
Abstract
The innate immune system is an integral component of the inflammatory response to pathophysiological stimuli. Toll-like receptors (TLRs) and inflammasomes are the major sensors and pattern recognition receptors (PRRs) of the innate immune system that activate stimulus (signal)-specific proinflammatory responses. Chronic activation of PRRs has been found to be associated with the aggressiveness of various cancers and poor prognosis. Involvement of PRRs was earlier considered to be limited to infection- and injury-driven carcinogenesis, where they are activated by pathogenic ligands. With the recognition of damage-associated molecular patterns (DAMPs) as ligands of PRRs, the role of PRRs in carcinogenesis has also been implicated in other non-pathogen-driven neoplasms. Dying (apoptotic or necrotic) cells shed a plethora of DAMPs causing persistent activation of PRRs, leading to chronic inflammation and carcinogenesis. Such chronic activation of TLRs promotes tumor cell proliferation and enhances tumor cell invasion and metastasis by regulating pro-inflammatory cytokines, metalloproteinases, and integrins. Due to the decisive role of PRRs in carcinogenesis, targeting PRRs appears to be an effective cancer-preventive strategy. This review provides a brief account on the association of PRRs with various cancers and their role in carcinogenesis.
Keywords
Introduction
Many pathological conditions that involve tissue damage, bacterial or viral infections, and metabolic disorders leading to chronic inflammation are known to develop as a consequence of chronic activation of innate immune pattern recognition receptors (PRRs). 1 The role of inflammation and innate immune function in cancer initiation and progression is well understood.2,3 Innate immunity is the first line of defense against infection and/or internal tissue injury and recognizes highly conserved sets of molecular structures called pathogen/damage-associated molecular patterns (PAMPs/DAMPs) via PRRs.1,4 Toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs) are the innate immune PRRs which, by their downstream signaling, activate multiple pro-inflammatory responses leading to an efficient antigen-specific acquired immunity. TLR signaling drives dendritic cell (DC) maturation, antigen presentation, as well as CD8+ T-cytotoxic effector functions, which are decisive for an efficient antitumor immunity. 5 TLRs are also expressed by intestinal epithelial cells (IECs), where they regulate tissue homeostasis by stimulating immune response to bacterial pathogens while attenuating the immune response against favorable microbes. 6 Furthermore, TLRs sense the breakdown of the protective intestinal barriers and trigger proliferative signaling. 6 An aberrant TLR expression or chronic stimulation can alter homeostasis and cause negative regulation of antitumor immunity, as evidenced by increased immune-suppressive functions such as enhanced regulatory T-cell proliferation.7,8 Similarly, upon activation, NLRs lead to the secretion of mature forms of interleukin (IL)-1β and IL-18, triggering multiple inflammatory signaling pathways, which contribute to the chronic inflamed tumor microenvironment. 3 DAMPs such as HMGB1 and ATP as well as microbial macromolecules such as double-stranded DNA (dsDNA) and double-stranded RNA (dsRNA) can act as intracellular or extracellular ligands to the innate PRRs.9,10 TLRs and NLRP3 function as critical sensors of metabolic disturbances caused by obesity, injury, and infection to augment the processes leading to the restoration of tissue homeostasis.11,12 TLRs and NLRs follow a coordinated signaling system that senses pathogenic invasion or tissue damage through various molecular signatures (PAMPs/DAMPs), triggering the expression of specific genes/signaling such as tumor necrosis factor (TNF)-α, IL-6, and IL-1β.1,13 Clinical studies suggest that the expression of PRRs is not limited to the immune cells, but they are also found to be elevated in various human epithelial cancers.14,15 Chronic activation of TLRs promotes carcinogenesis through pro-inflammatory responses, which augments the proliferative, anti-apoptotic, and pro-fibrogenic signals in the tumor microenvironment as well as in the tumor cells. 15 Thus PRR homeostasis and their aberrant expression confer a dual role, ie, an immune-enhancing role that potentiates antitumor immunity, and a tumor-promoting role triggered by the aberrant and dysregulated expression. This review presents the current understanding of the functions of PRRs in promoting carcinogenesis and metastasis.
TLRs: From Pathogenic Sensors to Role in Cancer
Pathogenic Sensor
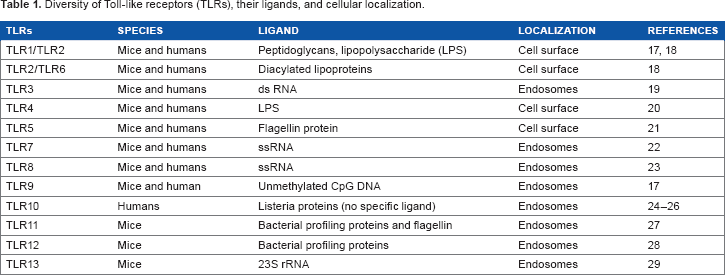
Diversity of Toll-like receptors (TLRs), their ligands, and cellular localization.
Associaton with Cancer
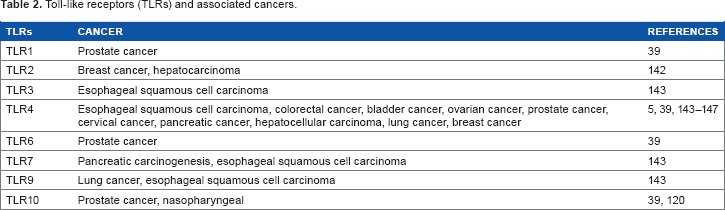
Toll-like receptors (TLRs) and associated cancers.
Signaling
Stimulation of TLRs is known to trigger the expression of several genes that are involved in various inflammatory and immune responses. Binding of ligands leads to the dimerization of TLRs, TLR2 forming heterodimers with TLR1 or TLR6 while all other TLRs forming homodimers (Fig. 1).17,41,42 Dimerization of TLRs initiate signaling events (originating from the cytoplasmic TIR domain of the receptor) and progresses through adaptor proteins involving two major pathways (Fig. 1). In the first, the activated MyD88 adapter protein recruits IRAK-4 to TLRs, facilitating IRAK-4-mediated phosphorylation of IRAK-1, which then binds to TRAF-6 leading to the activation of two subpathways. One of them involves MAP kinases leading to the activation of AP-1 and its recruitment to the nucleus, while the other, where the TAK1/TAB complex is activated, enhances the activity of the IKB kinase (IKK) resulting in phosphorylation and degradation of IKB, thereby increasing the localization of NF-κB to the nucleus.17,43 MyD88-dependent TLR signaling involves a second TIR domain molecule, namely TIRAP/Mal. Further, the MyD88-independent TLR signaling involves a third TIR domain involving a TRIF-dependent pathway, where signaling is executed with the recruitment of IRF to the nucleus via TBK-1. IRF-3 regulates the transcription of interferon (IFN)-β and is therefore called TIR domain containing adaptor-inducing IFN-β (TRIF).
17
TRIF-dependent pathways are pivotal during the stimulation through TLR3 and TLR4.2,44 All TLRs utilize the MyD88-dependent signaling pathway to induce the expression of pro-inflammatory cytokine genes with the exception of TLR3, which exclusively uses the TRIF pathway.
2
Both MyD88-dependent cascades and TRIF pathways have been associated with tumor growth and metastasis.
45
Eventually, TLR functions are regulated by the transcription factors AP-1, NF-κB, and IRF-3.2,17,46
TLR diversity, ligands, and signaling. TLRs are expressed on the epithelial and immune cells on the plasma membrane (TLR1, TLR2, TLR4, and TLR5) and endosomal membrane (TLR3, TLR7, and TLR9). Pathogenic PAMPS are the natural ligands of TLRs: eg, bacterial lipid-derived macromolecules are ligands for TLR1, TLR2, TLR4, and TLR6, while nucleic acid derivatives like dsDNA (TLR3) nucleotide complexes with other macromolecules (TLR7) and the CpG nucleotide sequence (TLR9). TLR signaling in an MyD88-dependent and TRIF-mediated (MyD88-independent) pathway leads to the translocation of AP-1, NF-κB, and IRF-3, respectively, to the nucleus, which induces the transcriptional activation of proinflammatory genes.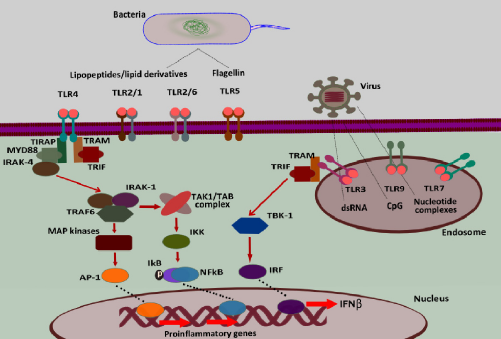
TLR signaling coupled with other inflammatory pathways contributes to the inflamed tumor microenvironment and leads to the inflammation-driven disease progression; eg, chronic infection by Helicobactor pylori leads to the development of gastric cancer. 47 In spite of the inability to stimulate TLR4 on its own, H. pylori actively promotes inflammation by upregulating TLR4 expression via the TLR2 and MEK1/2-ERK1/2 signaling pathway.48,49 IL-6 and other TLR-induced effector cytokines, such as TNF-α and IL-17A, strongly promote mucosal and hepatic cancer. Another associated signaling is the IL-6-mediated STAT3 activation, which is known to be highly pro-oncogenic and also contributes to the radioresistance of tumor cells.50,51 Similarly, STAT3-mediated T-helper type 17 (TH-17) expression has been shown to facilitate tumor development in APC–/+ mice. 52 Recent studies have shown the involvement of nearly all the TLRs in increased cancer incidence, disease severity, and poor prognosis, and thus can be exploited as targets for the cancer-preventive approaches.53–55
Inflammasomes: Inflaming Tumors
Inflammasome Assembly
Inflammasomes are multi-scaffold proteins with an interacting N-terminal homotypic protein–protein interaction motif called caspase-1 activation recruitment domain (CARD), where procaspase-1 undergoes a conformational change required for its cleavage and activation.
56
The release of mature IL-1β and IL-18 from myeloid cells is dependent on activation of procaspase-1 to active 10- and 20-kDa caspase-1 peptides.
57
Inflammasomes are formed with NLR proteins containing an N-terminal CARD or pyrin domains required for homotypic protein–protein interaction, an intermediate nucleotide binding with self-oligomerization potential, the NACHT domain, and a C-terminal domain containing leucine-rich repeats (LRRs).
58
NLRP3 inflammasomes (NACHT, LRR, and pyrin domain-containing protein) are involved in the sensing of DAMPs such as extracellular ATP, monosodium urate (MSU) crystals, asbestos, silica, and β-amyloid.59–62 Inflammasome activation requires the interaction of pyrin domain (PYD) of ASC (apoptosis-associated speck-like protein containing C-terminal CARD) with PYD of NLRP3, forming a functional inflammasome complex through CARD–CARD interaction of ASC with procaspase.
63
Classical inflammasome activation has two steps: The first step involves the induction of mRNA followed by the expression of pro-IL-1β and pro-IL-18 followed by NF-κB translocation to the nucleus, which requires either TLR/NOD stimulation or signaling through TNF-α or IL-1β receptor.
64
The second and the most critical step is the sensing of PAMPs or DAMPs by NLRs, which leads to the autocatalytic cleavage of caspase-1 (Fig. 2).
65
Coordinated action of TLRs and NLRP3 inflammasomes in tumor progression, angiogenesis, and metastasis.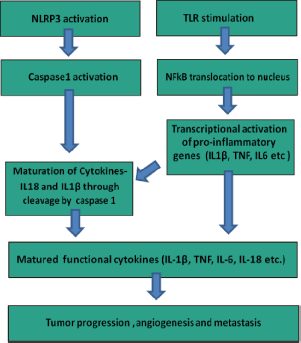
Association with Cancer
The major products of inflammasome activation are the pro-inflammatory cytokines IL-1β and IL-18, which are pro-tumorigenic in inflammation-induced gastrointestinal (GI) cancers. 3 Chronic inflammation in the stomach due to H. pylori infection or other causes is mediated by the upregulation of pro-inflammatory cytokines, including IL-1β.66,67 While stomach-specific expression of IL-1|3 in mice induces inflammation and gastric carcinogenesis, in colitis-associated cancer (CAC) IL-1β may promote tumor growth and invasion by inducing an epithelial to mesenchymal transition (EMT) as well as stem cell phenotype in colon tumor cells.68,69 Further, IL-1β stimulates COX-2, IL-6, IL-8, and CCL5 production, which leads to an increase in the proliferative and invasive capabilities of colon cancer cells.70–72
DAMPs as PRR Activators
TLR-mediated carcinogenesis was earlier thought to be associated with organs that directly or indirectly get exposed to the bacterial TLR ligands, such as the GI tract, skin, and liver.
73
However, with the discovery of the DAMPs as endogenous TLR ligands, the possible role of TLRs in carcinogenesis has extended beyond the pathogenic ligand-associated organs. DAMP–PRR interaction leads to the smoldering chronic inflammation, which nurtures transformed and tumor cells with mitogenic, anti-apoptotic, and proliferative factors (Fig. 3). DAMPs are either released from the dying cells in the injured/inflamed tissues or actively secreted from the lysosomes.
74
Activation of TLRs and inflammasomes by DAMPs released by apoptotic and necrotic cells. DAMPs from nucleus (HSPs, histones, HMGB1), cytosol (uric acid, ATP), and mtDNA activate PRR signaling in tumor and myeloid cells, which leads to the secretion of pro-inflammatory mediators such as TNF-α, IL-1-β, and IL-6. TLR signaling eventually promotes the transcriptional activation of pro-inflammatory cytokines via localization of NF-κB, while inflammasome activation guides caspases-1 to cleave pro IL-1-β to mature IL-1β, altering integrin and chemokine expression leading to the tumor growth and metastasis by promoting angiogenesis and tumor cell migration.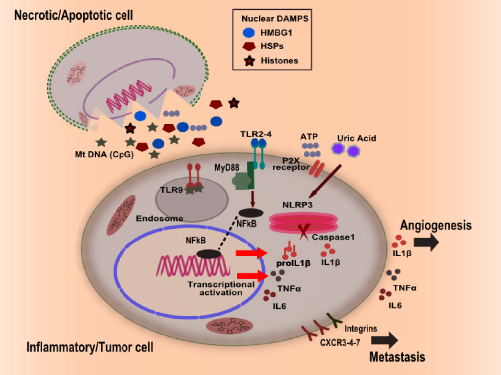
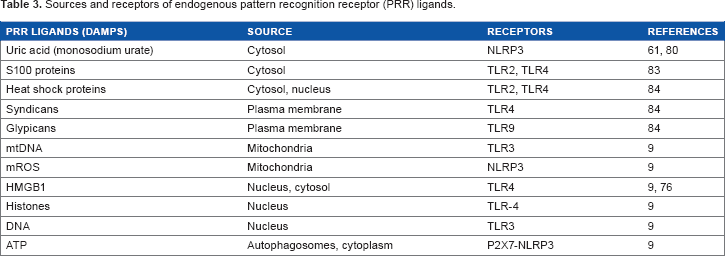
Sources and receptors of endogenous pattern recognition receptor (PRR) ligands.
PRRs in Tumor Growth and Metastasis
PRR expression in tumors has been linked to increased severity and metastasis. PRR signaling promotes tumor growth and metastasis by altering the integrins and tumor cell motility. PRR activation leads to immune tolerance, which helps in evading the antitumor immunity. Furthermore, release of angiogenic factors leads to the neo-vascularization and favors tumor cell growth and migration.
Invasion and Metastasis
TLRs along with intracellular signaling (NF-κB) regulates the integrin expression (B1) and motility in the tumor cells.87–89 TLR4 stimulation by lipopolysaccharide (LPS) has been shown to induce EMT and invasive phenotypes in human breast cancer cell lines such as MCF-7 and MDA-MB-231.89,90 Metastatic invasion involves the chemokine-mediated active transport of metastatic cells through the subcapsular sinus epithelium.91,92 TLR activation is known to induce the expression of CCR7 and CCR8, which are major chemokines involved in the tumor-mediated lymphatic remodeling of peritumoral lymph vessels and draining lymph nodes, thereby promoting metastasis.93,94 Many metastatic tumors appear to use the expression of these chemokine molecules to colonize in a new organ/tissue, while defective expression of the homing receptors such as CXCR3, CXCR4/CXCR7, and CCR6 has been observed in liver and lung metastases of colon cancer.95,96 Further, the ligands of CXCL19, SDF-1, and CCL20 receptors are highly expressed in the liver and lungs of metastatic colorectal cancer (CRC) patients.97,98 Integrin signaling is another metastatic mechanism, that is altered by the activation of TLRs. Integrins are required to facilitate immune cell trafficking in melanoma and other tumors. 99 TLR activation leads to the acquisition of various integrins, chemokine expression, and trafficking properties similar to the leukocytes. 100 Overexpression of PD-L1, TLR, TGF-β, CCL4, and CCL5 has been found to facilitate invasion and proliferation of such leukocytic tumor cells within the lymph nodes. 100 Leukocytic tumors show upregulated E/P-selectins, L-selectin ligands, alpha-4B1, ICAM-1, and VCAM-1 required for trafficking and homing to a distal tissue.6,100,101 IL-18 and IL-1P are inflammasome effector pro-inflammatory cytokines, which upregulate the expression of adhesion molecules and therefore play a role in hepatic metastasis. 102
Immune Evasion
TLRs also help tumor cells in immune evasion, which in turn leads to the increased aggressiveness of the tumor, and increased TLR expression in different cancers appear to confer advantages of both immune evasion and immune suppression.103,104 TLR signaling alterations lead to the production of IL-10 and TGF-β, which are the major immunosuppressive cytokines.105,106 Further, TLR activation is accompanied by an increased expression of suppressive costimulatory molecules such as PD-L1 and HLA-G, which leads to the development of the immune tolerance.107–109 These suppressive functions regulate the hyperactive immune system during auto-immune disorders. TLR4 signaling leads to the increased expression of suppressive costimulatory markers B7-H1 and B7-H2 and inducible nitric oxide synthase (iNOS) by providing resistance against cytotoxic T-cell (CTL) attack. 109 TLR-stimulated tumor cell supernatant has been shown to inhibit both T-cell proliferation and natural killer (NK) cell activity, while blocking of the TLR4 pathway reverses the tumor-mediated suppression of T-cell proliferation and NK cell activity and delays tumor growth. 107
Angiogenesis
The role of inflammasomes in metastasis and cancer progression is demonstrated by the role of IL-1β and IL-18 (matured by activated NLRP3 inflammasome), which are shown to facilitate tumor angiogenesis. Elevated levels of active IL-1β in melanoma cells have been shown to act both in paracrine and autocrine manners, promoting angiogenesis, and thus contributing to invasion and metastasis. 110 IL-1β induces the expression of angiogenic factors such as VEGF and IL-8, which promote tumor growth. 110 Lewis lung carcinoma (LLC) cells overexpressing with IL-1β displayed increased angiogenesis and secretion of VEGF, and macrophage inflammatory protein-2 (MIP-2/CXCL2) has been correlated with faster tumor growth, while IL-1β-KO mice showed decreased angiogenesis and growth of melanoma tumors.111,112 Further, NLRP3 activation by the DAMPs in the tumor milieu is also known to induce the production of active IL-1β continuously, thereby contributing to the neovascularization. 113 IL-1β signaling is also involved in the release of several angiogenic factors such as VEGF and IL-8 from tumor and stromal cells, thereby further promoting tumor growth. 113
PRRs as Cancer Biomarkers
PRR expression has been found to be upregulated in many cancers and can be exploited as a biomarker. TLR4 expression has been shown as a cancer stem cell marker in hepatocellular carcinoma. 96 The predictive role of TLR2, TLR4, and TLR9 has been established in gastric tumors and oral tongue squamous cell carcinoma. 55 Immunohistochemical analysis and expression studies in adenocarcinoma patients have suggested that the high expression of TLR3, TLR4, and TLR9 levels is associated with prostate cancer recurrence.114,115 NLRP3 products IL-1β and IL-18 in body fluids are correlated with a high risk of pancreatic carcinoma and poor prognosis. 116 IL-1β in cyst fluid appears to be a good predictor of enhanced risk of intraductal papillary mucinous pancreatic neoplasms. 116 Plasma IL-1β expression is generally elevated in advanced cancers and correlates well with clinical features such as infirmity, sarcopenia, and loss of appetite as well as weight. 117 Similarly, increased IL-18 levels in ovarian carcinoma patients appear to be a good predictor of disease severity. 118
The ability of TLRs in predicting various cancers appears to be linked to the single-nucleotide polymorphisms (SNPs) at the loci of PRRs and their regulator genes. 119 For example, patients carrying specific polymorphisms (enhanced TLR10 expression) show higher risk of prostate and nasopharyngeal carcinoma. 120 Further, serum or ascite concentrations of IL-1RA, as well as the SNPs affecting these concentrations, can be used as important biomarkers. 121 While SNPs in regulatory elements of NLRP3 determine susceptibility to inflammatory bowel disease, polymorphisms in IL-18 and IL-18R encoding genes can be used as biomarkers for Crohn's disease.122,123
PRR Signaling as Potential Targets to Inhibit Carcinogenesis
Chronic stimulation of PRR-induced signaling by PAMPs or DAMPs leads to mitogenic and anti-apoptotic signals, which aid the tumor-promoting inflammatory events that ultimately result in malignant transformation. Modifiers of PRR signaling can be useful in targeting the process of carcinogenesis. Although PRR antagonists could be useful as tumor-preventive anti-inflammatory agents, only a few of them such as TLR4 antagonists CRX-526 (synthetic lipid A mimetic) and 1A6 (monoclonal antibody to TLR) have been investigated for preventing murine colitis.124,125 While TLR blockade as a strategy has failed to reduce the chronic T-cell transfer colitis, administration of 1A6 during the recovery phase of colitis has been found to prevent tumor development in the azoxymethane/dextran sulfate sodium (AOM-DSS) model.125,126 TLR4 antagonists are also being evaluated in clinical trials for treating sepsis.127,128 Inhibitors of STAT3 (downstream to PRR activation) such as OPB-31121 are already in clinical trials as therapeutics, suggesting that targeting secondary signaling to PRR could also be a potential antitumor therapy.129,130 In vivo manipulation of NLRs has been shown to delay the onset of colitis and cure the associated carcinogenesis. Glyburide (glibenclamide), a sulfonylurea drug used for the treatment of type-2 diabetes, is known to inhibit ATP-sensitive K+ channels and can thus prevent NLRP3 inflammasome activation.131,132 Compounds that target TLR signaling and inflammasomes, such as IL-1P neutralizing antibodies, recombinant IL-1R antagonist (anakinra), and IL-18 binding proteins, have already been established as therapeutics, while cancer-preventive strategies using small-molecule inhibitors that target caspase-1 needs to be established.133,134 ST2825, a carboxyamide derivative and novel inhibitor of MyD88 dimerization, is currently under evaluation in preclinical studies for the treatment of chronic inflammatory diseases. 135 Other peptide inhibitors such as hydrocinnamoyl-l-valyl pyrrolidine (compound 4a) and Pephinh-MyD88 are also being developed to target MyD88 dimerization for the treatment of lymphoma with MyD88 mutations. 136 Selective inhibition of NF-κB in cancer cells can block the stimulatory effect of TNF and markedly increase susceptibility to TRAIL-induced cell death, resulting in tumor regression. 137 NF-κB inhibition and anti-TNF therapy, together with the administration of IFN or TRAIL, might offer an attractive strategy for immune-modulation-based cancer therapy. Anti-IL-1 monoclonal antibodies, soluble IL-1RII, and caspase-1 inhibitors are currently being exploited for the treatment of various inflammatory disorders, and can also be employed in tumor preventive strategies. PRR induction is fueled by various metabolites and dietary components.138,139 Metabolic interventions by dietary modification and manipulation of metabolism using modifiers such as metformin and 2-deoxy-D-glucose (2-DG) can also be useful as an effective preventive strategy against PRR-induced carcinogenesis. Indeed, dietary 2-DG has been shown to impair inflammation (DMBA-TPA)-induced tumorigenesis and radiation as well as tumor-induced angiogenesis, while enhanced local tumor control has been found to be associated with the stimulation of inflammation following therapeutic administration of 2-DG.140–143
Although TLRs and inflammasomes appear to be promising targets for cancer therapy and prevention, further studies are required to determine the context-specific functions of PRRs during cancer development and progression. Efficacy of therapies targeting inflammasomes will depend on our ability to determine when such therapies will be beneficial and when they will be detrimental due to the context-specific function of the inflammasomes in cancer.
Author Contributions
Prepared the preliminary draft of the manuscript: SP. Contributed to the writing of the manuscript: SS and VA. Jointly developed the structure and arguments for the paper: SP, BSD and ANB. Made critical revisions and approved final version: BSD, ANB and KN. All authors reviewed and approved the final manuscript.