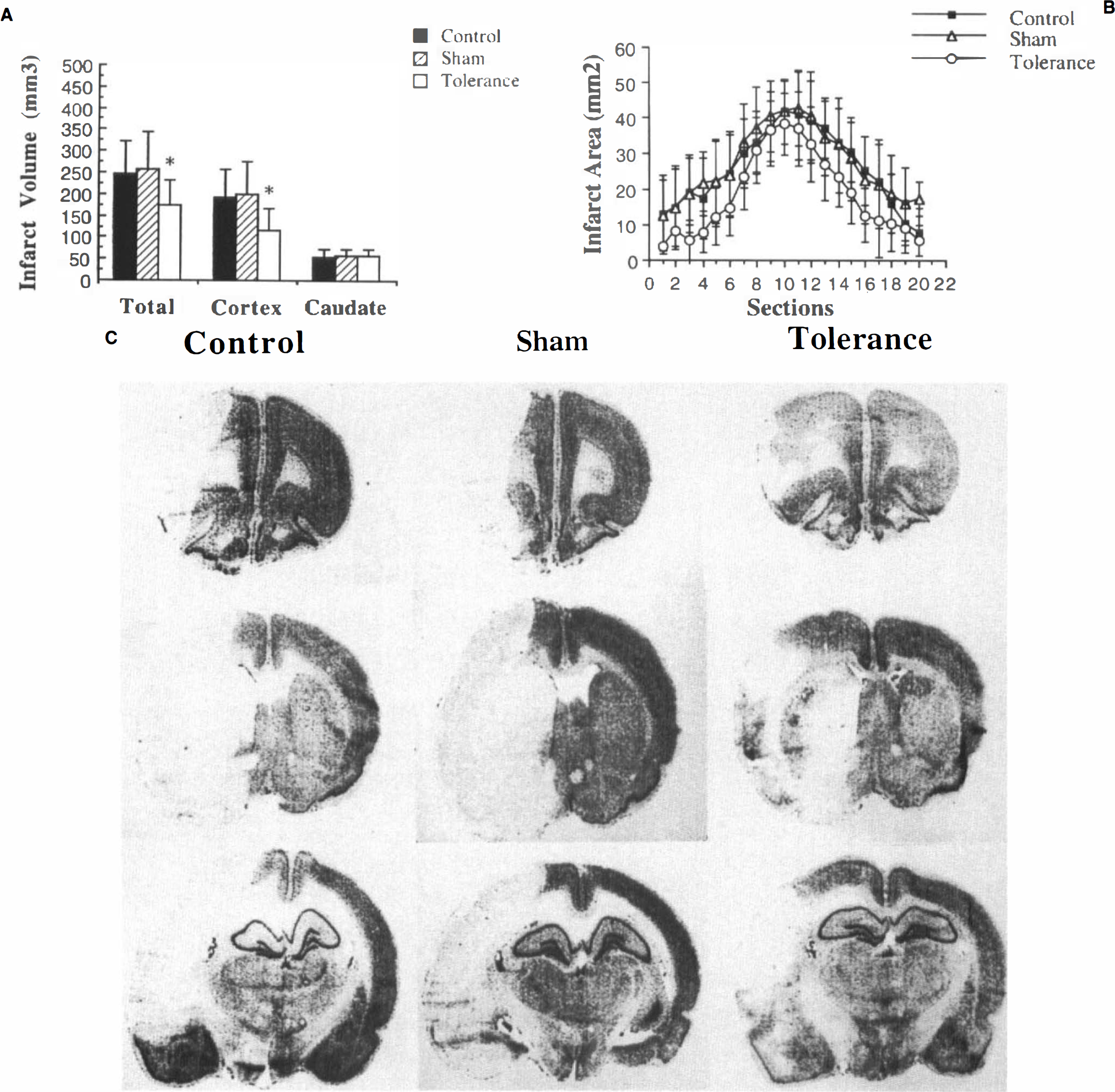
To confirm results from the TTC method, cresyl violet histology was done. Three groups (n = 6 per group) were established: a control group without preconditioning ischemia or sham operation, a sham-operated group, and a group given 3 × 10-isc. Seventy-two hours later, all three groups were subjected to 100 min of ischemia. Seventy-two hours after that, the rats were killed and the brains were frozen, processed for sectioning (20-μm slices), and stained with cresyl violet to measure the infarct volume. Figure 2 illustrates infarct volume in the penumbra (cortex) and the ischemic core (caudate) and, section by section, shows the effects of tolerance on infarct size. There was no significant difference between sham-operated and control animals, but the total and cortical areas of infarction were smaller in each section of the preconditioned animals than in either of the other two groups. The difference was due entirely to salvage of the cortex, as pretreatment ischemia did not significantly lessen the area of caudate infarcted (Fig. 2).
Among the 260 rats used in the infarct volume experiments, 33 died of either carotid hemorrhage during the surgical procedure for reperfusion (n = 3) or severe brain edema during reperfusion after 100 min of MCA occlusion (n = 30) and were excluded from the study. There was no difference in mortality rates between sham-operated animals (n = 14) and rats treated with preconditioning ischemia (n = 16). In 227 surviving rats, there were no significant differences in temporalis muscle temperature, arterial blood pressure, blood glucose, or blood gases between sham-operated and ischemia-treated rats before, during, or after MCA occlusion (data not shown).
Histopathology
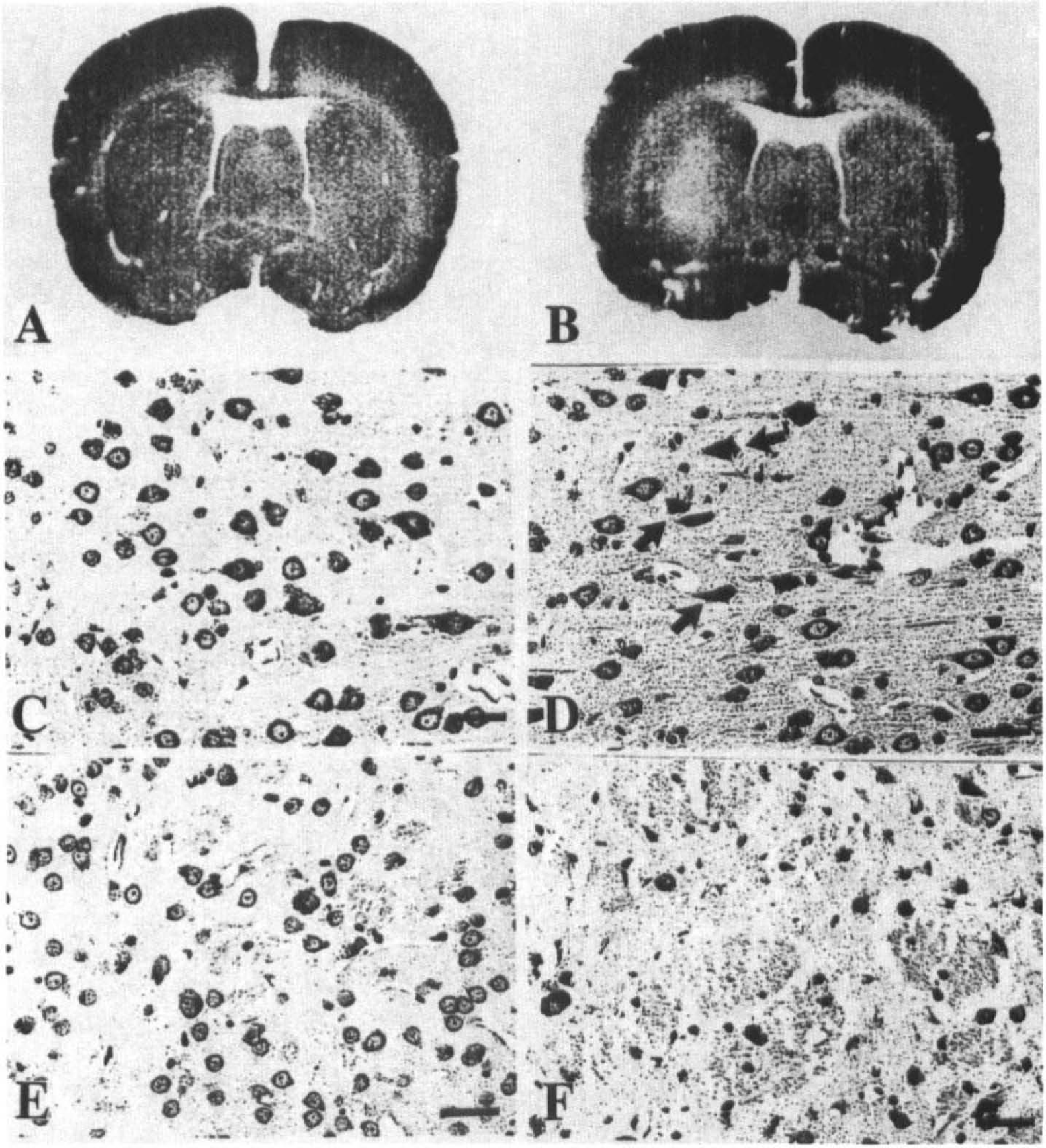
To determine the effect of the brief ischemia alone on histopathological outcome, 10 rats were subjected to 3 × 10-isc and killed either 24 or 72 h later (Fig. 3). At 24 h, little or no effect was seen at gross or microscopic levels in cortex or caudate. However, by 72 h there was evidence of patchy infarction of the caudate at the gross and microscopic levels. In addition, in some rats occasional selective neuronal necrosis was seen, predominantly in layers 3 and 5 of the cortex.
Blood flow
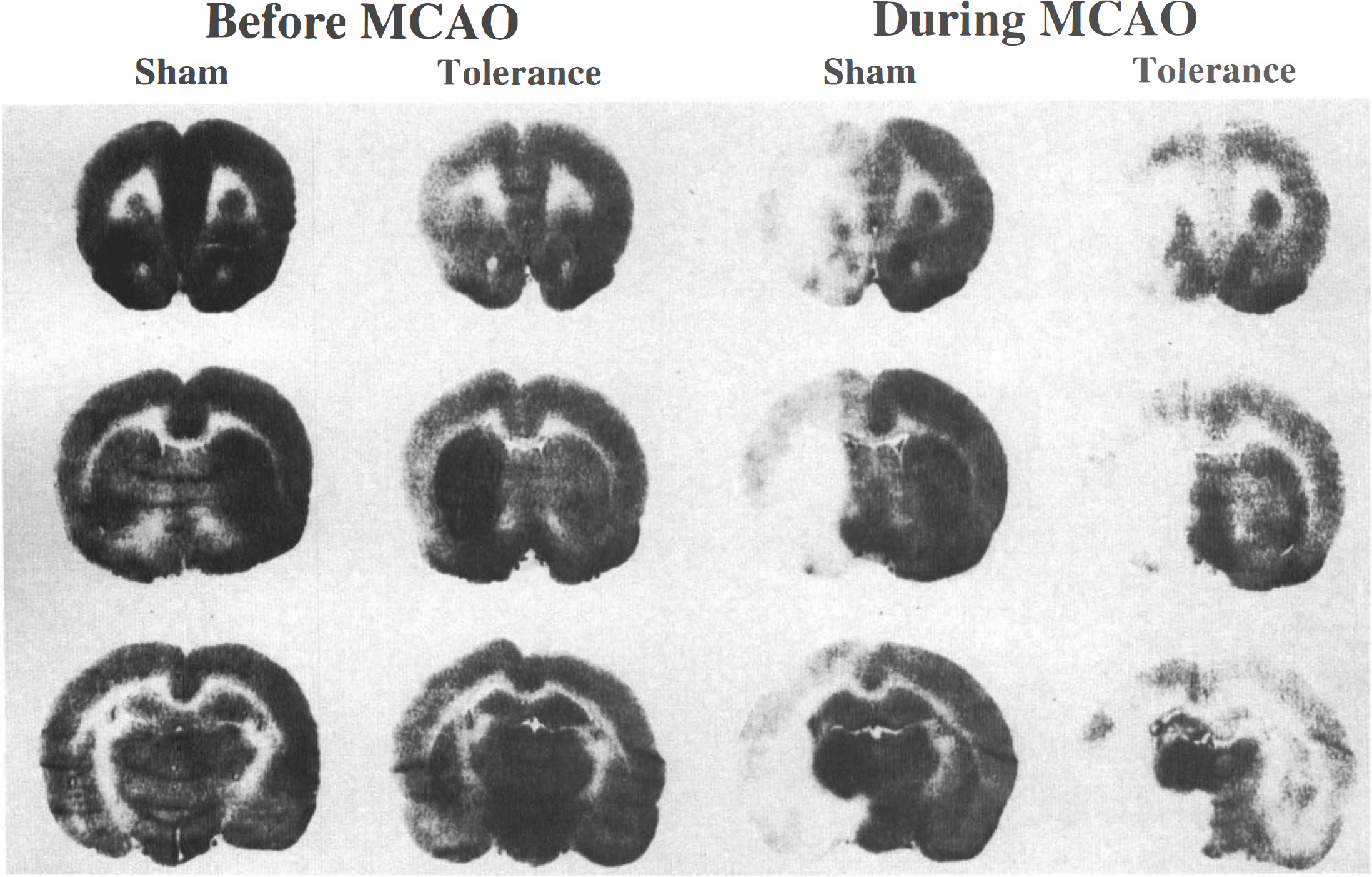
One logical explanation for the development of ischemic tolerance is an increase in collateral blood flow. Quantitative rCBF was studied 72 h after sham operation or 3 × 10-isc either before or during MCA occlusion by the iodoantipyrine method. The results of this experiment are summarized in Table 1, and examples are shown in Fig. 4. There was no significant change in rCBF in any region during MCA occlusion. The preconditioned group showed a significant increase in blood flow, which was restricted to the caudate in those animals studied just before occlusion. Since the caudate is never spared from infarction in the model used, the increased flow in this region could not explain the phenomenon of tolerance.
Regional cerebral blood flow in rats before and during 100-min MCA occlusion
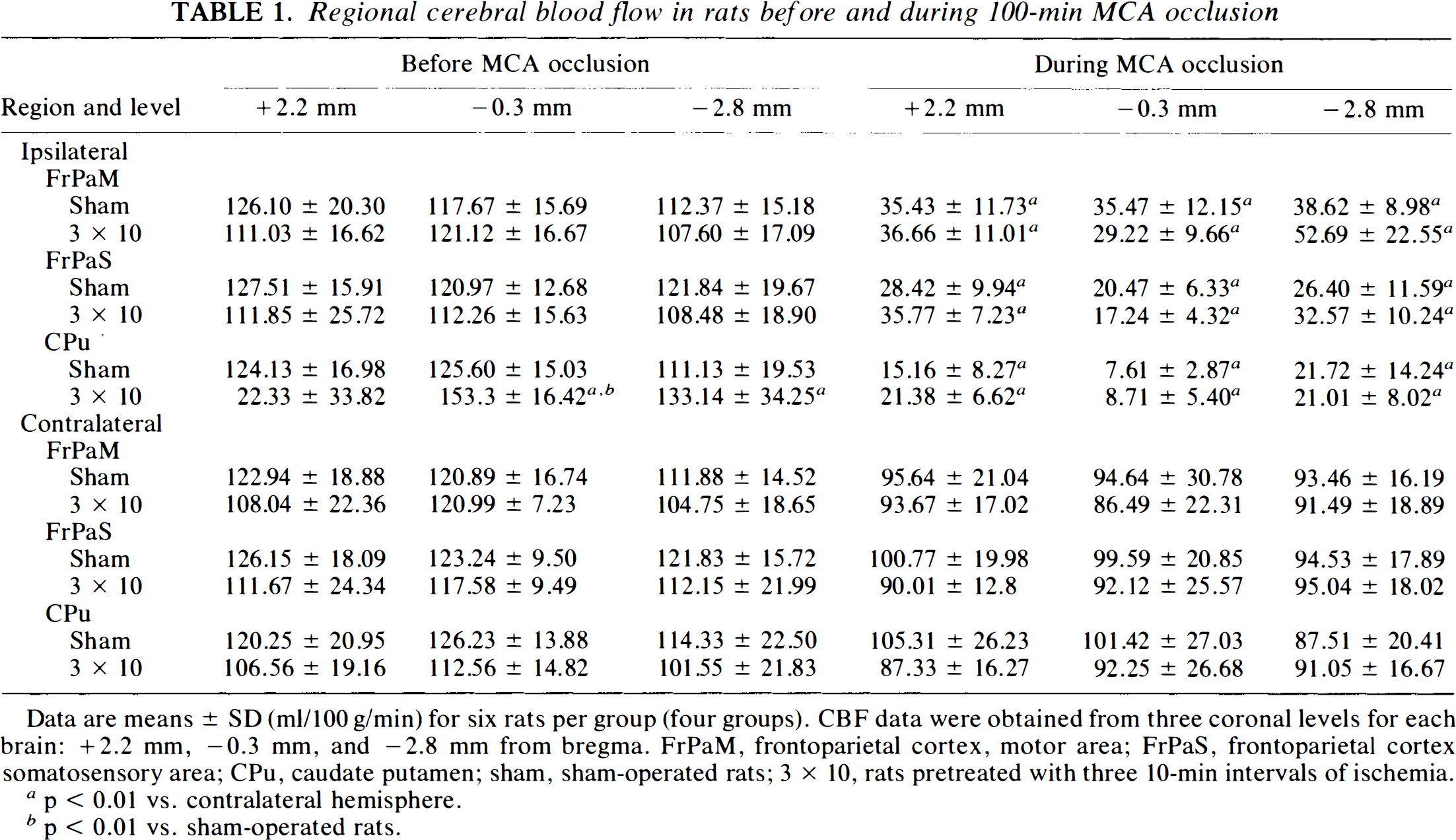
Data are means ± SD (ml/100 g/min) for six rats per group (four groups). CBF data were obtained from three coronal levels for each brain: +2.2 mm, −0.3 mm, and −2.8 mm from bregma. FrPaM, frontoparietal cortex, motor area; FrPaS, frontoparietal cortex somatosensory area; CPu, caudate putamen; sham, sham-operated rats; 3 × 10, rats pretreated with three 10-min intervals of ischemia.
p < 0.01 vs. contralateral hemisphere.
p < 0.01 vs. sham-operated rats.
Expression of stress proteins
Rats were subjected to 3 × 10-isc and then killed at 1, 2, 3, 5, 7, or 14 days after ischemia (three rats per time point). Cortical tissue from a region spared from infarction after preconditioning ischemia (Fig. 5A) was dissected for Western blot analysis of hsp70, grp75, and grp78 protein expression. Hsp70 expression was increased in ischemic animals versus expression in nonischemic controls at 1, 2, 3, and 5 days after 3 × 10-isc but was not significantly increased at 7 and 14 days (Fig. 5B and C). The stress proteins grp75 and grp78 were increased 1.5–2-fold at all time points studied, but the difference did not reach statistical significance (Fig. 5B and C).
An additional 12 rats were subjected to 3 × 10-isc and killed at 1 or 3 days (n = 6 per group) after ischemia. Two sham-operated rats were used as controls. The brains were removed and processed for immunohistochemistry analysis. Grp75 and grp78, but not hsp70, were constitutively expressed in low amounts in cortical neurons in control brains. Following preconditioning ischemia, all three stress proteins were increased in neurons with no difference seen between 1 day and 3 days (Fig. 6). Although there was no difference in the amount of hsp70 protein expressed at 24 and 72 h after 3 × 10-isc, there were some differences in the cellular distribution of hsp70 at these times. At 24 h, when tolerance was not yet present, hsp70 was expressed exclusively in neurons in the dorsal cortex (Fig. 6a); however, by 72 h, hsp70 was expressed both in glia (Fig. 6b) and in neurons (not shown) in regions where tolerance was present. The morphology of the cells and the observation that cells with this morphology in adjacent sections stain with GFAP suggest that the majority of these glial cells are astrocytes.
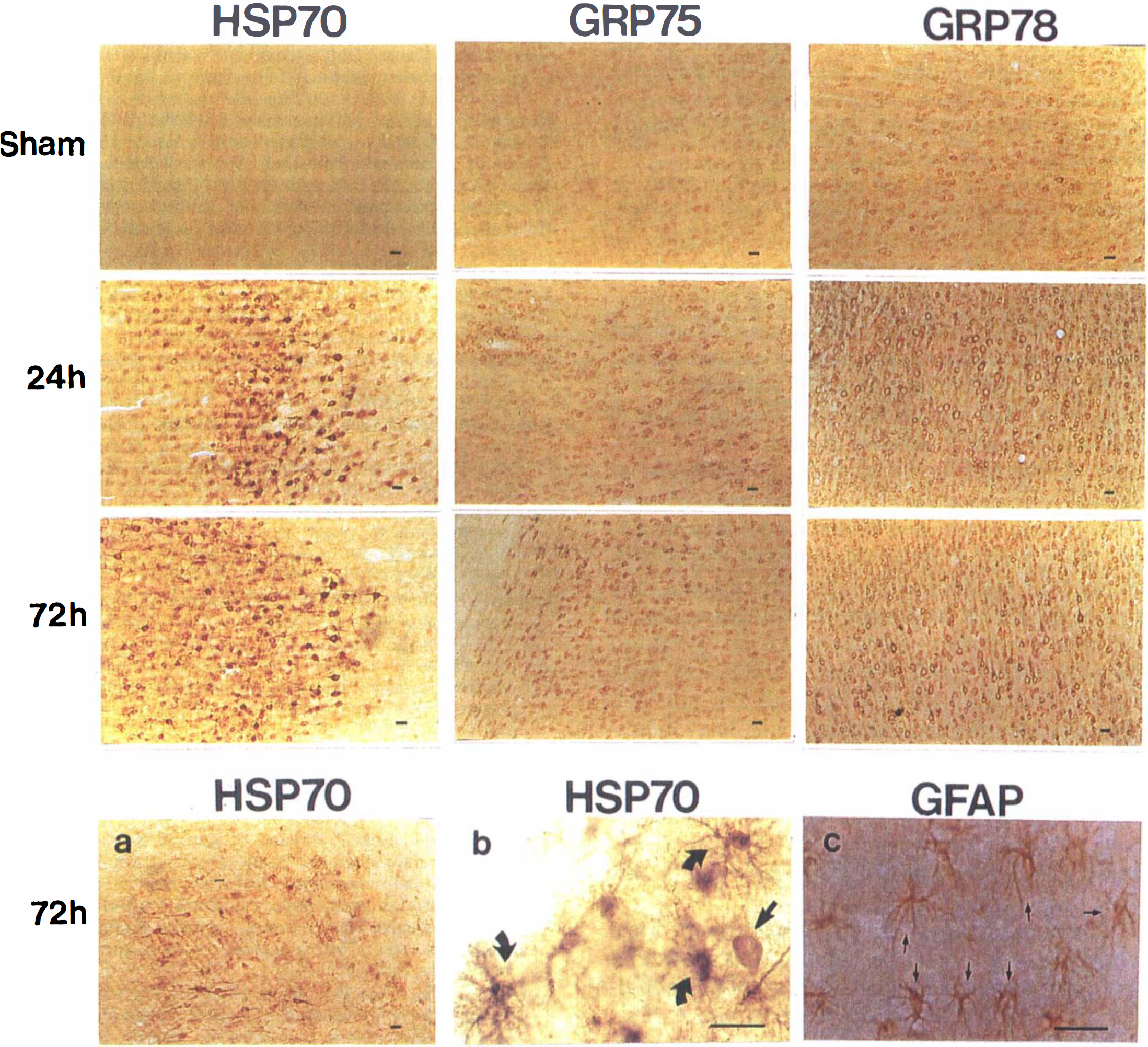
Hsp70, grp75, and grp78 immunocytochemistry from sham-operated rats or rats 1 or 3 days after 3 × 10-isc. Grp 75 and grp78 are constitutively expressed in cortical neurons in control brains, and all three stress proteins are increased in cortical neurons 24 h and 72 h after preconditioning. Hsp70 was also expressed in glial cells at 72 h after 3 × 10-isc:
DISCUSSION
The experiments presented here document and characterize the phenomenon of ischemic tolerance in focal ischemia: that is, in a brain region destined for infarction, prior exposure of that same brain region to transient focal ischemia renders the resulting stroke smaller. This observation complements our previous report demonstrating that brief periods of global ischemia result in smaller strokes following subsequent permanent MCA occlusion in the rat (Simon et al., 1993). These data suggest that tolerance occurs in cortical as well as hippocampal neurons and that the concept of tolerance is relevant not only to delayed selective neuronal death but also to frank infarction.
Transient ischemia does not produce tolerance by increasing collateral blood flow and thereby ameliorating hemodynamic abnormalities during MCA occlusion. Iodoantipyrine rCBF audioradiographs were performed in sham-operated and tolerant animals just before and during induction of stroke. In the tolerant animal, an increase in CBF was seen in the caudate and putamen ipsilateral to the transient ischemic insult. However, this brain region does not demonstrate tolerance in this model. No change in CBF was seen before or during induction of stroke in the cortical mantle, the area in which tolerance is induced (Fig. 4 and Table 1). Accordingly, changes in rCBF do not explain ischemic tolerance.
The parameters necessary for the induction of tolerance in focal ischemia in this experimental model have dose-response characteristics and a time window of efficacy (Figs. 1–4). The dose of transient ischemia that best induces tolerance in the rat is 30 min, which can be delivered either as a single insult or in three separate 10-min periods. Lesser durations of ischemia, either in divided or single doses, are inadequate (Fig. 1). The degree of prior ischemia necessary to induce tolerance is substantial, resulting in minimal injury to cortical neurons and moderate injury in the caudate and putamen (Fig. 5).
Time is necessary for the brain to develop tolerance. A 24-h interval between transient ischemia and the induction of stroke is insufficient to produce tolerance in focal ischemia (Fig. 1). A 48-h interval between the prior ischemia and the stroke does result in tolerance when the transient ischemia is 30 min in duration; tolerance persists at days 3 and 5 but is no longer expressed by day 7 (Fig. 1).
In the heart, where tolerance can develop in minutes, modification of existing proteins and, in some models, desensitization of adenosine receptors has been proposed as a mechanism of tolerance (Lawson and Downey, 1993). The longer time interval required for tolerance to develop in the brain than in the heart could be the result of different underlying mechanisms. This extended time is sufficient for the occurrence of alterations in gene expression, which could produce tolerance through at least two mechanisms: (a) reduced expression of proteins involved in cell injury, and (b) increased expression of proteins that confer tolerance to ischemia. The most frequently proposed explanation for ischemic tolerance in brain has been the latter.
The protein believed to be protective has been hsp70, which is increased after brief ischemia, since glutamate toxicity in neuronal cultures is attenuated by preincubating the cells with sublethal concentrations of NMDA or glutamate, and this protection requires new protein synthesis (Rordorf et al., 1991). Further, preinduction of hsp70 protects cultured neurons from glutamate neurotoxicity (Lowenstein et al., 1991). Many studies have shown that hsp70 is expressed after brief preconditioning global ischemia in tolerance models (Kirino et al., 1991; Simon et al., 1993; Glazier et al., 1994). Intraventricular anti-hsp70 antibodies and a nonspecific inhibitor of hsp synthesis interfere with the induction of tolerance after global ischemia (Nakata et al., 1993). However, a recent report suggests that tolerance may occur without increased hsp70 expression (Abe and Nowak, 1994).
Our results show that hsp70 protein is expressed during the times when tolerance is present in this model. Further, hsp70 protein is no longer expressed after the tolerant period. Thus, its expression correlates with the time course of the induction of tolerance in this model. On the other hand, hsp70 is also expressed at 24 h, before tolerance is present. There are several possible explanations for the lack of reduced infarction volume at 24 h when hsp70 protein is present. Hsp70 could be the first step in a cascade of events leading to tolerance instead of being a direct effector of tolerance. If so, it would necessarily be expressed before tolerance is present. Our results show that hsp70 is not expressed in glia at 24 h. Expression of hsp70 in glia has been associated with neuronal tolerance to oxygen and glucose deprivation (Snider and Choi, 1995). Hsp70 could exert a protective effect at 24 h, which could be masked by other events that increase the susceptibility of the brain to ischemia. Finally, hsp70 expression could be an epiphenomenon to other mechanisms producing tolerance.
Grp75 and grp78 are constitutive proteins that are homologous to hsp70 and that are upregulated by anoxia and hypoglycemia (Pelham, 1986). Grp78 is a soluble protein within the endoplasmic reticulum and appears to act as a molecular chaperon; it has been shown to have a protective role in cellular injury (Li et al., 1992). Grp75 is an inner mitochondrial protein that has significant homology to hsp70 (Mizzen et al., 1989). It has been proposed to transport proteins across the mitochondrial membrane and prepare them for final folding. Grp75 and grp78 mRNAs are upregulated within 24 h after cerebral ischemia and seizures (Lowenstein et al., 1994; Massa et al., 1995). Our results show that grp75 and grp78 expression were increased after preconditioning ischemia, although the time course did not correlate closely with that of ischemic tolerance.
A number of other candidate endogenous neuroprotectants are upregulated in ischemic brain as well; these include hsp-27, superoxide dismutase, and calcium-binding proteins (Simon et al., 1991; Kato et al., 1994; Liu et al., 1994; Lowenstein et al., 1994), as well as the antiapoptotic oncogene bcl-2 (Shimazaki et al., 1994; Chen et al., 1995). Whether these genes are involved in the induction of tolerance remains to be determined.
Beside induction of protective proteins, decreased expression of toxic proteins after preconditioning ischemia could also contribute to the induction of tolerance. Overall protein synthesis depression is a well-known sequel of brain ischemia (Dienel et al., 1980; Thilmann et al., 1986; Widmann et al., 1991), including ischemia that induces tolerance (Kato et al., 1994). However, selective inhibition of some class(es) of proteins may particularly affect the susceptibility of the brain to ischemic injury. For example, Pellegrini-Giampietro et al. (1992) showed selective downregulation of the GR2 subunit of the non-NMDA glutamate receptor in CA1 neurons following global ischemia. This change could make non-NMDA receptor channels permeable to calcium (Hollmann et al., 1991), thus increasing the intracellular calcium overload associated with ischemia. The finding that blockade of non-NMDA receptors prevents delayed neuronal death after global ischemia (Sheardown et al., 1993) is consistent with a role for non-NMDA receptors in the development of ischemic injury. Another example of selective inhibition of protein translation affecting ischemic injury is the NR1 subunit of the NMDA receptor. Blocking NR1 protein expression by antisense oligonucleotides decreases ischemic damage after MCA occlusion (Wahlestedt et al., 1993). Therefore, future studies to identify those proteins that are selectively depressed by preconditioning ischemia will help elucidate the molecular basis for tolerance induction.
A number of issues that require further study in this and other models of tolerance need to be addressed: (a) The induction of tolerance possibly involves many interdependent mechanisms. To definitively determine which specific changes in gene expression, if any, mediate tolerance in this model requires that protein expression be altered by the use of antisense oligonucleotides, transgenic technology, or viral transfection vectors, (b) If hsp70 protein expression underlies the induction of tolerance, differences in the cellular distribution of hsp70 may be important. The cellular localization of hsp70 protein expression in this model may be further characterized by double-labeling hsp70 with cell specific antibodies. In vitro experiments may provide insight as to whether hsp70 expression in glial cells is protective (Snider and Choi, 1995). (c) The observation that 2 × 10-isc is protective, with 24-h survival after 100-min ischemia but not 72-h survival, suggests that tolerance could delay infarction rather than provide absolute protection. Other factors, including differences in rat populations and in the specificity of TTC at different survival times, could also explain the discrepancy. Further experiments with longer survival times may clarify this issue.
These data demonstrate that tolerance to focal ischemia in cortex can be induced and show that there are alterations in gene expression but not alterations in blood flow in the tolerant region. This model of ischemic tolerance may allow both known and novel gene products with potential neuroprotective effects to be identified.