
Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a glycolytic enzyme, has been recently identified to be involved in the initiation of neuronal apoptosis. To investigate the serial changes and cellular localization of GAPDH expression, and its role in ischemia/reperfusion-induced neuronal apoptosis, the authors analyzed immunohistochemically brain areas of rats subjected to middle cerebral artery occlusion (MCAO) and reperfusion. Nuclear overexpression of GAPDH was noted in the ischemic core area after 2 hours of MCAO without reperfusion. During the subsequent reperfusion, nuclear accumulation of GAPDH in this area decreased in a time-dependent manner. However, cytoplasmic and nuclear GAPDH immunoreactivity was detected in neurons of the penumbra area of the parietal cortex, in rats subjected to 2-hour MCAO followed by 3-hour reperfusion. The increase of nuclear GAPDH immunoreactivity was persistently noted up to 48 hours of reperfusion, whereas cytoplasmic immunoreactivity correlated inversely with the duration of reperfusion. Moreover, double staining revealed colocalization of nuclear GAPDH and TUNEL in the penumbra area. The authors' study demonstrated that overexpression of GAPDH and nuclear translocation occurred in both the ischemic core and penumbra area soon after focal ischemia. These processes could be viewed as an early marker of ischemia/reperfusion-induced apoptotic neuronal death. The results suggest that GAPDH may play a critical role in the progression and spread of ischemic neuronal damage.
Keywords
Apoptosis, a form of programmed cell death, plays an important role in neuronal death, particularly in patients with degenerative neurologic diseases and ischemic stroke. In animal models of stroke, the morphologic changes of apoptosis are observed in neurons, such as shrinkage of total cell volume and nuclear condensation or DNA fragmentation (MacManus et al., 1994; Li et al., 1995). Other biochemical evidences of neuronal apoptosis can be identified in models of ischemic stroke, including the expression of Bcl-2 family proteins (Gillardon et al., 1996), release of mitochondrial cytochrome c (Fujimura et al., 1998), and activation of one or more members of the caspase family of cysteine proteases that regulate apoptotic cell death (Namura et al., 1998; Velier et al., 1999).
Recent studies have identified another mechanism of apoptotic cell death. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; EC 1.2.1.12) has long been considered a housekeeping enzyme, and its role in the glycolytic pathway has been well characterized. In addition to playing a prominent role in glycolysis in the cytosol, GAPDH binds to membranes to regulate endocytosis, probably via perturbation of the cytoskeletal structures (Caswell and Corbett, 1985; Robbins et al., 1995). In the nucleus, GAPDH participates in tRNA transport (Singh and Green, 1993), stimulates transcriptional activity (Morgenegg et al., 1986), and controls DNA replication and DNA repair (Meyer-Siegler et al., 1991; Baxi and Vishwanatha, 1995). Among these nonglycolytic activities, one of the most intriguing roles is as an integral part in one or more of the apoptotic death pathway. Overexpression of GAPDH and its subsequent accumulation in the nucleus is involved in neuronal apoptosis induced by several stimuli, such as aging in cultures, cytosine arabinoside, and low K+/serum deprivation, where GAPDH antisense oligodeoxynucleotides specifically prevent neuronal death in these apoptotic paradigms (Ishitani et al., 1996a,b,c, 1997). Moreover, it has been demonstrated by several independent laboratories that GAPDH is a general mediator of neuronal and nonneuronal cell death as a proapoptotic protein and uses nuclear translocation as a signaling mechanism (Sawa et al., 1997; Saunders et al., 1997; Ishitani et al., 1998; Shashideharan et al., 1999).
Recently, immunocytochemical examinations of postmortem tissues from patients with Parkinson's disease (Tatton, 2000) and Machado-Joseph disease (Ishitani et al., 2000) have clearly revealed nuclear accumulation of immunoreactive GAPDH in neurons of the affected brain regions. In addition, gene products of several neurodegenerative disorders, such as Huntington disease (Burke et al., 1996), spinocerebellar ataxia (Koshy et al., 1996), and Alzheimer disease (Schulze et al., 1993), have been reported to selectively interact with GAPDH. However, to our knowledge, there is no direct evidence of any relationship between GAPDH expression and ischemia/reperfusion-induced apoptosis of neurons, and there are also no reports on in situ detection of nuclear accumulation of GAPDH in neurons after an ischemic insult. The present study was designed to assess the serial changes and cellular localization of GAPDH during neuronal death as well as to determine whether the expression level of GAPDH correlates with ischemia/reperfusion-induced apoptosis of neurons in a rat model of transient middle cerebral artery occlusion (MCAO). The results presented here indicate that overexpression of GAPDH and its nuclear translocation are key processes in ischemia/reperfusion-induced apoptosis of neurons.
MATERIALS AND METHODS
Permanent and transient middle cerebral artery occlusion
All animal procedures were performed using a protocol approved by the Animal Care Committee of Juntendo University School of Medicine. Middle cerebral artery occlusion was performed in adult male Fischer 344 rats weighing 240 to 270 g (Charles River Japan Inc., Kanagawa, Japan) by the intraluminal suture method as described previously (Urabe et al., 1996).
The animals were anesthetized with 2% halothane–30% oxygen via a face mask, and the right common carotid artery and right external carotid artery were exposed through a midline neck incision. After the right external carotid artery was dissected free, an 18-mm-long silicon-coated 4–0 nylon suture was introduced into the internal carotid artery through the external carotid artery stump to occlude the middle cerebral artery. After 2 hours of right MCAO, the thread was removed to allow reperfusion. During this procedure, rectal temperature was kept at 37.0 ± 0.5°C using a heating pad (Unique Medical, Tokyo, Japan) and a heat lamp. As a control, sham-operated rats (n = 3) were subjected to the same procedure except for MCAO. Rats were killed at 2 hours after MCAO without subsequent reperfusion or after 3, 6, 12, 24, or 48 hours of reperfusion (n = 4, each). Under anesthesia with pentobarbital (50 mg/kg, i.p.), the brain was perfusion-fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and then removed rapidly. Postfixation was performed in the same fixative for 2 days at 4°C. Thereafter, each brain was frozen in powdered dry ice and coronal sections (30-μm thick) were cut on a cryostat (Leica CM1900; Leica Microsystems, Nussloch GmbH, Germany). The sections were stained with cresyl violet to evaluate the infarct area and neuronal damage.
Cerebrocortical cell culture
Cerebrocortical cell cultures were prepared from embryonic day 14 Wistar rats as described previously (Mochizuki et al., 1994) with some modifications. The cells were seeded at a density of 3.0 × 106 cells/well in 6-well tissue culture dishes precoated with polyethylenimine. After 7-day culture, these cells were treated with cytosine arabinoside for 48 hours. The cultured cells were incubated at 36°C and medium was refreshed twice weekly.
Characterization of ONO-3 anti-GAPDH monoclonal antibody
A monoclonal antibody (termed ONO-3) was raised against GAPDH purified from rabbit skeletal muscle. Briefly, the GAPDH antigen was prepared from rabbit skeletal muscle as described previously (Amelunxen and Carr, 1975) and injected into BALB/c mice. The immunized mice were then killed, and their spleen cells were fused to NE-1 myeloma cells. Hybridomas were used to generate ascitic fluid according to the standard method, and the resultant immunoglobulin G (IgG) was purified from the fluid by using a Protein A-diethylaminoethyl column. The specificity of this antibody was determined by immunoblotting. For this purpose, Fischer rat brain (frontoparietal cortex) was homogenized in RIPA buffer containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate in PBS, and a mixture of protease inhibitors (Protease Inhibitor Cocktail Set I; Calbiochem, Darmstadt, Germany) using a Teflon homogenizer. After centrifugation at 18,000g for 1 hour at 4°C, the supernatant was collected. Near-confluent cortical neurons in a 45-mm culture dish were washed twice with PBS and lysed with RIPA buffer and a mixture of protease inhibitors. The resultant lysate was centrifuged at 18,000g at 4°C for 1 hour and the supernatant was recovered. Chicken muscle GAPDH was purchased from Sigma (St. Louis, MO, U.S.A.). The respective lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis under reducing conditions. The separated protein bands were transferred to a nitrocellulose membrane (Hybond ECL; Amersham Life Science, Buckinghamshire, U.K.) and incubated with ONO-3 (1:1,000) for 1 hour, followed by incubation with the horseradish peroxidase–conjugated secondary antibody for 1 hour. The immunoreactive proteins were visualized by the enhanced chemiluminescence Western blotting detection system (Amersham).
Immunohistochemical staining
Immunostaining of cryostat sections was performed by the avidin-biotin-peroxidase complex method. The sections were incubated for 30 minutes with 0.3% H2O2 at room temperature, and then incubated overnight at 4°C with 10% normal goat serum (Dako Corporation, Carpinteria, CA, U.S.A.) and ONO-3 anti-GAPDH monoclonal IgG antibody at a dilution of 1:200. After washing in PBS, the sections were incubated for 2 hours at room temperature with biotinylated goat anti-mouse IgG (1:100 dilution) (Vector Laboratories, Burlingame, CA, U.S.A.). Then the sections were incubated with avidin–biotin–peroxidase (Vectastain ABC kit; Vector Laboratories) at a 1:1:100 dilution for 1 hour at room temperature. After washing in PBS, the sections were finally incubated for 10 to 20 minutes with a solution containing 0.02% 3,3-diaminobenzidine tetra-chloride and 0.03% H2O2 in PBS. Sections were dehydrated in serially concentrated ethanol solutions, immersed in xylene, and coverslipped with Malinol (Muto Pure Chemicals Co., Ltd., Tokyo, Japan).
Double immunofluorescence staining
For double staining of microtubular-associated protein 2 (MAP-2), glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter molecule 1 (Iba 1), and GAPDH, coronal brain sections were first incubated overnight at 4°C with 10% normal swine serum (Dako) and anti-MAP-2 polyclonal antibody (dilution at 1:200; Bio-Rad Technologies, Richmond, CA, U.S.A.), anti-GFAP polyclonal antibody (dilution at 1:10,000), or anti-Iba 1 polyclonal antibody (diluted at 1:400, a kind gift from Dr. Kohsaka) in PBS-Tween 20. After washing in PBS, the sections were incubated with fluorescein isothiocyanate–conjugated swine anti-rabbit IgG (1:40 in PBS, Dako) for 1 hour at room temperature and then washed in PBS. Next, each section was incubated overnight at 4°C with 10% normal rabbit serum (Dako) and ONO-3 (1:200 in PBS-Tween 20). After washing with PBS, sections were incubated for 1 hour at room temperature with tetramethylrhodamine-conjugated rabbit anti-mouse IgG (1:40, Dako), followed by another washing with PBS. Nuclear staining was carried out using Hoechst 33258 (Wako Pure Chemical Industries, Osaka, Japan) after staining for ONO-3 as already mentioned. TUNEL staining was performed using a commercially available kit (In Situ Cell Death Detection, POD; Boehringer Mannheim, Mannheim, Germany) and double staining for GAPDH and TUNEL was also performed. The respective colocalization of GAPDH and MAP-2, GFAP, Iba 1, Hoechst 33258, or TUNEL in the brain sections was observed using a confocal laser scanning microscope. The sections were first examined with a conventional fluorescence microscope (EclipseTE300; Nikon, Tokyo, Japan), and then with the confocal laser scanning microscope (Axiovert 100M) using the LSM 510 system (Zeiss, Tokyo). All images were obtained from individual optical sections. Data were minimally processed to generate superimposed images by using Laser Sharp processing and Photoshop software (Adobe System, San Jose, CA, U.S.A.).
Cell count and statistical analyses
Sections were examined by confocal microscopy and stained cells in 0.25 mm2 of each MCA territory (Fig. 1) were counted. The stained sections were assessed independently by two investigators (R.T. and T.U.) and data were expressed as mean ± SD of four independent experiments. Statistical analysis was performed using one-way analysis of variance followed by Fischer's protected least significant difference test. P < 0.05 was considered to indicate significance.
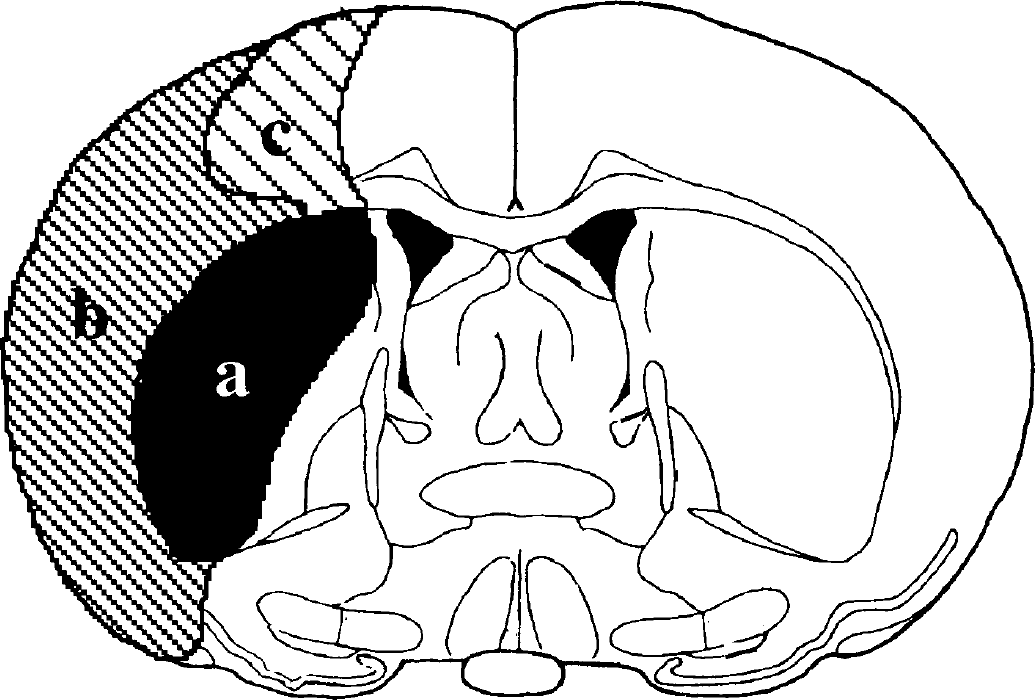
Schematic representation of the distribution of neuronal damage in rat brain after transient focal ischemia delineated by cresyl violet staining. Three areas subjected to analysis of immunohistochemistry are illustrated.
RESULTS
Cresyl violet staining in the lateral part of the striatum had already disappeared after 2 hours of MCAO. In rats subjected to ischemia followed by 6 hours of reperfusion, areas of infarction were noted in the striatum as well as in MCA-related areas of the parietal cortex. The extent of infarction further widened at 48 hours after reperfusion to include the entire MCA territory of the parietal cortex. Schematic representation of the extent of ischemia is shown in Fig. 1.
To determine the specificity of ONO-3 anti-GAPDH monoclonal antibody, Western blots were prepared using different lysates, including rat brain lysate (Figure 2, lane 2) and rat cortical neuron lysate (lane 3). The blots revealed a single band at a molecular mass of 38 kd, which comigrated with the purified GAPDH from chicken muscle (Fig. 2, lane 1). These findings indicated that ONO-3 could react with monomeric GAPDH of rat brain and neurons. Results of preliminary studies of immuno-dot blot analysis conducted in our laboratories indicated that this antibody could detect nuclear accumulation of GAPDH in cultured rat cerebellar granule cells before apoptosis (R. Ishitani and T. Kuwae, April 2001, unpublished observations). In this context, previous studies have indicated that nonglycolytic GAPDH protein in the nucleus is present preferentially in monomeric form (Sirover, 1999).
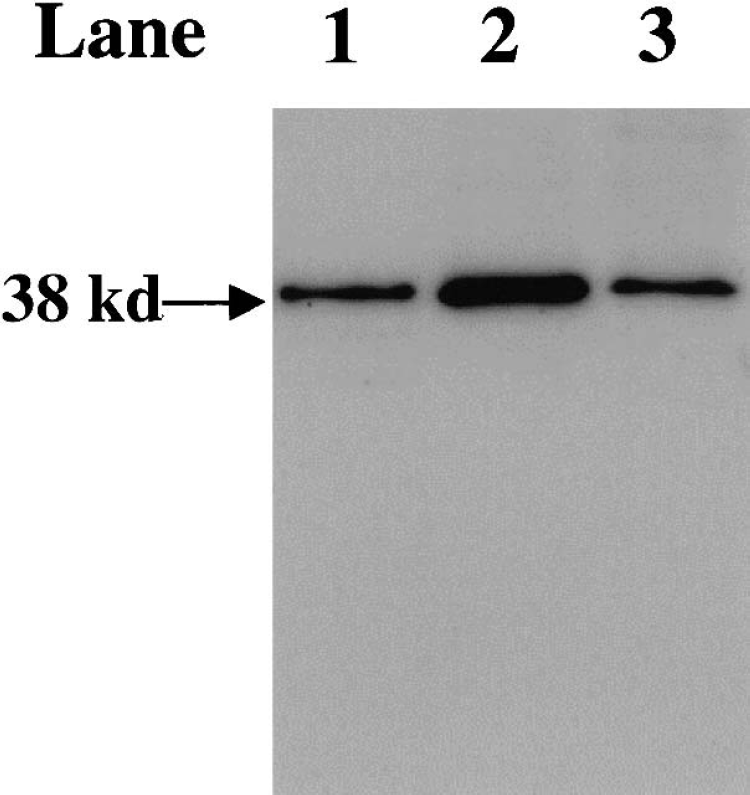
Specificity of ONO-3, an immunoglobulin G monoclonal antibody raised against glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Immunoblotting patterns of lysates (lanes 2 and 3) and purified GAPDH (lane 1). Samples were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. After protein transfer, those blots were incubated with ONO-3 as described in Materials and Methods. Lane 1: chicken muscle GAPDH. Lane 2: rat brain lysate. Lane 3: cultured cortical neuron lysate. The blots reveal a single band at a molecular mass of 38 kd.
Overexpression of GAPDH was not observed in brains of sham-operated normal rats or in the contralateral hemispheres of MCA-occluded rats (Fig. 3A). After 2 hours of MCAO without subsequent reperfusion, few GAPDH-positive cells were seen in the striatum (data not shown), but immunoreactivity for GAPDH was prominent in the ischemic core of the parietal cortex and the immunoreactivity was noted in nuclei and/or both in nuclei and cytoplasm (Fig. 3B). However, nuclear accumulation of GAPDH decreased proportionately with the duration of the succeeding reperfusion (Fig. 3C). At 48 hours after reperfusion, a number of round- or irregular rod-shape GAPDH-positive cells were detected in this area (Fig. 3D).
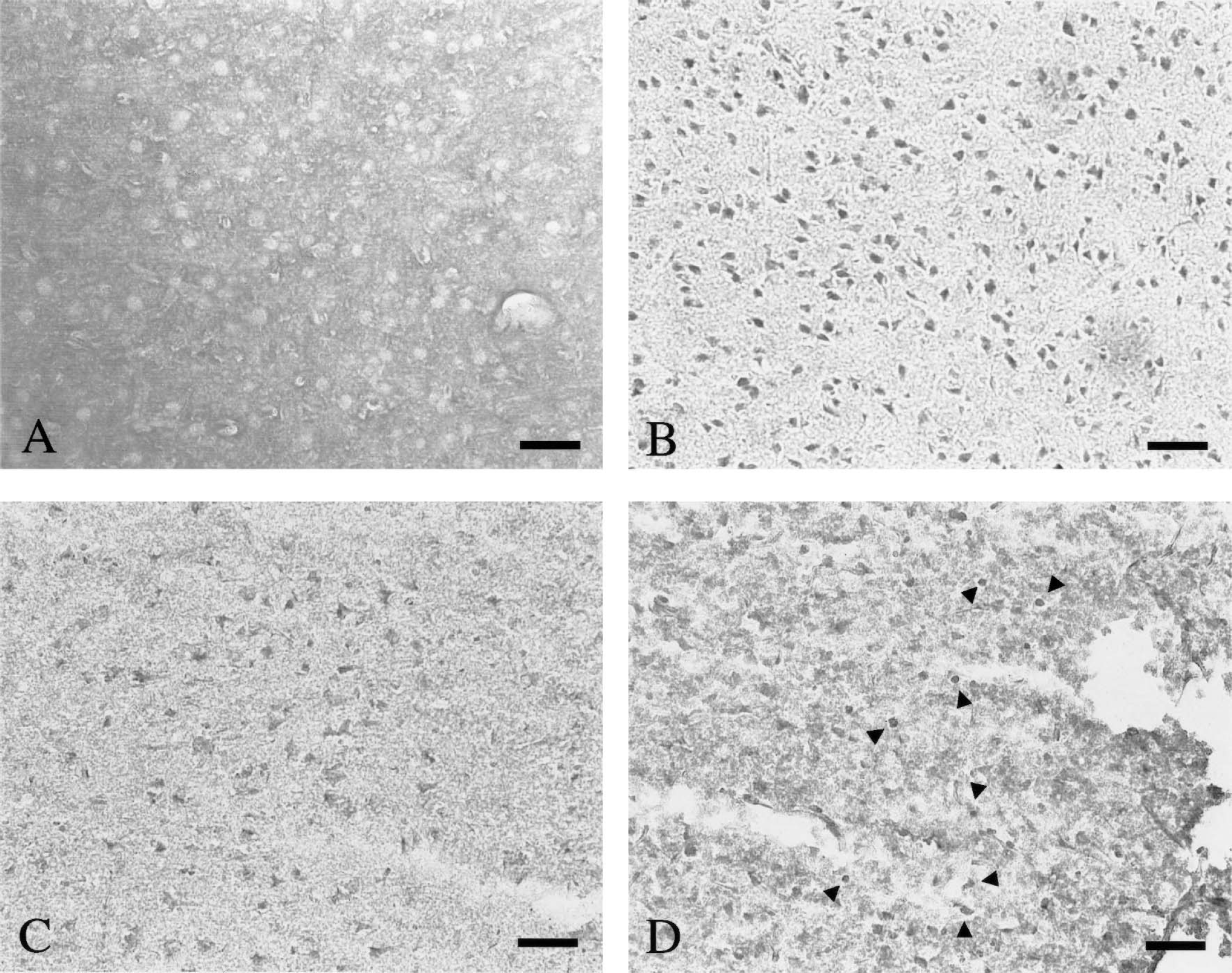
Immunostaining for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in rat brains after middle cerebral artery occlusion (MCAO) and reperfusion. GAPDH immunoreactivity is scarcely detected in the contralateral side of parietal cortex
After 3 hours of reperfusion, a significant number of nuclear GAPDH-positive neurons became evident in the penumbra area (Fig. 4B). Some cytoplasmic GAPDH-positive neurons were also detected in this area (Fig. 4B), but such neurons were abundant in parts distant from the ischemic core (Fig. 4A). Nuclear GAPDH immunoreactivity was markedly increased in this area and was persistently seen at after 12 hours and up to 48 hours of reperfusion (Figs. 4C and 4D). In contrast, cytoplasmic GAPDH decreased in a time-dependent manner. Quantitative cell count data of GAPDH-positive cells in each area during cerebral ischemia and ischemia followed by reperfusion are shown in Fig. 5.
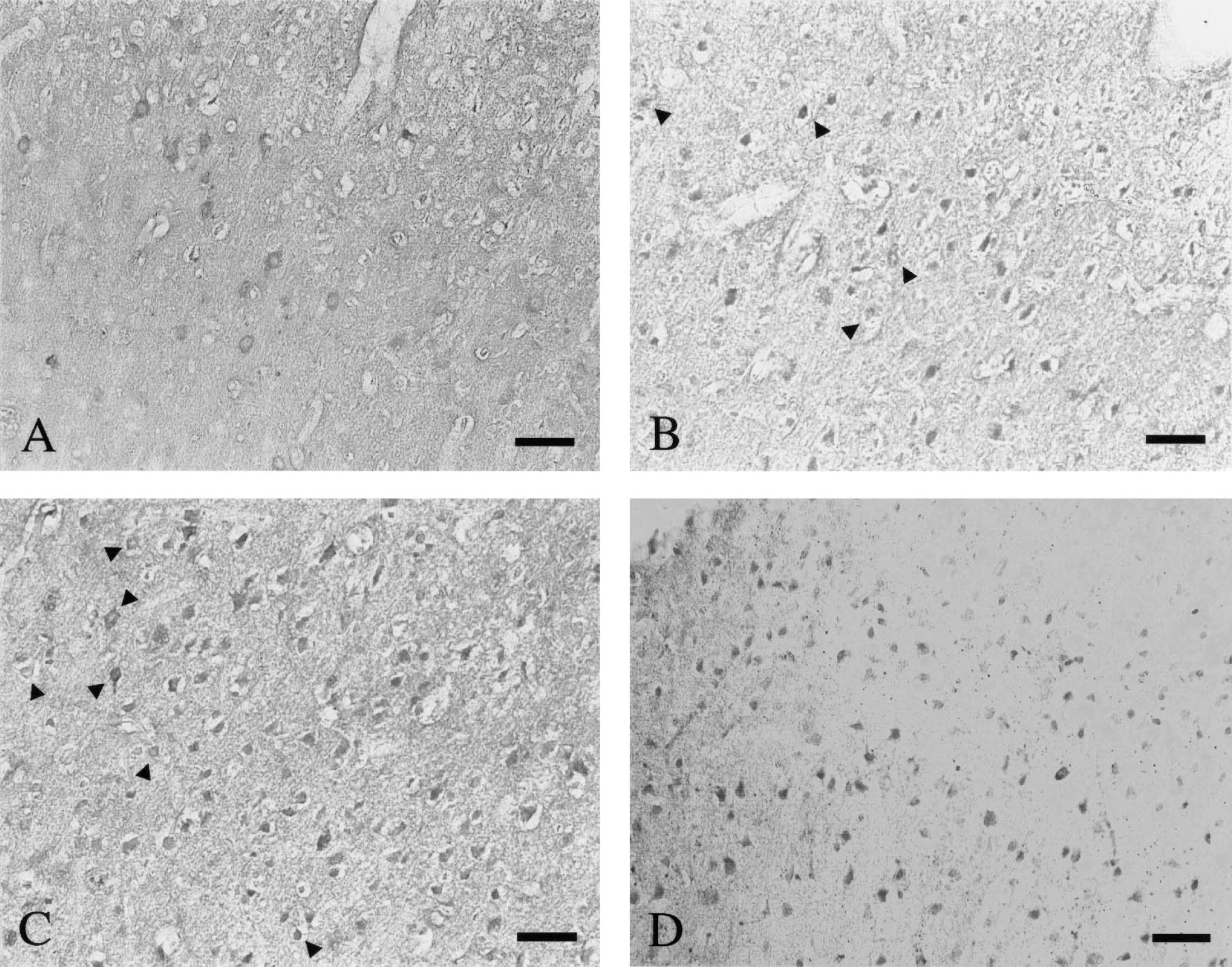
Immunostaining for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the parietal cortex at the penumbra zones after middle cerebral artery occlusion followed by reperfusion. A number of cells with nuclear accumulation of GAPDH are observed
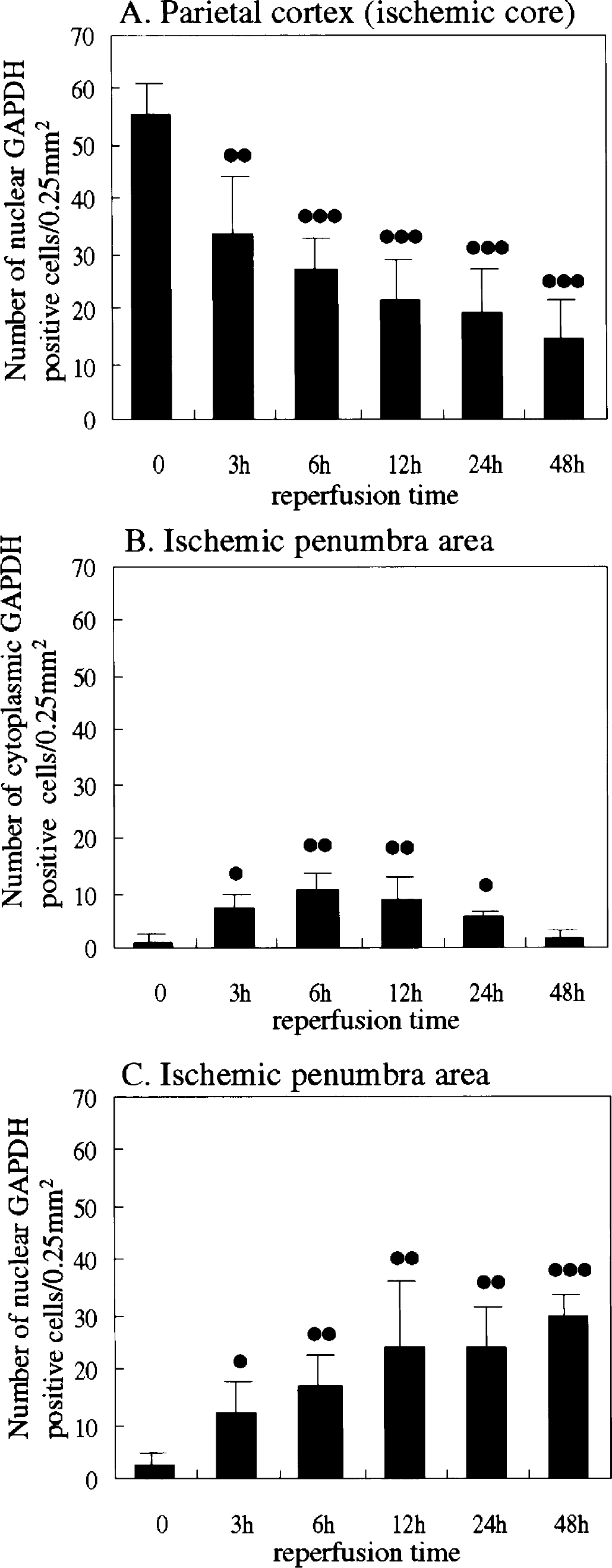
The number of glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-positive neurons was counted in each parietal cortex of middle cerebral artery areas including ischemic core
To identify positively stained cells and the location of their inherent organelles, we performed double staining for GAPDH and MAP-2, GFAP, Iba 1 or Hoechst 3325. MAP-2-positive neurons showed GAPDH immunoreactivity within both the ischemic core (Figs. 6A to 6C) and penumbra area (Figs. 6D to F). At 48 hours of reperfusion, a few GAPDH-positive neurons were seen in the ischemic core area, whereas a certain number of Iba 1–positive microglia/macrophages were detected in this area (Figs. 6G to 6I). However, colocalization of GAPDH with GFAP could not be recognized (data not shown). Double staining for GAPDH and Hoechst 33258 showed the presence of nuclear GAPDH in the ischemic core and penumbra area (Figs. 6M to 6O), whereas cytoplasmic GAPDH immunoreactivity was detected predominantly in the penumbra area (Figs. 6J to L).
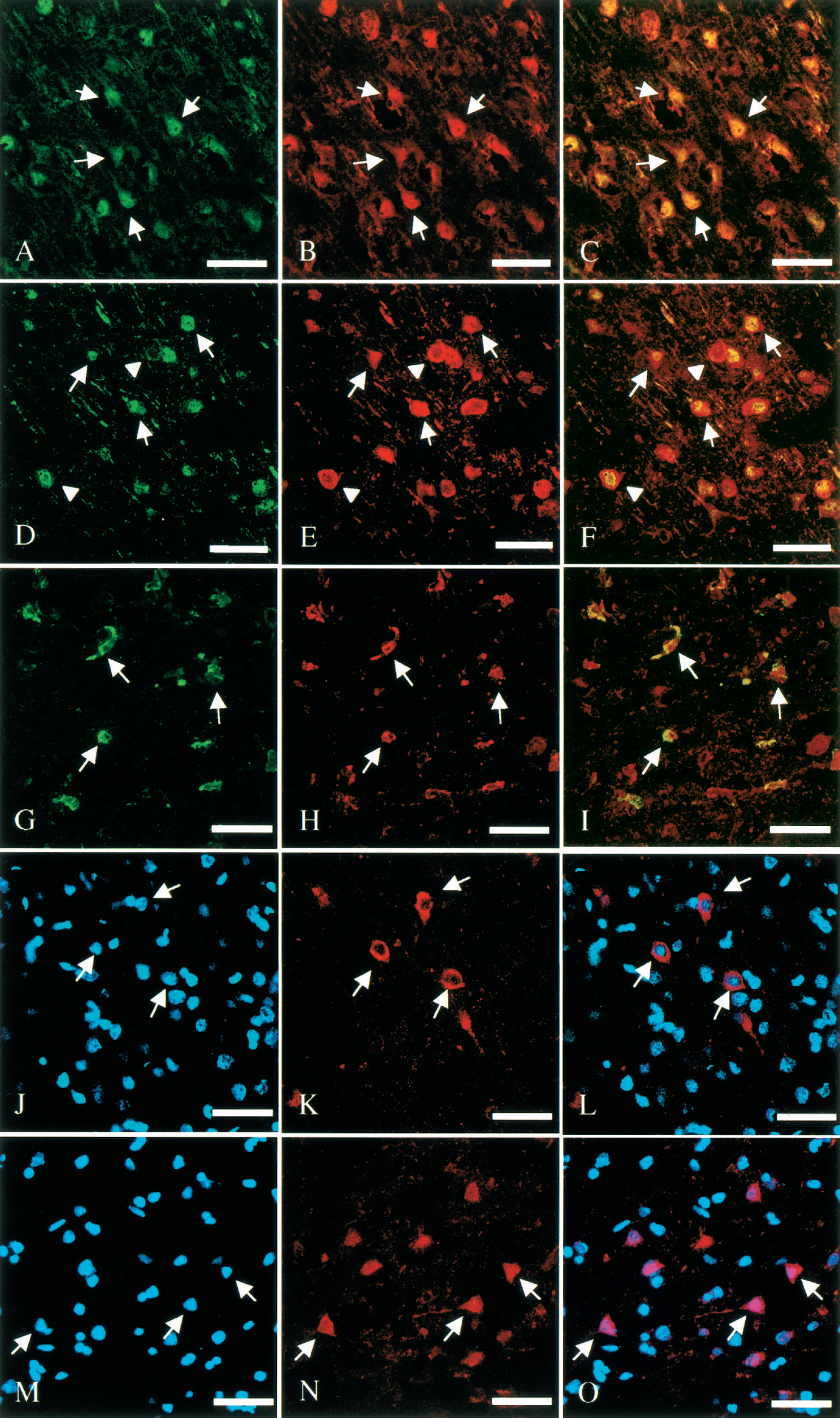
Photomicrographs showing double immunofluorescence labeling and confocal images of the rat brain focal cerebral ischemia followed by reperfusion. Microtubular-associated protein 2 (MAP-2) (
A few TUNEL-positive cells were detected in the lateral area of the caudate/putamen after 6 hours of reperfusion. These cells increased in number in a time-dependent manner; thus, abundant TUNEL-positive cells were identified at 48 hours of reperfusion in the ipsilateral caudate/putamen and in the infarct zone of the parietal cortex. Colocalization of GAPDH with TUNEL-positive cells was scarcely seen in the ischemic core of parietal cortex, whereas these types of cells were present in a considerable number in the penumbra area (Fig. 7).
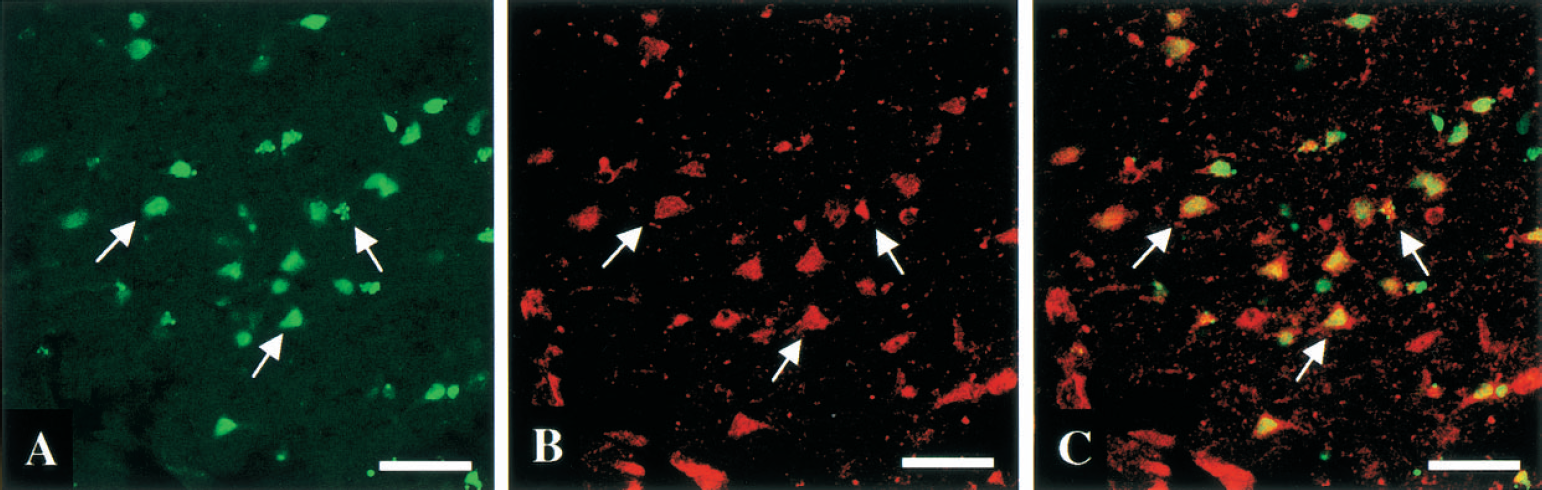
Photomicrographs showing double immunofluorescence labeling and confocal images of the penumbra area after 48 hours of reperfusion. TUNEL-positive cells are green
DISCUSSION
The present study clearly demonstrated early nuclear translocation of GAPDH (i.e., nuclear accumulation), a marker of apoptotic neuronal death, in the ischemic core and penumbra region after focal ischemia/reperfusion. These results are well consistent with the working hypothesis that overexpression and nuclear translocation of GAPDH may be an important trigger of apoptotic neuronal death in the ischemic brain. Recent studies provided a compelling evidence that the cells overexpressing GAPDH, which are transfected with GAPDH cDNA, execute apoptotic death, and GAPDH overexpression is strikingly associated with nuclear translocation of GAPDH (Tajima et al., 1999; Shashidharan et al., 1999). To our knowledge, however, the present study is the first report describing in situ detection of overexpression and subsequent nuclear accumulation of GAPDH and its implication in neuronal apoptosis in ischemia/reperfusion–injury.
In this type of murine focal ischemic stroke model, the occurrence of necrotic death has been reported in the ischemic core zone, whereas apoptotic death can be seen in the periinfarct zone (Li et al., 1995). It has also been reported that TUNEL-positive neurons in the ischemic penumbra are mainly apoptotic cells, whereas those in the ischemic core tend to be necrotic cells (Charriaut-Marlangue et al., 1996). In our study, a marked nuclear accumulation of GAPDH at 2 hours of permanent MCAO was detectable transiently in the ischemic core of the parietal cortex but disappeared time-dependently at progressive stages of ischemia. However, we also observed a few GAPDH-positive cells in the lateral part of the striatum where infarction had already occurred. These results suggested that the nuclear translocation of GAPDH in ischemic neurons might be induced as some signaling mechanism preceding the determination of cell death, and finally be necrotic but not apoptotic cell death because of the large reduction in cerebral blood flow. In this regard, glutamate elicits neuronal necrosis or apoptosis, depending on mitochondrial function and energy supply (Nicotera and Lipton, 1999) and it should be noted that secondary necrosis also involves overexpression of GAPDH at its early stage (Ishitani et al., 1997). However, another interesting observation supports the involvement of GAPDH overexpression in the apoptotic neuronal pathway of this paradigm. Because increased levels of GAPDH mRNA have been observed both in in vivo and in vitro models of ischemia/hypoxia (Graven et al., 1994; Feldhaus and Liedtke, 1998), it seems likely that the ischemic insult in our study induced upregulation of GAPDH at the transcription level, similar to several classical apoptotic death paradigms reported previously (Ishitani et al., 1996a,b, 1997).
Ionized calcium-binding adapter molecule 1 is one of the specific markers of microglia/macrophages, and enhanced expression of this molecule has been reported in the ischemic core and periischemic area of the rat brain after transient focal ischemia (Ito et al., 2001). In the present study, a certain number of GAPDH-positive microglia/macrophages were detected in the ischemic core area at 48 hours after reperfusion. Because macrophages within the infarct tissue express Bax protein, which results in nuclear DNA fragmentation (Mabuchi et al., 2000), we might also be able to detect GAPDH-positive apoptotic macrophages in this area. However, further studies are needed to confirm the presence of such cells.
In the periinfarct penumbral region, we provided evidence implicating overexpression of GAPDH protein in ischemic cell death. After a short period of reperfusion, an increase in nuclear and cytoplasmic GAPDH immunoreactivity was detected in this zone. However, a number of cytoplasmic GAPDH-positive cells were still detected at a later period of ischemia. Although we did not have direct evidence for the translocation of overexpressed cytoplasmic GAPDH to the nucleus in our in vivo model and there is no information about the role of cytoplasmic GAPDH, serial intracellular localization and distribution of GAPDH-positive cells in this area suggested overexpression and subsequent nuclear translocation of GAPDH in ischemia/reperfusion injury, and that these two processes may contribute to the evolution of neuronal toxicity. Moreover, after 48-hour reperfusion, many neurons showed colocalization of GAPDH and TUNEL in this area (Fig. 7). This change in intracellular distribution of neuronal GAPDH at the penumbra zone occurred before the onset of cell death–related morphologic changes. These observations suggest that oxidative stress induced by reperfusion after cerebral ischemia might lead to serial changes in GAPDH immunoreactivity. Interestingly, it has been demonstrated that oxidative stress induces a marked increase of GAPDH mRNA in the rabbit aorta without alteration of its glycolytic activity (Ito et al., 1996). By using the GAPDH-green fluorescent cDNA chimera, it has been demonstrated that exposure to oxidative stress induces nuclear translocation of the fusion protein in transfected cells (Shashidharan et al., 1999). Although the molecular mechanisms underlying the nuclear translocation of overexpressed GAPDH is still unclear, this is the first observation suggesting that transient overexpression of cytoplasmic GAPDH, associated with nuclear accumulation of GAPDH, represent subcellular apoptotic signals in the early stage of reperfusion.
Finally, our findings suggest that overexpression of GAPDH is one of the key mediators of ischemia/reperfusion-induced neuronal signaling for apoptotic death, and that GAPDH immunostaining may be a useful tool for detecting neuronal apoptosis induced by an ischemic insult. Although additional studies are necessary, the present results suggest that proapoptotic GAPDH protein could be viewed as a putative molecular target for prevention of ischemic neuronal death.
Footnotes
Acknowledgments:
The anti-Iba1 polyclonal antibody was a gift from Y. Imai and S. Kohsaka at Department of Neurochemistry, National Institute of Neuroscience, Tokyo, Japan.