
Abstract
Apoptosis is an important contributor to the pathophysiology of lung diseases such as acute lung injury (ALI) and chronic obstructive pulmonary disease (COPD). Furthermore, the cellular environment of these acute and chronic lung diseases favors the delayed clearance of apoptotic cells. This dysfunctional efferocytosis predisposes to the release of endogenous ligands from dying cells. These so-called damage-associated molecular patterns (DAMPs) play an important role in the stimulation of innate immunity as well as in the induction of adaptive immunity, potentially against autoantigens. In this review, we explore the role of apoptosis in ALI and COPD, with particular attention to the contribution of DAMP release in augmenting the inflammatory response in these disease states.
Keywords
Introduction
Homeostatic cell death is essentially non-inflammatory. Furthermore, programmed cell death contributes to tolerance of host antigens made accessible to the immune system during normal physiologic processes, such as organ development. These anti-inflammatory and immunosuppressive effects depend on several factors that drive the biology of apoptosis, including the specific cell type undergoing death, the cause of cell death, the microenvironment surrounding the dying cell, and the final clearance of apoptotic debris. 1
Efficient removal of apoptotic cells, also known as efferocytosis, 2 is central to controlling potentially damaging inflammation and hastening tissue repair. 3 Ineffective efferocytosis can cause apoptotic cells to undergo delayed “secondary” necrosis, with release of potentially damaging intracellular and cell-surface molecules. These endogenous cellular components constitute the damage-associated molecular patterns (DAMPs). In a manner similar to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), DAMPs can activate dendritic or other target cell pattern receptors (e.g. Toll-like receptors (TLR)), influencing the process of immunity and tissue responses to injury. 1 Thus, the release of DAMPs from necrotic cells can engender a second wave of tissue damage during acute processes and, if chronic, potentially trigger auto-immune processes. Indeed, DAMPs have been implicated in the pathogenesis of a variety of acute and chronic pulmonary diseases (Fig. 1).4–30
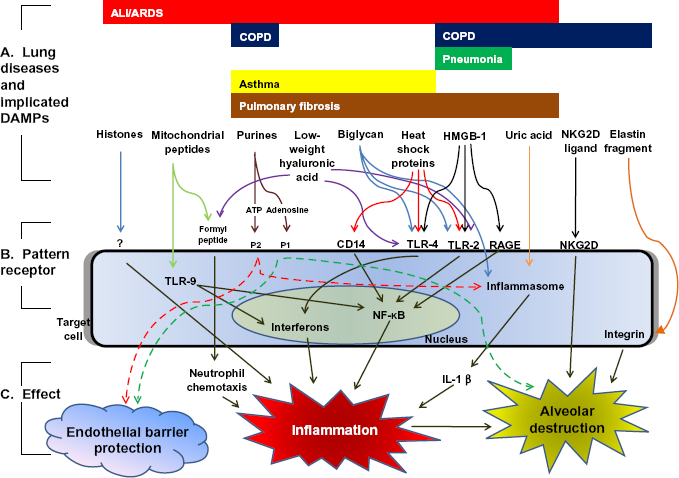
DAMPs implicated in lung diseases.
There is growing evidence of the presence of apoptosis in lung diseases such as emphysema, a chronic obstructive pulmonary disease (COPD) caused by cigarette use.31,32 The increased expression of apoptosis markers in emphysema, however, does not translate to an anti-inflammatory state: COPD is characterized by chronic inflammation that persists even after smoking cessation. 33 This paradoxical finding in emphysema may, in part, reflect cigarette smoke-associated impairment in macrophage-mediated efferocytosis. 34 The consequent increase in secondary necrosis may lead to release of DAMPs, triggering inflammation and autoimmunity. 35 An expanding body of experimentation in humans and animal models has advanced the concept that auto-immunity against lung cells contributes to the persistent inflammation found in COPD. 35 Both apoptosis and cellular senescence/aging may participate in the breach of immune tolerance, ultimately accounting for the chronic inflammation seen in COPD. 36
Excessive apoptosis and/or deficient efferocytosis may also affect the outcome of acute lung diseases, such as acute lung injury (ALI). ALI and its more severe variant, the acute respiratory distress syndrome (ARDS) are associated with excessive apoptosis.37–39As with smoking-induced emphysema, ALI is also associated with factors (e.g. tumor necrosis factor—alpha (TNF-α)) that impair efferocytosis. 40 DAMPs released from apoptotic and secondarily necrotic cells may thus contribute to the pathophysiology of ALI.
In this review, we will focus on ALI and COPD, diseases paradigmatic of the influence of apoptosis on lung disease. We propose the hypothesis that excessive apoptosis and ineffective efferocytosis lead to release of inflammatory mediators from apoptotic and/or secondary necrotic cells, exacerbating lung dysfunction. Acutely, these processes lead to activation of innate immunity, contributing to inflammatory diseases such as ALI. Chronically, these processes trigger adaptive immunity against self structures, leading to persistent lung inflammation as seen in COPD. It is in this light that DAMPs exposed from apoptotic cells or from tissue injury are promising therapeutic targets in acute and chronic lung diseases.
Apoptosis in Acute Lung Disease: ALI
Alveolar cell apoptosis and ALI pathophysiology
ALI is a common critical illness, affecting over 190,000 patients annually in the United States. 41 This syndrome has a greater than 30% mortality and significant morbidity: those who survive are often left with prolonged functional impairment. 41 The pathophysiology of ALI is characterized by aberrant pulmonary endothelial and epithelial barrier function. This barrier dysfunction leads to pulmonary edema, producing acute hypoxemia. 42
Despite over 40 years of study, the mechanisms underlying ALI pathogenesis remain unclear. ALI is associated with increased pulmonary concentrations of proinflammatory cytokines and neutrophil extravasation into the alveolar space,42,43 suggesting a role for innate immunity in disease onset and propagation. The influence of adaptive immunity in ALI is unknown, although recent work has demonstrated an important role for regulatory T-cells in lung injury resolution and repair. 44 Indicative of this lack of a clear understanding of ALI pathogenesis, no disease-specific therapy has been shown to improve survival in ALI. 45
Alveolar septal cell apoptosis likely contributes to ALI pathogenesis in response to various environmental stimuli, 46 perhaps by induction of endothelial and epithelial barrier dysfunction. 39 Analysis of bronchoalveolar lavage fluid collected from ARDS patients demonstrated increased concentrations of Fas/Fas Ligand, sufficient to induce epithelial apoptosis in-vitro. 47 Prevention of Fas/Fas Ligand-induced alveolar cell apoptosis attenuated ALI and improved survival in a mouse cecal ligation-puncture model of sepsis. 48 Furthermore, inhibition of alveolar apoptosis with the pan-caspase inhibitor z-VAD-fmk prevented sepsis-induced 49 and ventilator-induced 39 lung injury.
While apoptosis may induce barrier dysfunction early in ALI, increasing evidence suggests that apoptosis plays a beneficial role during ALI resolution. Apoptosis may limit the duration of pulmonary inflammation by curtailing neutrophil lifespan. 50 The beneficial influence of apoptosis in ALI can be further explained by the proregenerative role of clearance of apoptotic cells. This beneficial effect is mediated via the production of growth factors, including vascular endothelial growth factor (VEGF) and hepatocyte growth factor, from macrophages engulfing apoptotic cells. 3 Phosphatidylserine-mediated apoptotic cell removal triggers production of anti-inflammatory transforming growth factor (TGF)-β and prostaglandins, 3 allowing for confinement of the extent of septal injury and hastening recovery in ALI. 44 More recent evidence linked efferocytosis with upregulation of regulatory T cells and improved recovery from LPS-induced lung injury. 44
Systemic DAMP release as a cause of ALI/ARDS
Systemic illnesses (such as sepsis or massive hemorrhage) may trigger ALI, presumably through elaboration of circulating mediators capable of inducing barrier dysfunction. The search for these circulating mediators has led to increased interest in the role of DAMPs in the pathogenesis of lung injury. (Fig. 2) Systemic inflammatory conditions associated with ALI (e.g. sepsis, burn injury, trauma) are often characterized by extensive multi-system tissue necrosis and apoptosis.51–53 As the cytokine milieu of these inflammatory conditions can impair effective efferocytosis,38,40 secondary necrosis may additionally contribute to increased levels of circulating DAMPs. In contrast to rapidly-released inflammatory cytokines (such as TNF-α and IL-1β), the release of DAMPs during systemic illness is often delayed. 54 Given that patients at risk for ALI often present to medical attention well into the course of their initial systemic inflammatory illness, therapies aimed at blocking late-acting DAMPs may have greater clinical relevance than therapies aimed at more rapidly released mediators.55,56
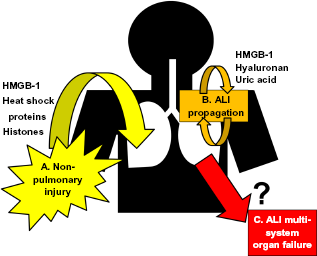
Potential contribution of DAMPs to ALI onset, propagation, and associated multisystem organ failure.
High mobility group box 1 (HMGB-1), a late-acting mediator of sepsis and other systemic inflammatory conditions, has thus attracted interest as a potential therapeutic target in the prevention of non-pulmonary triggered ALI.55,57 HMGB-1 is a ubiquitous nuclear protein released into the extracellular space by activated inflammatory cells or cells undergoing primary necrosis—cell types common to conditions capable of triggering ALI. Interestingly, it was believed that HMGB-1 is not elaborated by cells undergoing secondary necrosis after failed efferocytosis.58,59 However, recent studies have challenged this concept, suggesting that the release of HMGB-1 during secondary necrosis varies according to cell type. 60 Once released, HMGB-1 is a potent mediator of lung inflammation, 10 acting on pattern receptors such as TLR-4, TLR-2, TLR-9 and the receptor for advanced glycosylation end-products (RAGE). 61 In animal models of systemic injury, inhibition of HMGB-1 is associated with a significant attenuation of pulmonary endothelial barrier dysfunction and lung injury (Table 1).10,56,62–82 In human patients with severe traumatic injury, serum HMGB-1 concentrations at initial presentation predict the risk of ALI development during the subsequent hospitalization. 83
HMGB-1 as a therapeutic target in the prevention of ALI/ARDS.
Heat shock proteins (HSPs) have been similarly implicated as triggers of ALI during systemic illness. HSPs are highly conserved, abundant cytosolic proteins induced during environmental stress; this induction typically provides protection against lung injury. 9 During sepsis 84 and other systemic inflammatory conditions, however, HSPs are released into the extracellular space via cellular necrosis or active secretion. 9 Extracellular HSPs function in a dramatically different fashion than intracellular HSPs. Extracellular HSP70 triggers innate immunity via activation of TLR2, TLR4, or CD14,85–87 potentially initiating lung injury. 88 Extracellular HSP60 may similarly induce ALI after traumatic injury via TLR2/4 signaling.89,90
While less well studied, other DAMPs may contribute to the development of ALI from non-pulmonary illness. Mitochondrial contents can be released from dying cells after trauma; these contents activate neutrophils via TLR-9 and formyl peptide receptor-1, inducing pulmonary inflammation and edema in a rat model. 5 Similarly, nuclear histones are released into the extracellular space during sepsis; these histones potently contribute to sepsis-induced mortality and ALI. 4 Additionally, sepsis is marked by degradation of the endothelial glycocalyx lining the systemic vasculature, releasing proteoglycans and gly-cosaminoglycans into the circulation.91–93 The soluble proteoglycan biglycan contributes to lung inflammation in animal models of sepsis via TLR-2 and TLR-4 signaling. 8 Furthermore, septic degradation of hyaluronic acid (a glycocalyx-associated glycosamin-oglycan) into circulating low weight fragments could potentially induce lung injury via activation of lung TLR-4.7,94 The consequence of the systemic release of other glycosaminoglycan fragments (e.g. heparan sulfate) is uncertain.
Local production of DAMPS in the injured lung
Lung pathology in ALI is defined by extensive necrosis as well as apoptosis of a variety of cell types.95,96 As the cytokine milieu of an injured lung includes mediators (such as TNF-α) known to hamper efferocytosis, apoptotic cells may ultimately undergo secondary necrosis.40,43 Thus, observed increases in pulmonary DAMP concentrations during lung injury likely reflect not only primary cellular necrosis (capable of elaborating HMGB-1) 97 but also secondary necrosis of apoptotic cells, with consequent elaboration of non-HMGB-1 DAMPs such as hyaluronic acid98,99 or uric acid 11 (Fig. 2).
As described previously, much of the attention to HMGB-1 has focused on its role as a cause of “indirect”, non-pulmonary triggered acute lung injury. However, HGMB-1 may also be directly produced within a distressed lung by activated inflammatory cells or necrotic alveolar cells, leading to additional tissue damage and a propagation of lung injury.10,81 This production is directly damaging to the lung, as evidenced by the injurious effects of intratracheal or intrabronchial HMGB-1 administration to rats 100 and mice. 101 Antagonism of HMGB-1 attenuates pneumonia- and ventilator-induced lung injury (Table 1).
Other DAMPs released during primary or secondary necrosis may also participate in the propagation of lung injury. Animal models of VILI are marked by increased production of low molecular weight HA, 98 potentially via extracellular matrix fragmentation. Administration of a hyaluronic acid-blocking peptide decreased alveolar inflammation after bleomycin instillation. 7 Uric acid, a primarily intracellular molecule that forms inflammatory monosodium urate once released into the extracellular space, is also increased in the alveolar fluid of patients with ALI.11,58 Alveolar uric acid may directly activate the NALP3 inflammasome, inducing IL1-β and further augmenting lung inflammation. 26 Furthermore, extracellular matrix degradation during lung injury may reveal cryptic peptide sequences in matrix proteins such as lami-nin α-5, activating inflammatory cell production of matrix metalloproteinases and potentially augmenting ALI. 102
While pulmonary production of some DAMPs may exacerbate lung injury, others mitigate ongoing injury. Nucleotides released from dying cells may provide endothelial barrier protection (and oppose lung injury) via activation of P2 purinergic receptors.103,104 Additionally, extracellular ATP is converted into adenosine, a molecule that improves endothelial barrier function in models of VILI. 6 These protective effects of extracellular purines, however, may be opposed by ATP activation of the purine receptor P2X(7), stimulating dendritic cell production of inflammatory JL-1β.105,106
Pulmonary DAMP production as a cause of ALI-induced multisystem organ failure
ALI-associated barrier dysfunction facilitates access of alveolar cytokines to the systemic circulation. 107 Thus, DAMPs produced by the distressed lung may become systemically distributed, amplifying its pathogenetic effects. (Fig. 2) Indeed, in a cohort primarily composed of patients with pneumonia-induced ALI, serum HMGB-1 concentrations were markedly elevated in comparison to healthy controls. 108 In a mouse model of pneumonia-induced ALI, plasma levels of HMGB-1 were significantly elevated. 81 Increased levels of circulating DAMPs may be recognized by other organ systems via pattern receptors such as RAGE (receptor for advanced glycosylation end products) and TLRs. HMGB-1 induced RAGE activation has been associated with renal dysfunction in models of systemic lupus erythematosis. 109 Activation of renal TLR-4 and TLR-2 by endogenous DAMPs (including HMGB-1 and hyaluronic acid) is known to mediate renal ischemia-reperfusion injury.110,111 Systemic administration of HMGB-1 alters gut mucosal barrier function, leading to translocation of bacteria out of the intestinal lumen. 112 In a severe acute pancreatitis model of systemic HMGB-1 release, renal and hepatic dysfunction may occur. 113 As multisystem organ failure is thought to be the primary cause of death in ALI, 42 understanding the role of DAMPs in mediating this systemic injury has high clinical relevance. Surprisingly, no studies have specifically addressed the role of DAMPs in mediating non-pulmonary organ failure following ALI.
Apoptosis in Chronic Lung Disease: COPD
Among the >16 million Americans with COPD, approximately 120,000 deaths occur yearly. COPD is the only disease with a progressive increase in incidence, morbidity, and mortality amongst the most frequent diseases affecting the USA. 114 Ongoing cigarette smoking is the strongest determinant of clinical deterioration in smokers. Although inflammation is critical in COPD, the triggers of inflammation in the smoker's lungs and the role of this chronic inflammatory state in the initiation and progression of the disease remain unclear. Research in emphysema, the most common form of COPD, has largely revolved around the interaction between CS, lung inflammation, and protease/antiprotease imbalance, with little or no insight into how the alveolar cell compartment is affected by CS.
Apoptosis and efferocytosis as contributors to copd pathogenesis
The lung is continuously exposed to potentially injurious environmental agents. Cigarette smoke, for instance, is replete with potent oxidants. Infections also impart a significant oxidant burden as part of the initial activation of inflammatory cells. 115 This assault triggers several molecular signals involved in stress responses, many of which may engage the apoptotic process, such as p38 MAP kinase, JNK, and RTP801 (an inhibitor of mTOR signaling).116,117
The impact of apoptosis on COPD development is supported by the observation of increased expression of active caspase 3 in human emphysematous lungs. 118 Blockade of apoptosis in experimental emphysema with a broad-spectrum caspase inhibitors prevented the development of emphysema. 32 These findings were later validated by an attenuation of cigarette smoke-induced emphysema in caspase 3 null mice. 119 A potentially proinflammatory action of alveolar cell death was suggested by the observation that blockade of apoptosis in caspase 3 knockout mice led to secondary decreases in inflammation from chronic cigarette smoke exposure. 119
It has been postulated that the increased detection of apoptotic cells in emphysematous lungs also arises from decreased efferocytosis.2,34 Several mechanisms of impaired efferocytosis described in diseases such as cystic fibrosis 3 have also been observed in COPD, including excessive levels of extracellular matrix proteases 120 and oxidative stress. 121 Impaired efferocytosis has been noted in alveolar macrophages retrieved from smokers, 34 largely due to the effect of cigarette smoke. 122 The prolonged residence of apoptotic cells in smokers’ lungs may be an additional factor for release on cellular contents via secondary necrosis (though this hypothesis has not been yet investigated). This delayed or impaired clearance process may lead to auto-immunity and persistent inflammation if the apoptotic cells stimulate immune responses or lead to release of DAMPs that mediate inflammation.
Oxidative stress amplifies the effect of apoptosis in COPD
Oxidative stress is an integral element of the pathogenesis of COPD. Macromolecular damage of proteins and DNA by oxidative stress has been documented in COPD lungs.123,124 Moreover, oxidative stress is a common denominator of several lung diseases (including ALI and emphysema), which also present with apoptosis and the potential for dysfunctional efferocytosis. 125
We observed that lung cell apoptosis mutually interacts with oxidative stress in a model of emphysema caused by vascular endothelial growth factor receptor blockade. 126 This interaction was observed in subsequent experimental studies of rodent emphysema.127–129 Apoptosis and oxidative stress create mutually interactive feedback loops, 126 leading to alveolar destruction and emphysematous airspace enlargement.
Further studies have provided support of the interaction between oxidative stress and apoptosis. Activation of terminal caspases leads to cleavage of the iron-sulfur component p75 NDUSF1 of mitochondrial complex 1, causing the generation of oxidants during mitochondrial respiration.130,131 Oxidative stress causes the activation of novel mediators of mito-chondrial-driven apoptosis such as cofilin, which is normally involved in regulation of the cytoskeleton but localizes to the mitochondria once oxidized. 132 Finally, the process of oxidative stress may alter protein processing, including proteasome function, as documented by decreased function of the 20s proteasomal subunit in ALI 133 and the recent evidence of endoplasmic reticulum stress and dysfunctional proteasome subunit expression in COPD lungs.134,135 This ER stress is associated with increased apoptosis.
Extracellular DAMPS and COPD development
Despite increasing evidence of increased apoptosis and ineffective efferocytosis in COPD, relatively little is known about the influence of extracellular DAMPs upon COPD development. COPD is marked by increased matrix metalloproteinase activity, leading to degradation of the alveolar extracellular matrix and consequent release of potential DAMPs. Elastin fragments released during matrix degradation have been shown stimulate macrophage chemotaxis into the alveolus. 16 In turn, antagonism of elastase fragments attenuated emphysema in an animal model of protease overactivity. 16 Moreover, the tripeptide proline-glycline-proline, released from degraded extracellular matrix, may also drive acute inflammation, via binding to CXCR2. 136
COPD lungs have increased expression of other endogenous ligands, such as MHC-class I polypeptide associated sequence A (MICA) and B (MICB). These ligands bind to the NKG2D receptors expressed in NK, NKT, and Υζ T cells. 15 Activation of NKG2D (via overexpression of the mouse paralog retinoic acid early transcript 1 (RAET1) in alveolar type II cells) led to alveolar enlargement associated with alveolar cell apoptosis. 15
Recent studies have confirmed an association between pulmonary HMGB-1 and COPD development. Patients with COPD had elevated BAL HMGB-1 levels, perhaps via secretion by bronchial epithelial cells or alveolar macrophages. 13 Alveolar HMGB-1 correlated inversely with FEV1 and diffusion capacity—suggesting a relationship between HMGB-1 and COPD severity. 13 Elevated HMGB-1 was associated with increased alveolar concentrations of the proinflammatory cytokine 1 L-1β. As smokers had elevated airway and alveolar macrophage expression of RAGE, the authors hypothesized that the proinflammatory effects of HMGB-1 in the COPD lung were mediated through this pattern receptor. 13
Extracellular purines (ATP, adenosine) have received increasing attention as potential contributors to airspace destruction in emphysema. While ATP and adenosine have a protective effect in acute lung injury (as described previously), chronic levations of extracellular purines are associated with obstructive lung disease.18,137 Chronic activation of the adenosine (P1) receptor A2BR in mice induced macrophage production of osteopontin, contributing to airspace enlargement and emphysema. 12 Similarly, chronic cigarette smoke exposure was associated with increased ATP in BAL fluid; this ATP induced elastase and chemokine production by neutrophils. 138
Studies have also examined the influence of viral PAMPs on COPD pathogenesis. Double-stranded RNA and influenza virus infection were found to activate the intracellular pattern receptor Inducible (RIG) I-like helicase; this activation greatly potentiated alveolar destruction caused by exposure to cigarette smoke in mice. 139 This amplification of emphysema was associated with increased alveolar cell apoptosis and inflammation, potentially mediated through eI2F-α signaling. 139
Of note, potentially inflammatory endogenous ligands in COPD are not limited to the lung. Serum uric acid concentrations are increased in COPD patients, correlating with airflow obstruction and severity of dyspnea. 14
Apoptosis, cell-surface DAMPS, and autoimmunity
The new concept that COPD may have an autoimmune component was developed approximately 7 years ago 140 and revised in 2009. 35 Recent investigations have documented the presence of oligoclones of T cells in COPD lungs, 141 auto-antibodies bound to emphysematous tissues, 142 and elastin-reactive T cells clones isolated from advanced emphysema lungs. 143 The process of auto-immunity can be initiated or enhanced by the exposure of non-tolerogenic peptides as self antigens, possibly expressed at the cell surface of apoptotic cells; 144 an example is the apoptosis triggered by the cytotoxic T-cell granzyme B in scleroderma. Granzyme B activates the apoptotic machinery in target cells and uncovers an alpha-helical coiled-coil N-terminal domain in the histidyl-transfer RNA synthetase (HisRS, Jo-1). 145 The expression of its granzyme B-cleavable form is enriched in the lung and localized to the alveolar epithelium, suggesting lung specificity of the auto-immune process as defined by a specific cleavage epitope of a self antigen. 145 In addition, cell death may precede several additional examples of auto-immune diseases, including type I diabetes and post-myocardial infarct pericarditis and potentially graft rejection following graft injury due to ischemic damage. 146
Oxidative stress generated through the process of apoptosis may have opposing roles in the induction of tolerance vs. immunogenicity due to apopto-sis. Oxidation of surface phosphatidylcholine (PC) species on the surface of apoptotic cells may lead to generation of auto-reactive IgM antibodies directed against oxidation-specific epitopes; 147 this process may underlie the pathogenesis of atherosclerosis as oxidized PC leads endothelial cells to become proinflammatory. This pathogenetic process may also occur in smokers with generation of oxidation-specific epitopes in apoptotic cells since these individuals have increased evidence of increased lung and muscle cell death, 148 are at increased risk of atherosclerosis, have increased expression of markers of oxidative stress, 123 and exhibit systemic inflammation. 149
However, the detrimental effect of oxidative stress in driving inflammation due to apoptosis is far from clear. Both caspases and oxidative stress were required for tolerance induced by apoptotic cells (fibroblasts and spleen cells) against the hapten trinitrophenol (TNP). 131 The latter studies showed that oxidant-mediated modification of HMGB-1 was required for the tolerogenic signal conveyed by apoptotic cells. The anti-inflammatory action promoted by apoptosis-generated oxidative stress is at odds with the observation that oxidative stress may rather impair efferocytosis 150 and therefore lead to progression to frank necrosis. 151 Further clarification of the fate, levels, and oxidation modification of HMGB-1 in emphysema are therefore pressing to clarify its potential role in the induction of persistent lung inflammation.
Conclusion
In the nearly 20 years since Polly Matzinger first proposed the concept of danger signaling in immunity, 152 it has become apparent that numerous disease states are driven by the proinflammatory effects of endogenous ligands. As both acute (ALI) and chronic (COPD) lung diseases are marked by cellular environments which favor apoptosis and delayed efferocytosis, it is likely that the pathophysiology of these disease states is shaped by the presence of DAMPs. Indeed, the role of DAMPs in the induction of ALI by systemic illness has been the source of intense investigation. However, much remains to be learned about the role of DAMPs in propagating existing lung injury and in the induction of the multisystem organ dysfunction that often defines survival in ALI. Similarly, there is a need to further explore the role of DAMPs in COPD—with regards to the induction of both innate and adaptive immunity. Future advances in these topics hold great promise in the development of therapies for ALI and COPD.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.