
Abstract
We previously showed that initiator caspases-2 and −8 are prominently activated in ischemia/reperfusion (I/R)-induced injury in cardiomyocytes, but while blockade of caspase-2 activity enhanced cell survival, blockade of caspase-8 activity did not protect cardiomyocytes. Because apoptotic death in these cells is characterized by a burst of reactive oxygen species (ROS) at reperfusion and their survival by inhibition of this burst, we examined the effects of blocking caspase-2 and caspase-8 activities on ROS production. Caspase-2 inhibition blocked the reperfusion-induced ROS burst, while inhibition of caspase-8 did not. We also examined effects of caspase inhibition on mitochondrial membrane potential (ΔΨm) and mitochondrial function and found that blocking caspase-2, but not caspase-8, allowed recovery of ΔΨm and mitochondrial functionality. Furthermore, knockdown of caspase-2 by small-interfering (si)RNA confirmed caspase-2 participation in cytochrome c release, which correlates with loss of ΔΨm and cell death in these cardiomyocytes.
Introduction
Ischemia/reperfusion (I/R) injury in cardiomyocytes is associated with initiation of the mitochondrial pathway of cell death, including cytochrome (cyt) c release, loss of mitochondrial membrane potential (ΔΨm), and enhanced generation of reactive oxygen species (ROS) production.1,2 We recently demonstrated that reperfusion rapidly induces the mitochondrial pathway of apoptosis in chick cardiomyocytes after 1 h of simulated ischemia, while ischemia alone did not. 3 Subsequently, we demonstrated that among several caspases, caspase-2 was most rapidly activated, and that its activation appeared to be required for initiation of the mitochondrial pathway of cell death. 4
Caspase-2 is one of the most conserved caspases across animal species, and it shares structural and functional features of both the “initiator” caspases and the more downstream “effector” caspases such as caspase-3.5,6 Caspase-2 more closely resembles CED-3, the prototypical C. elegans caspase, than other mammalian caspases, 7 and it has been suggested that caspase-2 represents a primordial protease that may serve several pathways. 8 Involvement of caspase-2 in the mitochondrial pathway of cell death seems to depend on a number of factors, including the nature of the stimulus and the cell type. 9 This link between caspase-2 and apoptotic mitochondrial events still needs to be fully elucidated, 10 and while evidence is accumulating for a role for caspase-2 in stress-induced cell death, 11 its role in apoptotic injury following I/R in the heart or other organs remains poorly defined.
An early study of I/R-induced cell death in the heart implicated caspase-2 activation in a rabbit model of myocardial infarction, 12 but there was no indication of the role of this caspase in the apoptotic cascade. Following our initial study identifying a role for caspases in reperfusion-induced death of ischemic cardiomyocytes, 3 we examined individual caspases in this model, and our data suggested that caspase-2 activity may play an important role in initiation of I/R-induced cell death; these studies also noted the prominent later activation of caspase-8. Indeed, caspase-2 and caspase-8 have been shown to have sequential roles13–16 or roles upstream of mitochondria in a number of models of apoptosis,17–20 but few studies have compared their relative contributions to the initiation of mitochondrial events, and to our knowledge, none have compared their effects on mitochondria during I/R-induced cell death in cardiomyocytes. Because it is therapeutically more advantageous to block an apical or initiating caspase than a downstream caspase, 6 it is important to differentiate between these caspases, particularly in the I/R-induced mitochondrial pathway. Thus, the aims of the present study were to compare the effects of blocking caspase-2 and caspase-8 activity on mitochondrial events associated with I/R-induced death in these cells; namely, ROS generation, loss of membrane potential, and cyt c release. Results from these studies showed that inhibition of caspase-2 activity, but not that of caspase-8, rescued ΔΨm and mitochondrial function and blocked the ROS burst. Knockdown of caspase-2 via siRNA confirmed a specific role for this caspase in cyt c release and I/R-induced cell death in these cardiomyocytes.
Materials and Methods
Materials
The caspase-2 inhibitor, benzyloxycarbonyl-Val-Asp(Ome)-Val-Ala As-(Ome)-CH2F (zVDVAD-fmk), and the caspase-8 inhibitor, z-Ile-Glu(Ome)-Thr-Asp(Ome)-Ch2F (zIETD-fmk), were purchased from Calbiochem (LaJolla, CA) and the caspase-3 inhibitor Ac-Asp-Gln-Thr-Asp-H (Ac-DQTD-CHO) was obtained from Peptides International (Louisville, KY). Optimal time and dose of these inhibitors were determined in our previous study. 4 Time of dose was chosen for therapeutic relevance (at reperfusion rather that all-course or administration during ischemia), and dose was determined by efficacy and specificity in our flow-through reperfusion model. 4 For western blot analyses of cyt c release, anti-cytochrome c antibody was purchased from BD Biosciences (San Diego, CA). The fluorescent dyes, propidium iodide (PI) for assessing cell viability, tetramethylrhodamine ethyl ester (TMRE) for analyses of ΔΨm, and the reduced form of MitoTracker Red (CM-H2XRos) for mitochondrial imaging and functional assessment (see below) were obtained from Molecular Probes, Inc. (Eugene, OR). Other buffer and media reagents were obtained from Sigma (St. Louis, MO).
Chick Primary Cardiomyocyte culture
Embryonic chick ventricular myocytes were isolated as previously described.1,3 Briefly, hearts of 10 -day -old chick embryos were removed; the ventricles were minced and enzymatically digested with 0.025% trypsin (Invitrogen, Grand Island, NY) for 4-5 cycles with gentle agitation at 37 °C. The resultant cell suspension was centrifuged, and the cell pellet was resuspended in media. Cells were pre-plated for the removal of fibroblasts, and re -plated (0.7 × 106) onto 25 mm glass coverslips. Cardiomyocytes were cultured in a humidified incubator for 3 days at which point the cells demonstrated synchronous contractions. Experiments were performed on day 4 or 5 using spontaneously contracting cells (viability > 99%).
Perfusion System and Perfusate composition
Synchronously contracting cells on glass coverslips were placed in a 1.2 ml Sykes-Moore perfusion chamber (Penn Century Co., Philadelphia, PA) as previously described.1,3 Tubing to these chambers was stainless steel (Bellco, Vineland, NJ) and the chambers were sealed with Kynar-gaskets to minimize any potential diffusion of ambient O2 into the system. The normoxic solution pumped with a perfusion rate of 0.25 ml/min consisted of an oxygenated balanced salt solution (BSS) with 149 torr PO2, 40 torr PCO2, pH 7.4, 4.0 mM [K+], and 5.6 mM glucose. Simulated ischemic BSS consisted of 20 mM 2-deoxy-glucose and 8.0 mM [K+]. It was bubbled with 80% N2 and 20% CO2 to produce a PO2 of 3-5 torr, a PCO2 of 144 torr, and a final pH of 6.8.1,3
Video/fluorescent microscopy
For cell imaging, a Nikon TE 2000-U inverted phase/epifluorescent microscope was used, and a charged-coupled device camera was used to monitor contractions and measure fluorescence over time in the same field (−70 μm × 90 μm) of cells. Fluorescent images were acquired from a cooled Cool-SNAP-ES camera (Photometrics, Tuscon, AZ) and changes in fluorescent intensity over time were quantified with MetaMorph® software (Molecular Devices Corp., Downington, PA). 4
Measurement of Intracellular ROS
Carboxy-H2DCF-DA (6-carboxy −2′,7′-dichlorodihyd rofluorescein diacetate, 1 μM; Invitrogen, Carlsbad, CA), the carboxylated and more cell-permeant 21 form of the classic H2DCF-DA, was used to monitor intracellular ROS. This non-fluorescent cell permeable dye is oxidized to the highly fluorescent car-boxy-dichlorofluorescein (DCF); DCF fluorescence was measured at excitation 488 nm/emission 520 nm and expressed as arbitrary units (a.u.).
Viability Assay and Contraction analyses
Viability of cardiomyocytes was assessed by the fluorochrome propidium iodide (PI, 5 μM) at excitation 540 nm/emission 590 nm. PI is a fluorescent exclusion dye that binds to chromatin upon loss of cell membrane integrity. Cell death was quantified throughout the experiment in a randomly selected field of cells. At the end of each experiment, all cells in this field were permeabilized with digitonin (300 μM). Percentage cell death (PI uptake) was expressed as PI fluorescence relative to the maximal value seen after 1 h of digitonin exposure (100%). Cell contractions were assessed as previously reported. 1 A return of contraction was indicated when synchronous contractions were seen throughout the field of cells.
Mitochondrial Integrity and function
To monitor mitochondrial function in treated cardiomyocytes, the reduced form of MitoTracker Red (CM-H2XRos) was used. This dye is taken up specifically in the mitochondria where it fluoresces red after it is oxidized by actively respiring mitochondria, and thus provides a useful measure of functional mitochondria and a selective visual indicator of constitutive ROS production by functional mitochondria.22,23 Cells were treated essentially according to manufacturer instructions. Briefly, cardiomyocytes were incubated with CM-H2XRos (200 nM) at 37 °C for 45 min and fluorescence was imaged during I/R and after caspase inhibitor treatments. CM-H2XRos has also been reported to be suitable for determining mitochondrial membrane potential changes during apoptosis in some cell types.24,25 In preliminary experiments utilizing cyanide 3-chlorophenylhydrazone (CCCP; 5 μM) to dissipate mitochondria membrane potential in chick cardiomyocytes, we confirmed CM-H2XRos to be potential sensitive in these preparations. In cardiomyocytes preloaded with CM-H2XRos (200 nM; 30 min) treatment with 5 μM CCCP resulted in rapid and significant loss of fluorescence (not shown). Nevertheless, to confirm and quantify loss and/or rescue of ΔΨm, we utilized a standard TMRE analysis as previously described. 26 Briefly, coverslips with confluent cardiomyocytes were preloaded with TMRE (100 nM) for 45 min before the I/R protocol and inhibitor treatments. Cells were imaged at 37 °C (excitation 535 nm/emission 610 nm), and changes in fluorescence were expressed in arbitrary units (a.u.).
siRNA Silencing of Caspase-2
The chicken (Gallus gallus) caspase-2 mRNA sequence was obtained from the National Center for Biotechnology Information (NCBI) database (NCBI reference sequence NM001167701.1). Active (sense: 3′-GGAAGCUCUGAAGAAGAAC −5′) and scrambled (sense: 3′-GGGUCUAAGGAGAAACAAC-5′) sequences were designed from this sequence using the siRNA Target Designer (v1.6) available from Promega (Madison, WI). These sequences were checked against the NCBI database to ensure they did not correspond to other chicken genes that could affect cardiomyocyte function or survival. Caspase-2 active and scrambled siRNA sequences were submitted to Qiagen, Inc. (Valencia, CA) for synthesis. Cells were transfected using the RNAiFect kit (Qiagen); briefly, the caspase −2 siRNA or scrambled siRNA were mixed with the RNAiFect transfection reagent (Qiagen) to produce the transfection complexes. After determination of an optimally effective ratio of siRNA:RNAiFect, the cardiomyocytes were seeded to reach ∼75% confluence after 24 h under normal growth conditions (see above). The medium was then removed and replaced with fresh media containing the transfection complexes containing either the caspase-2 or scrambled siRNA. A fluorescein-conjugated control siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) was used to monitor transfection, and because a reliable antibody to chick caspase-2 did not exist, we utilized RT-PCR to confirm specific knockdown of caspase-2 message in these cells. Primers for these confirmatory experiments were as follows: Chick caspase-2: Forward 5′-AAGAACCGGGTGATGCTGGCG-3′, reverse 5′-CTGGAGATTGGGGCAGTTTGC-3′; chick caspase-8: forward 5′-GAGCACGTCACTGTAAGGAAG-3′, reverse 5′-ATTTTCTTCAACAGGCTCTTG-5′; chick β-actin: forward 5′-ATG GATGATGATATTGCTGCGCTC-3′, reverse 5′-TAT CCACATCACACTTCATGATGG-3′.
Data analysis
Each individual experiment on a field of 500 cells represented an n of 1. All data are expressed as mean ± SEM. Statistical analysis was performed using commercially available statistic software (Sigma Stat 3.0). Data between groups recorded over an interval were compared using a two-way repeated measures ANOVA. When significant, Tukey's post-hoc test was performed for comparison between individual groups. A P-value < 0.05 was considered statistically significant.
Results
Inhibition of Caspase-2 Activity Blocks I/r-Induced Cell death
We previously reported that isolated chick cardio-myocytes exposed to 1 h of simulated ischemia followed by 3 h of reperfusion exhibited significant reperfusion injury,3,4 and in these new experiments, we confirm those results to establish a baseline for the following studies (Fig. 1). Furthermore, using the peptide caspase inhibitors only during the first 30 min of reperfusion, 3 we confirmed the protective action of the caspase-2 inhibitor zVDVAD-fmk, while demonstrating the lack of any protection provided by the caspase-8 inhibitor zIETD-fmk (Fig. 1), despite the prominent (≥7-fold at 3 h of reperfusion) increase in caspase-8 activity confirmed from previous studies. 4 Our simulated I/R protocol resulted in 50.2 ± 4.31% cell death (in vehicle-only controls) by 3 h of reperfusion whereas inhibition of caspase-2 activity significantly reduced cell death within 1 h post-inhibitor treatment and decreased cell death to 27.5 ± 1.75% by 3 h of reperfusion (P < 0.05).
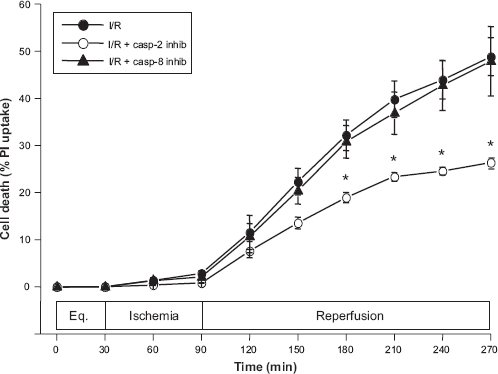
Effects of caspase (casp) inhibition, using selective casp-2 inhibitor (inhib), zVDVAD-fmk, and casp-8 inhibitor zlETD-fmk (each at 100 μM), on I/R-induced cell death in chick cardiomyocytes. Treatment of cells with the peptide inhibitors only at the point of reperfusion significantly blocks cell death when casp-2 activity is targeted (27.5 ± 1.75 vs. 50.2 ± 4.31, *P < 0.05), while inhibition of casp-8 activity fails to provide any protection (47.9 ± 7.34% cell death, P = NS).
Inhibition of Caspase-2 Activity Blunts Reperfusion-Induced Ros burst
In a previous study, we examined the sensitivity of the reperfusion-induced oxidant burst to caspase inhibition using the broad-spectrum caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-CH2F (zVAD-fmk) and found little effect of this inhibitor on augmented ROS production. 3 However, since that time, several studies have reported the ineffectiveness of this inhibitor in blocking caspase-2 activity.27–29 Therefore, using the same I/R protocol with a 30-min administration of caspase inhibitors (100 μM) at the point of reperfusion, we examined the effects of more selective inhibition of caspase-2 on the reperfusion-induced ROS generation. These studies showed that only the caspase-2 inhibitor significantly blocked ROS generation as measured by DCF fluorescence (Fig. 2). While fluorescence reached a maximum of 2.51 ± 0.13 a.u. as early as 10-15 min after reperfusion (P < 0.01) in the control I/R experiments, DCF fluorescence reached a maximum of only 1.50 ± 0.22 a.u., and at a later time point (30 min after reperfusion) than in control I/R experiments (Fig. 2).
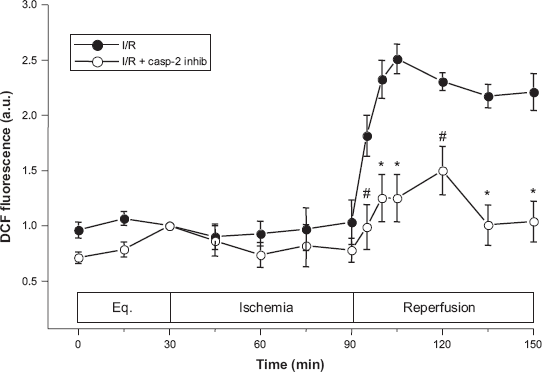
Effects of inhibition of caspase-2 activity on intracellular ROS generation as measured using the oxidation of 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCHF-DA) to fluorescent marker carboxy-DCF. Treatment with the VDVAD inhibitor significantly blunted I/R-induced DCF fluorescence from a measured maximums of 2.51 ± 0.13 a.u. (at 15 min repefusion) in vehicle control to 1.50 ± 0.22 a.u. (at 30 min reperfusion) with caspase-2 inhibitor (#P < 0.01; *P < 0.05).
Inhibition of either Caspase-8 or-3 Activities Does Not Affect Reperfusion-Induced Ros burst
The prominent activation of caspase-8 in this model of I/R-induced injury led us to examine the effects of inhibition of caspase-8 activity on the reperfusion-induced ROS burst using the caspase-8-selective inhibitor, zIETD-fmk. As shown in Figure 3, in contrast to the effects of the caspase-2 inhibitor, administration of predetermined (see above) caspase-8 activity-blocking doses 4 of zIETD-fmk had no effect on either extent or duration of ROS generation as detected by DCF fluorescence, and there were no significant differences at any time points measured. Because caspase-3 can also target mitochondria and lead to augmented ROS production,30,31 we tested a caspase 3-selective inhibitor to examine effects of inhibition on DCF fluorescence under identical conditions. Because the well-known and widely used caspase-3 inhibitor, Ac-DEVD-CHO, was recently shown to inhibit caspases-3, −7 and −8 equally,32,33 as well as caspase-2 in some systems, 34 we utilized the more selective caspase-3 inhibitor, Ac-DQTD-CHO, 33 to perform these experiments. As with caspase-8 inhibition, neither the level nor duration of reperfusion-induced ROS production was affected significantly by the inhibition of caspase-3 activity with this inhibitor (Fig. 3, inset).
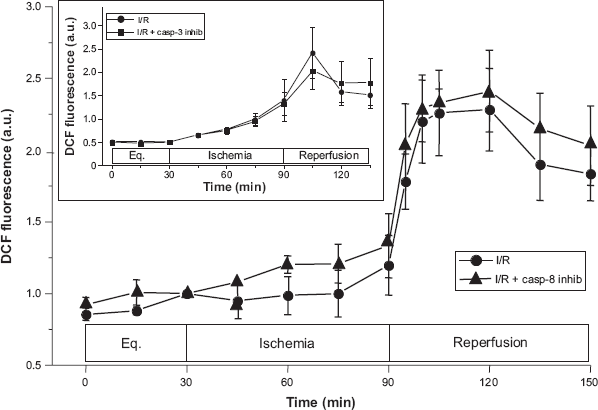
Effects of inhibition of caspase-8 activity on intracellular reactive oxygen burst as measured using 6-carboxy-DCHF-DA. Treatment with the IETD inhibitor had no effect on I/R-induced DCF fluorescence (peak fluorescence = 2.28 ± 0.29 a.u. for control and 2.41 ± 0.28 a.u. with caspase-8 inhibtion; P = NS) INSET: Effect of the caspase-3 inhibitor, zDQTD-fmk, on ROS generation showing DCF fluorescence measured at 15-min intervals. Addition of this inhibitor also failed to block ROS generation; there were no statistical differences at any time point (P = NS).
Inhibition of Caspase-2 Activity Rescues ΔΨm and Mitochondrial function
To assess mitochondrial function and loss of ΔΨm during I/R protocols we utilized two methods using well-characterized, mitochondria-specific dyes. As outlined above, the reduced form of MitoTracker Red (CM-H2XRos) fluoresces when oxidized in living cells and is sequestered in the mitochondria. In addition to measuring constitutive ROS generation, this MitoTracker dye allows discrimination of apoptotic cells which poorly retain the dye.24,25 We qualitatively examined fluorescence in identical preparations of cardiomyocytes before the simulated I/R protocol, at the end of the 60 min ischemia and 1 h after the start of reperfusion. As in other experiments, caspase-2 and caspase-8 inhibitors were added for 30 min at the point of reperfusion. While there appeared to be a slight reduction in fluorescence from control levels (Fig. 4A) during ischemia (Fig. 4B), fluorescence was almost entirely lost in vehicle-treated controls after 1 h of reperfusion (Fig. 4C). This loss of mitochondrial integrity was rescued by the caspase 2-selective inhibitor (Fig. 4D), but not by caspase-8 inhibition (not shown). To quantify ΔΨm during reperfusion under these treatments, we preloaded cells with the cationic fluorescent probe TMRE, which allows detection of changes in ΔΨm via Nernstian distribution and changes in the fluorescence of the dye. 35 As previously reported, in our simulated I/R model system, ΔΨm falls during ischemia, and this fall is linked to augmented ROS generation at reperfusion in unprotected cells.26,36 While inhibition of caspase-2 activity allowed significant repolarization as measured by TMRE (Fig. 4E), blockade of caspase-8 activity did not significantly affect TMRE fluorescence at any time point (not shown). Overall, repolarization allowed by caspase-2 inhibition represented a 25%-30% recovery of the starting ΔΨm, which is sufficient for rescue from I/R-induced injury and enhanced survival in this model. 26
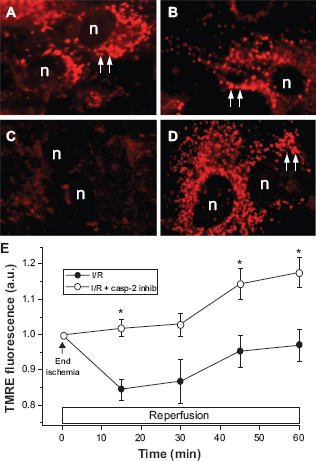
Effect of Inhibition of caspase-2 activity on mitochondrial function and membrane potential during our simulated ischemia/reperfusion protocol (see Methods) as measured by CM-H2XROS (A-D) and TMRE A) Equilibration period before ischemic treatment; CM-H2XRos fluorescence in normal, functional mitochondria (arrows); B) Ischemic period; C) Untreated, I/R-only cardiomyocytes exhibit loss of fluorescence caused by reperfusion; D) mitochondrial function is rescued in cells treated with caspase-2 inhibitor, zVDVAD-fmk, at the point of reperfusion (n = nuclei); E) zVDVAD-fmk treatment also rescues mitochondrial membrane potential as measured by TMRE fluorescence. For clarity, results of caspase-8 inhibition are omitted, but no significant (P > 0.05 at all points) rescue of mitochondrial membrane potential was observed.
Knockdown of Caspase-2 by Sirna Blocks I/r-Induced Cyt c Release and Cell death
To confirm a specific role for caspase-2 in mitochondrial induction of I/R-mediated apoptosis, we utilized RNAi technology, as described above, to knockdown the expression of caspase-2 in chick cardiomyocytes. Because we were unable to locate a reliable chicken caspase-2 antibody, we used RT-PCR to confirm the efficacy of our knockdown technology. As shown in Figure 5A, our siRNA protocol effectively abrogates caspase-2 mRNA expression by 24 h and continued to suppress expression through 72 h. All cyt c release and cell death experiments were performed on cells 48 h after transfection. Cyt c release was significantly inhibited at 1 h by knockdown of caspase-2, while transfection of scrambled siRNA had no effect (Fig. 5B). These effects correlated with cell death as measured by PI (as in Fig. 1) with knockdown of caspase-2 having effects very similar to those of the inhibitor zVDVAD-fmk; scrambled siRNA had no significant effect on cell death compared to untreated cells as in Figure 1.
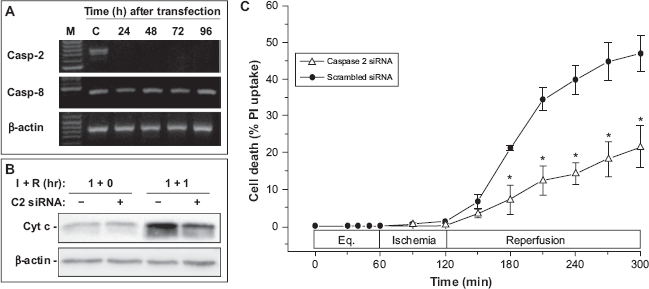
Effect of siRNA-mediated knockdown of caspase-2 on I/R-induced cyt c release and cell death in chick cardiomyocytes. A) RT-PCR for caspase-2 (casp-2) and caspase-8 (casp-8) mRNAs, confirming specific knockdown of mRNA for caspase-2 and lack of effect on caspase-8 or β-actin mRNA by caspase-2 siRNA transfection (M = marker lane; C = control, scrambled siRNA lane). B) effects of caspase-2 siRNA and scrambled siRNA on cyt c release into the cytoplasm. Low baseline levels of cytoplasmic cyt c are seen in both treatments prior to R, but cyt c release is significantly reduced by siRNA knockdown of caspase-2 (C2 siRNA; +) from that seen in control, scrambled (-) siRNA-treated cells (representative of 3 experiments). C) Levels of cell death reflect the effects of siRNA treatment on cyt c release shown in panel B, with significant reduction of cell death by caspase-2 siRNA, but no effect of scrambled siRNA on I/R-induced cardiomyocyte death (21.0 ± 6.81% vs. 45.8 ± 5.80%; *P < 0.05). The level of cell death in scrambled siRNA-treated cells is not statistically different than that seen for untreated control (or caspase-8 inhibitor-treated) cells in parallel I/R studies shown in Fig. 1 (45.8 ± 5.80% vs. 50.2 ± 4.31%; P = NS).
Discussion
We and others have reported that isolated cardiomyocytes exposed to simulated ischemia demonstrate a burst of ROS and accelerated cell death upon reperfusion,1,4,37 and results from several groups implicate the mitochondrial electron transport chain (ETC) as a likely source of ROS.1,2,38,39 Perturbation of the ETC in ischemic cardiomyocytes leads to the loss of ΔΨm36 and release of cyt c upon reperfusion unless rescued by antioxidants or mitochondrial drugs.40,41 The present studies have confirmed a critical role for caspase-2 in I/R-induced injury in chick cardiomyocytes, and extended these studies to show that caspase-2 acts on the mitochondria to induce or augment reperfusion-induced ROS generation, loss of ΔΨm, cyt c release and subsequently, cell death in ischemic cardiomyocytes.
In a few recent studies, an important role for caspase-2 has been implicated in cerebral ischemic injury,42,44 as well as in retinal reperfusion injury.45,46 I/R injury involving caspase-2 in murine germ cells 47 and in porcine proximal tubule cells 48 have also been reported, but few studies have addressed the role of caspase-2 in myocardial I/R injury. An early study of in vivo I/R injury implicated caspase-2 along with caspases-3 and −7 in a rabbit model of myocardial infarction and demonstrated protection with a broad spectrum caspase inhibitor, 12 but the sequence and specific roles of these caspases in the apoptotic pathway were not determined.
Evidence for a role for caspase-2 upstream of the mitochondria49–52 has stimulated a resurgence of interest in this caspase, and recent reviews summarize its potential roles in tumor suppression 6 and stress-induced apoptosis. 11 However, little information is available for the role of caspase-2 in pathological responses, such as myocardial I/R injury. 11 Subsequent to our study of caspase 2-mediated initiation of apoptosis in chick cardiomyocytes, 4 it was suggested that the protein ARC (apoptosis repressor with a CARD domain) 53 protected H9c2 rat myocytes from a caspase 2-mediated mitochondrial death pathway induced by oxidant stress. 54 These studies focused on ARC itself rather than the mitochondrial actions of caspase-2, however, and since ARC also binds other caspases, most notably including caspase-8, 53 it is difficult to differentiate the roles of caspase-2 and caspase-8 or to assign a definitive mitochondrial role for caspase −2 from these studies. Nevertheless, such reports support the critical involvement of caspase-2 in cellular responses to oxidative stress, particularly in skeletal or cardiac muscle.11,54
While there is evidence of caspase-8 participation in I/R injury in several organs,55–57 including the heart (see reviews),41,58 most studies do not distinguish upstream from downstream activities of this caspase and few compare it to caspase-2 with regard to mitochondrial events. Our previous studies showed a prominent (6- to 8-fold) increase in caspase-8 activity, but suggested that most of this activity resulting from reperfusion is seen following (by at least 2 hr) the rapid peak in caspase-2 activity that occurs within the first hour of reperfusion. 4 Caspase-8 activation is classically associated with ligation of and signaling through the death receptors (DR), most notably Fas/ CD95. Upon DR ligation, this pathway is initiated by formation of the so-called DISC (death-inducing signaling complex), leading to the cleavage and activation of pro-caspase-8. However, there are several reports that appear to link caspase-8 activation to caspase-2 activation. For example, both caspase-2 and caspase-8 appear to contribute to resveratrol-induced apoptosis of colon cancer cells. 17 Importantly, these studies also showed that caspase-2 inhibition by zVDVAD-fmk or siRNA could at least partially prevent caspase-8 activation, 17 as we had observed in our previous study. 4 Similarly, in certain cancer cells or cell lines, caspase-2 can cleave and activate caspase-8 and this activation can be blocked by zVDVAD-fmk or siRNA knockdown of caspase-2.13–16 While sequential activation of both caspases-2 and −8 was demonstrated upstream of the mitochondria in one of these studies, 13 our results suggest that only caspase-2 activity is necessary for initiating the reperfusion-induced mitochondrial ROS burst, cyt c release, and cell death. Similarly, the loss of ΔΨm, which accompanies ischemia in cardiomyocytes,26,36 remains low after untreated (vehicle-only) reperfusion. Protection of the cells with the caspase-2 inhibitor, but not the caspase-8 inhibitor, allowed partial recovery of the ΔΨm at reperfusion. This protection or “rescue” of these cardiomyocytes is similar to that seen, for example, by scavenging residual O2 36 or with NO treatment 26 and correlates with cell survival in this model.26,36
While caspase-3 (and the closely related caspase-7) are generally accepted as key downstream effectors or “executioner” caspases, there is evidence that caspase-3 can also target the mitochondria during apoptosis and cause the loss of ΔΨm and the enhanced generation of ROS.30,31 Studies in double knockout fibroblasts have suggested that caspases-3 and −7 are key mediators of the loss of ΔΨm in apoptosis stimulated by UV irradiation or staurosporine. 59 While caspase-3 is activated approximately 4-fold by 3 h of reperfusion in this model and its inhibition can at least partially inhibit cell death in our model,3,4 the results presented here suggest that the primary activities of caspase-3 (or caspase-7) are not upstream of mitochondrial dysfunction. This suggests that the documented activity of caspase-3 on the mitochondria either varies with cell- and stimulus-type or that it represents a downstream, amplifying event. Indeed, Ricci and colleagues 30 showed that in isolated mitochondria, caspase-3 induced ROS production through action on the ETC only in already-permeabilized mitochondria. Our results also suggest that in the (acute) timeframe of the ROS burst seen in I/R-induced cardiomyocytes, only caspase-2 directly affects ROS generation, and any mitochondrial actions of caspase-3 or caspase-8 are likely downstream or amplifying events. Indeed, downstream activation of caspases-3 and −8 have been previously demonstrated in several systems as likely amplifiers of apoptosis in a mitochondrial feedback loop.60,61
Finally, it is important to note limitations of the present study. As we have discussed in our previous reports,3,4 our model of simulated ischemia likely does not reproduce all the characteristics of “in vivo” ischemia, but we believe that this model recreates several critical components, including hypoxia, hypercarbia, hyperkalemia, and substrate deprivation.1–4 In comparison to some mammalian cell systems, there are also limitations to the avian cardiomyocyte system, but it also exhibits several important advantages, including reperfusion-induced oxidant generation, synchronous contractile function, and preconditioning protection.1–4 Moreover, with respect to the current study, we believe the well-documented evolutionary conservation of caspase-2,5–8,11,62 as well as remarkable conservation of other apoptotic molecules and mechanisms,63–66 make this model a useful one in such studies. In these studies, however, we were unable to assess the role of Bid, a BH3-only protein, which can be cleaved to its proapoptotic form by caspases-2 and −3, as well as by caspase-8,50,67 as we were unable to verify the reactivity of any available antibody for endogenous chicken Bid. Because of this limitation, we have recently moved some of our continuing studies of upstream activities of caspase-2 into our new murine model. 68 This model will permit more detailed analyses of Bid and other apoptotic proteins, as well as permit the use of (caspase-2) knockout animals to more rigorously examine these mechanisms of I/R injury. In addition, as previously discussed, caution must be exercised when interpreting results from the use of peptide caspase inhibitors, which may have some overlap in function. However, our previous studies 4 and studies from other groups indicate that of the caspases studied, caspase-2 may possess the most specific activity. 69 Together with the present data, including the lack of effect of the caspase-8 and caspase-3 inhibitors (which effectively block their respective caspases in these cells)4 on the mitochondrial events studied, provide strong evidence for caspase-2 activity in initiating these critical proapoptotic events.
In conclusion, these data confirm and extend our previous reports, which suggested that in I/R-induced apoptosis of isolated cardiomyocytes, caspase-2 functions upstream of the mitochondria and leads to the initiation of the mitochondrial pathway of cell death in these cells. Furthermore, they shed light on the mechanisms involved in caspase-2 actions on mitochondria in I/R-mediated cell death, with effects on critical mitochondrial apoptotic events such as ROS generation, loss of membrane potential and cyt c release. While our previous data demonstrated that caspase-8 was the most highly activated “initiator” caspase in I/R-induced cardiomyocytes in terms of fold-increase in activity from baseline or ischemia only levels, the present studies show that caspase-8 inhibition has little effect on these early mitochondrial events and confirm that blockade of caspase-8 activity does not protect cardiomyocytes from reperfusion-induced cell death. Nevertheless, these and similar data from other groups do not rule out potentially important downstream and perhaps amplifying roles for caspase-8 or caspase-3 in I/R-induced myocardial injury.30,31
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
This work was supported by NIH grants HL079641 (K.J. Hamann, P.I.), HL084643 (K.J. Hamann, P.I) and HL068961 (T.L. Vanden Hoek, P.I.) and by a DOD grant N00014-0401-0796 (T.L. Vanden Hoek, P.I.). We thank Isabelle Gagnon and Lisa Hoffman for additional technical support.