
Abstract
Systemic lupus erythematosus (SLE) manifests itself in the form of damage to various organs as a result of an autoimmune reaction to the organism's own molecules. Understanding the processes leading to the activation of the autoimmune response, as well as identifying potential autoantigens and their role in the pathogenesis of SIE, is an important task for understanding the mechanisms underlying the pathogenesis of SLE. Here we describe a model of SLE pathogenesis in which the induction of an autoimmune response is based on mitochondrial antigens. In addition to options for initiating an autoimmune response involving mitochondrial components, the model describes the role of pathways and immune cells of these pathways of adaptive immunity in SLE, as well as factors leading to the development of chronic inflammation in SLE. The creation of such models is important for a better understanding of the pathogenesis of SLE, as well as the identification of new therapeutic targets for this disease.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease. The etiology of SLE is not fully understood, it is believed that SLE is caused by the interaction of various factors: genetic, immunological, hormonal and environmental.1,2 The pathogenesis of SLE is primarily due to the state of distribution of various autoantibodies that accumulate and are deposited in the form of immune complexes. In SLE, various organ systems other than skin itself can be attacked by the immune system, such as the excretory, nervous, cardiovascular, or respiratory systems. Symptoms of the disease can occur unevenly over time, stretching for months or even years. 3 In this regard, the diagnosis of SLE is a difficult task for the modern medicine. SLE is a worldwide disease that most often affects women aged 15–45 years. 4 Although the disease occurs in people of different ethnicities, SLE is most common among non-Caucasians. While prevalence in Europe and the United States is higher among people of African descent, SLE is rare in Africa.4,5 In the United States, SLE is more common to African Americans, who also tend to have more severe disease. The Centers for Disease Control and Prevention announced an estimated prevalence more than 320 thousands cases of probable or certain SLE, more commonly in African Americans, American Indians, and Alaska Natives. 4
Treatment of this disease depends on the affected organs and systems, and on the disease stage. This may include topical use for skin problems, non-steroidal anti-inflammatory drugs (NSAIDs) for musculoskeletal disorders, and immunosuppressants. 6 Therapeutic improvements are still unable to significantly reduce mortality and the risk of comorbidities in patients with SLE. 7 Although there is currently an understanding of the pathological basis and risk factors for SLE, the exact pathogenesis is still unknown, which creates difficulties for effective treatment of this disease. Therefore, the study of the pathogenesis of SLE is an extremely urgent task today. A possible factor influencing the initiation and progression of autoimmune inflammation in SLE is mitochondrial components: structural macromolecules of mitochondria, as well as metabolites formed during cellular respiration. Consideration of the pathogenesis of SLE in this direction can provide a better understanding of the development of this disease, as well as contribute to the discovery of new potential therapeutic targets.
Overview of mitochondrial components that cause inflammation
Mitochondrial DNA and RNA
The mitochondrial genome is a double-stranded circular DNA molecule about 16 thousand base pairs in size. It is packed with nucleoids, which are slightly elongated, irregularly shaped structures approximately 80–100 nm in size. Nucleoids bind to the inner mitochondrial membrane (IMM) and are distributed throughout the mitochondria. 8 They contain relatively high levels of mitochondrial transcription factor A (TFAM), which is necessary for the maintenance of mtDNA, since it is responsible for its packaging. 9
Due to the α-proteobacterial origin of mitochondria, mtDNA has unique properties that are important for its role in innate immune responses and inflammation. Released mtDNA has been shown to cause pro-inflammatory state. 10 Methylation of CpG regions in mtDNA differs from nuclear DNA methylation and confers mtDNA immunogenic potential due to its similarity to pathogen DNA, especially bacteria. Some authors describe the absence of methylation in mtDNA, while others report that nuclear DNA methyltransferase (DNMT1) is found in mitochondria, suggesting some degree of methylation in mammalian cells. 11
In addition to hypomethylation, mtDNA can undergo oxidative damage, which is immunogenic and can be recognized by PRR receptors regardless of the degree of methylation. 12 Extracellular mtDNA binds a TLR9-mediated signaling pathway that seems to recognize unmethylated or undermethylated CpG DNA. In macrophages, the interaction of mtDNA with TLR9 induces the production of pro-inflammatory cytokines, chemotaxis, and phagocytic activation via a MyD88-dependent signaling cascade. 13 In the mouse model of acute liver failure, mtDNA has been shown to induce neutrophil activation, which increased inflammation and organ damage. 14 Intra-Peritoneal mtDNA injection causes lung damage and arthritis with mononuclear cell infiltration in mice. 15 These data support the view that mtDNA is involved in the development of inflammatory responses in vivo. 11 mtDNA also induces inflammation in microglial and neuronal cells of mice. 16 In addition, mtDNA stress induced by TFAM deficiency has been reported to trigger cytosolic antiviral signaling, aided by leakage of cytosolic mtDNA. 17 Thus, the degree of packaging, stability, localization, and the presence of modifications or mutations of oxidative damage are involved in mtDNA innate immunity signaling. 11 Also, in a number of studies,18,19 it was demonstrated that, in addition to mtDNA, mtRNA was also an important inducer of autoimmune inflammation. So, mtRNA can be considered as one of the main inducers of triggering interferon production. In study 18 it was shown that increased levels of mitochondrial RNA autoantibodies presented in a mouse serum as well as in a serum from patients with SLE. The level of autoantibodies to mtRNA positively correlated with the level of autoantibodies to mtDNA. In addition, using multivariate analysis, a positive correlation between the level of autoantibodies to mtRNA with the development of lupus nephritis was found. Thus, the level of autoantibodies to mtRNA can potentially become an effective biomarker for the onset and development of SLE. In a study, 20 it was demonstrated that mitochondrial DNA in SLE moved to the extracellular space, where it formed complexes with autoreactive antibodies, due to interaction with the pore-forming protein gasdermin D. Thus, gasdermin D can be considered as a potential therapeutic target in the treatment of SLE.
Succinate
Succinate is an intermediate in the tricarboxylic acid (TCA) cycle, which is formed from succinyl-CoA by succinyl-CoA ligase. However, succinate has been shown to be secreted into the extracellular environment in vitro, and this is stimulated by treatment with antimycin A, which inhibits electron transfer between cytochromes B and C1. 11 Indeed, extracellular succinate acts as a signaling molecule and is recognized by immune cells through its G protein pair receptor, namely succinate receptor 1 (SUCNR1, also referred to as GPR91). Activation of the receptor stabilizes hypoxia-inducible factor-1 alpha (HIF-1α), which promotes pro-inflammatory differentiation of T-lymphocytes. Succinate has also been described to have a synergistic effect with Toll-like receptor (TLR) ligands in dendritic cells for cytokine production. 21
Cardiolipin
Cardiolipin (CL) is a phospholipid that accounts for 20% of the total lipid content in IMM. 22 CL consists of two main phosphatidylglyceride chains and a glycerol group. Four fatty acid chains of varying length and degree of saturation are associated with CL. 23 This phospholipid plays a key role in many mitochondrial processes, including protein import, dynamics, respiratory chain functionality, and metabolism. 24 Cellular necrosis exposes CL to the extracellular environment, which can be sensed by T cells via CD1d. 11 In addition, CL can directly bind to NLRP3 and activate the inflammasome-mediated immune response, thereby inducing an inflammatory response. 25 Cardiolipin is one of the first autoantigens to initiate the formation of autoantibodies in SLE. In this case, the highest concentration of this antigen is formed in the liver, which provokes its damage by autoantibodies. 26 This observation was confirmed in a study on patients with SLE. 27
Mitochondrial transcription factor A
TFAM is a member of the HMG box protein family. TFAM interacts with mitochondrial DNA and regulates both its transcription and replication, thereby modulating mtDNA-encoded protein expression and mtDNA copy number. 28 Correct expression of TFAM is critical for mitochondrial function and therefore cellular homeostasis. The presence of extracellular TFAM has been described to induce an inflammatory response similar to that of another DAMP from the same protein family, namely HMGB1. TFAM also enhances the immunogenicity of mtDNA. 29 Being associated with mtDNA, it can interact with the plasma membrane receptor RAGE and induce mtDNA internalization, thereby facilitating its recognition by TLR9. In addition, TFAM enhances the secretion of pro-inflammatory cytokines. 30
The role of immune cells in the pathogenesis of systemic lupus erythematosus
Macrophages
One of the main functions of macrophages is the clearance of cellular debris after apoptosis by phagocytosis. Disruptions in the clearance of cellular components after cell death leads to the emergence of autoreactivity to accumulations of DAMPs, which begin to be perceived as autoantigens. 31 In support of this statement, patients with SLE showed a decrease in the number and activity of phagocytic macrophages, 32 especially in the marginal zone macrophages, which are the main participants in the clearance of apoptotic cell debris. 33 As it is known, there are two groups of macrophages: M1 macrophages, which are the one of the main initiators in the development of the inflammatory immune response and subsequent tissue damage, and M2 macrophages, which are responsible for tissue repair. 34 When comparing the expression profiles of myeloid cell genes in patients with SLE and healthy people, an increased expression of genes involved in the activation of M1 macrophages (STAT1 and SOCS3) and a reduced expression of genes involved in the activation of M2 macrophages (STAT3, STAT6 and CD163) were found. 35 This finding shows that the balance between pro-inflammatory and anti-inflammatory macrophages is shifted towards M1 macrophages, which is one of the potential causes of inflammation development in SLE.
Neutrophils
In the pathogenesis of SLE, disturbances in neutrophil functions were found, which included a decrease in phagocytic and lysosomal function, activation of adhesion molecules ICAM-1 and VCAM-1, an increase in oxidative activity, a reduced ability to be cleared, as well as increased aggregation.36,37 As a reliable diagnostic sign of diffuse proliferative lupus nephritis, which is one of the possible complications observed in SLE, is an increased concentration of tissue infiltrating neutrophils. 38 One of the reasons for the enhancement of the autoimmune response may be the formation of neutrophil extracellular traps (NETs) during a specific death of neutrophils, called netosis, which is one of the signs of the pathogenesis of SLE. 39 NETs are essential for eradicating bacterial pathogens, but under conditions of autoimmune inflammation, NETs, which are primarily made up of DNA, themselves begin to act as autoantigens, leading to an increase in inflammation. In the pathogenesis of SLE, netosis is enhanced as a result of exposure to antiribonucleoprotein complexes and cell debris in peripheral tissues formed during apoptosis. 37 In addition, the degradation process of NETs is disrupted due to the presence of inhibitors to DNase I inhibitors and antibodies to NETs fragments. 40 All this contributes to the further development of inflammation and its chronification.
Dendritic cells
The development of a stable autoimmune response is based on the prolonged presentation of the autoantigen and increased production of pro-inflammatory cytokines. 41 Together with macrophages, dendritic cells are the main phagocytic cells involved in the removal of cell debris formed during apoptosis. 41 Inhibition of dendritic cell maturation factors may be one of the reasons for the development of an autoimmune reaction, which was shown in a mouse model with a deficiency of the Blimp-1 transcription factor. 42 In the pathogenesis of SLE, plasmacytoid dendritic cells play an important role, producing large amounts of IFN I, which is a strong proinflammatory cytokine and is able to initiate apoptosis, thus contributing to the release of destroyed autoantigens from tumor cells. 43 IFN I can activate the proliferation and maturation of T cells. 44 Thus, dendritic cells are able to activate T-lymphocytes in two ways: through antigen presentation and indirectly through type I interferon production. High concentrations of plasmacytoid dendritic cells have been found in kidney and skin lesions in SLE patients, which confirms the involvement of dendritic cells in tissue damage. 45 An interesting property found in the pathogenesis of SLE was the ability of platelets to activate dendritic cells, which, in turn, led to increased production of IFN-a. 46
CD4+ T-lymphocytes
CD4+ T-lymphocytes are the major regulators of the immune response, which can modulate the immune response through the secretion of certain types of cytokines. In SLE, increased secretion of IL-17 and reduced expression of IL-2 were noted. 47 It has been shown that CD4+ T-lymphocytes are able to enter the kidneys of patients and induce increased inflammation by recruiting neutrophils, which contributes to the progression of SLE. 48 The mechanism for altering the expression of these cytokines is well known. Increased activity of the CaMKIV enzyme leads to the activation of the transcription factor CREMa, which interacts with the DNA methyltransferase DNMT3, which in turn binds to the IL2 locus in CD4+ T lymphocytes, resulting in an increase in CpG methylation and a decrease in IL2 expression, the opposite effect occurs relative to the IL17A locus, where the expression of the cytokine IL17 increases. 49 The transition to humoral immunity is facilitated by a subgroup of CD4+ T-lymphocytes, follicular T-helper cells (Tfh), which express the transcription factor BCL6 and the cytokine IL-21, which are necessary for B-lymphocyte maturation and antibody production. 50 Studies conducted on a mouse model have shown that inhibition of the actions of these molecules leads to an attenuation of the disease. 51 CD8 + T-lymphocytes
Some studies have demonstrated reduced cytotoxic activity of CD8+ T-lymphocytes in the pathogenesis of SLE. 52 The defect in nascent cytotoxicity may be of clinical importance, as a reduced cytotoxic immune response is associated with reduced protection against intracellular pathogens, and bacterial and viral infections are considered to be key factors in mortality in SLE patients. 53 A decrease in the function of CD8 + T-lymphocytes may be associated with impaired expression of a number of receptors and enzymes, such as: CD244, CD38, perforin, and granzyme. 54 At the same time, there is evidence that CD8 + T cells can accumulate in organs and cause tissue damage, but it is not clear whether this sublocalization of cytotoxic T lymphocytes is associated with the development of a T-cell immune response as a result of the presentation of autoantigens. 54
B-lymphocytes
The role of immune cells in the pathogenesis of SLE

Model of the systemic lupus erythematosus pathogenesis based on mitochondrial components
Triggering an inflammatory response
Exposure to various environmental factors (ultraviolet radiation, viral and bacterial infections), together with genetic factors that reduce antioxidant protection, can contribute to the development of oxidative stress. 58 Oxidative stress is a state of disturbance of the redox balance of the cell towards a decrease in pH, while reactive oxygen species (ROS) play a key role in its development. The development of oxidative stress, in turn, contributes to the occurrence of mitochondrial dysfunction, which is observed in SLE in peripheral blood T-lymphocytes. 59 Disruption of mitochondrial function leads to a decrease in ATP production, which affects the decrease in the efficiency of the oxidative phosphorylation process and, as a result, leads to a metabolic shift towards glycolysis. 60 Another important consequence of mitochondrial dysfunction is an increase in ROS production due to electron leakage in the respiratory chain, which leads to an additional increase in oxidative stress. In addition to the loss of mitochondrial function during oxidative stress to T cells in SLE patients, a disruption of mitochondrial dynamics is observed - an increase in mitochondrial biogenesis and a weakening of mitophagy, as a result of which the number of large dysfunctional mitochondria increases in the cell, the production of ATP in which is reduced.
One of the key factors underlying the inhibition of mitophagy in SLE is the overexpression of the HRES-1/Rab4 enzyme, which is responsible for endosomal traffic during autophagy. In a study, 61 it was demonstrated that overexpression of HRES-1/Rab4 led to the accumulation of dysfunctional mitochondria in CD4+ T-lymphocytes, which was demonstrated in a mouse model of SLE and lymphocytes isolated from patients with SLE. It was shown that the level of the Drp1 protein, which was the initiator of mitophagy, was markedly reduced. In turn, the introduction of the compound 3-PEHPC, which was an inhibitor of HRES-1/Rab4, led to an increase in the content of Drp1 and a decrease in mitochondrial accumulation. In view of this, 3-PEHPC may be considered as a potential therapeutic agent in the treatment of SLE. Another found inhibitor of mitophagy that affects the progression of SLE is the CD38 protein. It has been found that its increased expression in SLE patients led to impaired CD8+ T-lymphocyte function and greater vulnerability of patients to viral infections. 62 Thus, CD38 inhibitors may also be considered as potential therapeutic agents in the treatment of SLE.
Nitric oxide (NO), secreted by monocytes, is an important inflammatory mediator. At the same time, in the culture of T-cells of patients with SLE, it was found that NO was an inducer of mitochondrial biogenesis, while it also mediated ATP depletion, causing ETC complex IV dysfunction, and induced an increased calcium flux into the mitochondria of T-lymphocytes.
63
All this led to the accumulation of dysfunctional mitochondria and the development of oxidative stress. It has been noted that T-lymphocytes in SLE often died by necrosis, while releasing heat shock proteins, which stimulated monocytes to produce NO, thus creating a positive feedback in the pathogenesis of SLE. While the production of ROS increased, which further leads to the development of oxidative stress,
64
ROS subject biological macromolecules to oxidative modification, as a result of which tree radical molecules can be formed that are highly toxic to cells (for example, 4-hy droxy-2-nonenal, which occurs as a result of lipid peroxidation) and are able to propagate a radical chain reaction to other molecules, which is another reason for increased oxidative stress.
65
In addition, ROS destroy cellular structures, especially mitochondria that produce them, which leads to the release of a number of molecules that, under pathological conditions, become DAMPs (Damage-associated molecular patterns),
66
and under conditions of impaired autotolerance in SLE and other autoimmune diseases, they play the role of autoantigens, which are “hunted” by their own immune cells, perceiving them as foreign molecules. The involvement of many mitochondrial DAMPs has been identified in the development of SLE, including mitochondrial DNA, HSP60, cardiolipin and etc.
67
Mitochondrial components released from damaged mitochondria into the cytoplasm bind to NLR receptors of innate immunity, that activates the transcription factor NF-kB, an increases the expression of interferon and pro-inflammatory cytokines, which ultimately leads to the development of an inflammatory response. However, many types of cells, primarily immune cells, are able to secrete their own mitochondria into the extracellular space,
67
as a result of which mitochondrial DAMPs bind to TLR receptors on the surface of immune cells, which also triggers inflammation. In addition, mitochondrial components can be in the extracellular space as a result of cell death. Increased cell death by necrosis and apoptosis is confirmed in SLE, an important role in the initiation of which is played by the previously mentioned oxidative stress.
60
In SLE, a specific death of neutrophils is also noted (NETosis), which results in the formation of neutrophil extracellular traps (NETS), which leads to the release of mitochondrial DNA into the extracellular space and subsequent activation of interferon.
68
Oxidative stress in SLE may be caused by increased activity of complex I. A study
69
showed that N-acetylcysteine reduced oxygen consumption in T-lymphocytes of SLE patients. N-acetylcysteine also showed its effectiveness directly in a clinical study on patients with SLE.
70
Another oxidative stress inhibitor, rapamycin, has been shown to be effective in a clinical trial.
71
The general scheme for the development of autoimmune inflammation based on mitochondrial components is shown in Figure 1. Development of autoimmune inflammation in SLE in response to oxidative stress and mitochondrial dysfunction.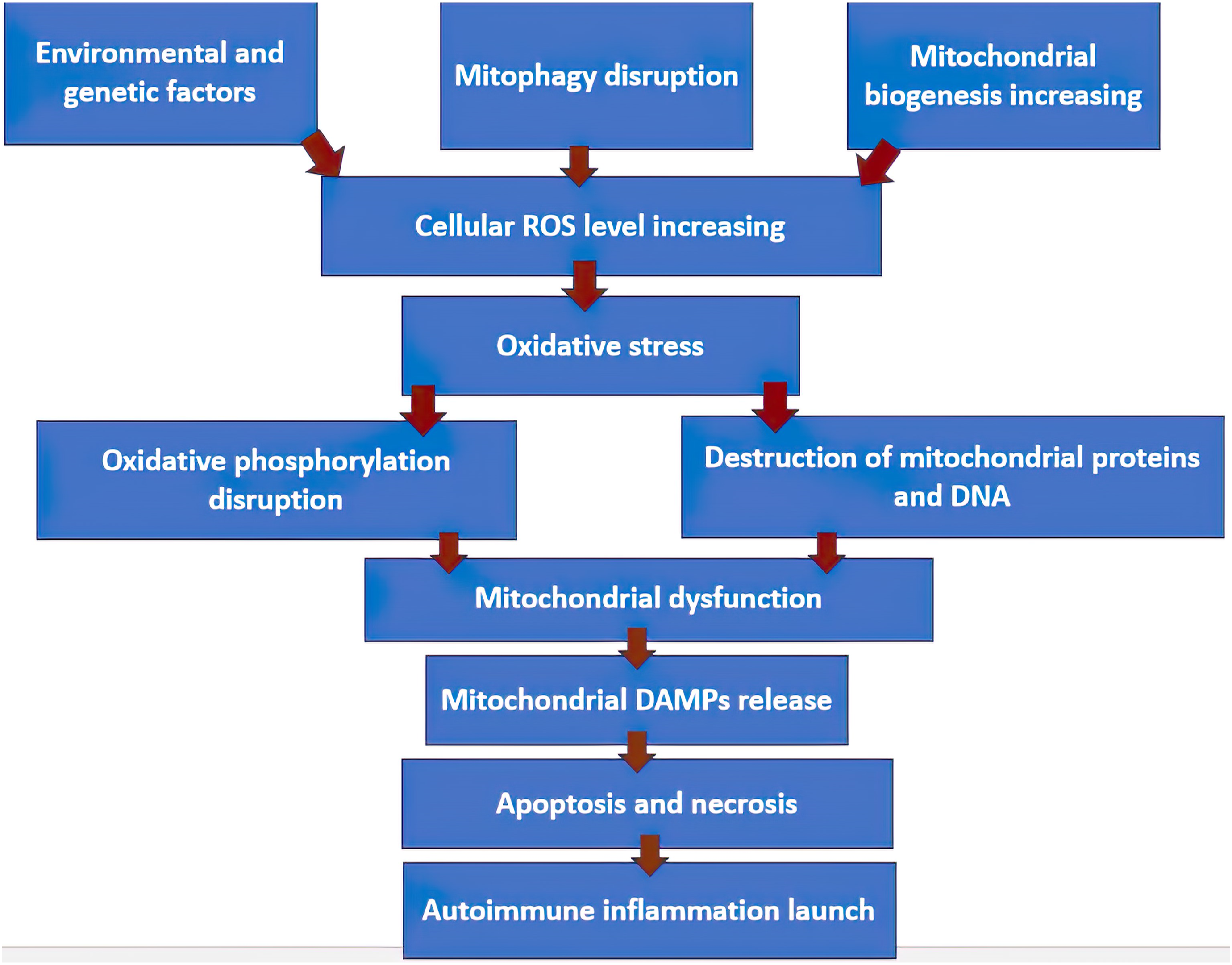
Development of an adaptive immune response
After triggering the innate immune response, mitochondrial antigensare presented on the surface of antigen-percentage cells, the main representatives of which in SLE are dendritic cells. 72 Attracted by inflammatory stimuli, T cells migrate from the lymph nodes and interact with dendritic cells; during this interaction, antigen presentation occurs, which triggers the maturation and proliferation of T lymphocytes. 60 It has been shown that follicular T-helpers, which are a subpopulation of CD4+ T-lymphocytes, which have a significant role in triggering and enhancing the humoral immune response, have a high activity in the pathogenesis of SLE. At the same time, it is noted that follicular T-helpers produce high levels of the cytokine IL-21, elevated levels of which are found in the plasma of patients with SLE. 73 It has been shown that IL-21, in addition to activating B cells, stimulates the activation of CD8+ T lymphocytes, which additionally enhances the autoimmune response in SLE. 74 Thus, follicular T cells are the central link in the regulation of the autoimmune response in SLE, activating both humoral and cellular adaptive immune responses. However, it is still worth noting that the humoral immune response plays a dominant role in the further progression of the disease. An important role in the development of an autoimmune reaction is played by the ratio of follicular and regulatory T-helpers, which in the initial state in the absence of an immune stimulus is 1:1, but when an immune response occurs, the number of follicular T-cells increases. 75 Proper functioning of regulatory T cells is an important factor in preventing the development of an autoimmune reaction. Follicular T cells regulate the activation, proliferation and differentiation of antigen-specific B cells through the interaction of CD40L and ICOS molecules on the surface of follicular T cells with their ligands on B cells: CD40 and ICOSL, as well as through the production of cytokines BCL6, IL21 and IL4. 75 After the activation of autoreactive B cells and their maturation into plasma cells, the production of autoantibodies begins. The production of antibodies to mitochondrial molecules has been shown in SLE, while the main characterized mitochondrial antigens in the pathogenesis of SLE are mitochondrial DNA, cardiolipin and heat shock protein HSP60, in addition, antibodies AMA-M3 and AMA-M5S are registered, the structure of mitochondrial molecules for which is still unknown. 76 It was found that the presence of antibodies to cardiolipin was associated with the risk of thrombocytopenia. 77 The combined presence of antibodies to cardiolipin and HSP60 is associated with the occurrence of vascular disorders. 78 It is noteworthy that autoantibodies, by binding to autoantigens in SLE, form an immune complex that activates innate immune leukocytes, thus creating a positive feedback loop that enhances the autoimmune response. 60
Chronification of inflammation
The involvement of mitochondrial autoantibodies in additional stimulation of innate immunity is one of the factors leading to the development of chronic inflammation in SLE. Additional activation of innate immune cells, primarily macrophages, causes re-inflammation, during which tissue damage occurs. The mitochondrial DAMPs released during the damage cause a new “wave” of the inflammatory response. Impaired clearance, which is noted in the pathogenesis of SLE, also makes its contribution in chronification of inflammation: as a result of necrotic or apoptotic cell death in peripheral tissues, autoantigenic molecules accumulate in the external environment, which under normal conditions are quickly absorbed by phagocytes, however, with impaired clearance, they become targets for increased inflammation.
79
Another reason for the development of chronic inflammation in SLE is the ability of autoreactive B cells to enhance the autoimmune response by activating autoreactive T cells through antigen presentation and costimulation, as well as through the production of the pro-inflammatory cytokine IL-6, whose action, in addition to directly increasing inflammation, is aimed at inhibiting the proliferation of regulatory T cells, which are the main suppressors of the development of an autoimmune reaction.
80
An important role in the development of chronic inflammation is played by mitochondrial DNA released by neutrophils in response to stimulation with autoantibodies to TLR7 agonists.
81
The release of elevated levels of mitochondrial DNA is associated with impaired DNA degradation in lysosomes as a result of inhibition of phosphorylation of mitochondrial transcription factor A (TFAM), one of the key participants in the regulation of mitochondrial DNA degradation.
81
The released mitochondrial DNA promotes the activation of interferon synthesis and induces the activation of dendritic cells.
81
In addition to mitochondrial DNA, additional activation of dendritic cells is caused by increased production of ROS, which activate the Myc-dependent metabolic signaling pathway, which in turn activates the mTORC1 pathway and leads to accelerated maturation of dendritic cells and increases the rate of T-lymphocyte activation in SLE.
82
Chronification of inflammation is the main reason for the incurability of this disease. Targeting one pathway that increases inflammation and leads to its chronification, such as blocking NETosis, will often not be successful in therapy because it does not limit other pathological pathways. The model described above is shown in Figure 2. The model of the pathogenesis of systemic lupus erythematosus based on mitochondrial components.
Discussion
As shown in the previous section, mitochondrial components play an important role in the initiation and progression of the autoimmune response, which, however, does not eliminate important unanswered questions. First of all, an important task is to identify the exact molecular structure of unknown mitochondrial DAMPs for which autoreactive antibodies have already been found, as well as to study in more detail the effect of autoantibodies on the re-induction of the innate immune response, which leads to chronic inflammation. A more complete understanding of the functions of the mitochondrial autoantigen-autoantibody complex in the pathogenesis of SLE will not only help to better understand the pathological picture in SLE, but also potentially allow the identification of promising therapeutic targets in the treatment of SLE, as well as the creation of a highly sensitive immunological test system based on this protein interaction, which will allow more accurate diagnosis and prediction of the course of the disease. Conducting comprehensive studies on the detection of new mitochondrial antigens in SLE, for example, as, 83 helps to find new effective disease biomarkers that can be useful in the patient stratification. Mitochondrial antigens can also find their use as molecular correlates of SLE complications, as shown in the development of neuropsychiatric SLE, which correlated with the level of autoantibodies to cardiolipin. 84 Another important issue is the study of the role of mitophagy impairment in the production of mitochondrial DAMPs, Identification of proteins participating in mitophagy, whose functions are impaired in the pathogenesis of SLE, will allow the development of new therapeutic strategies that will potentially help reduce the formation and accumulation of autoantigenic molecules, which will reduce the reactivity of autoimmune cells. Great importance is the study of autoantigens by the level of immunogenicity, understanding the “strength” of autoantigens will allow pinpointing the most powerful inducers of inflammation and determining the antigenic load for each mitochondrial component. Another direction worth paying attention to in the pathogenesis of SLE is the metabolic changes caused by immune cell mitochondrial dysfunction and how these changes affect the activation of autoreactive leukocytes. An important factor in the study of the pathogenesis of SLE is the discovery of new cell populations involved in the development of the disease. For example, erythrocytes have also been shown to be involved in the pathogenesis of SLE. 85 It was noted that in SLE in erythrocytes, the programmed death of mitochondria is disrupted as a result of a defect in the autophagic pathway. As a result of antibody opsonization of erythrocytes bearing mitochondria, the production of type I interferon (IFN) in macrophages is induced. 85 Based on the foregoing, mitochondria are the important link in the pathogenesis of SLE, and researchers should pay closer attention to the dysfunctions of mitochondrial proteins that occur during an autoimmune reaction. An important step is to identify specific groups of cells involved in the production of mitochondrial autoantigens: whether they are predominantly immune cells or other cell types not directly involved in the autoimmune reaction, but exposed to inflammation. Solving these problems can contribute to a strong development in scientific knowledge about the pathogenesis of SLE and cognate diseases that have a similar development scenario.
Conclusions
The occurrence of oxidative stress and subsequent mitochondrial dysfunction and disturbances in mitochondrial dynamics in SLE contributes to the spread and accumulation of mitochondrial components, which, when recognized by immune cells, are defined as autoantigens and initiate the development of inflammation. Mitochondrial components are released into peripheral tissues as a result of cell death or direct release of mitochondria by immune and some other cell types. The development of an adaptive autoimmune response proceeds predominantly in a humoral way with the production of autoantibodies, which, in combination with mitochondrial autoantigens, are able to re-induce innate immunity, which contributes to the development of chronic inflammation. Other factors leading to the chronification of inflammation are impaired clearance of autoantigens, activating effect of B-lymphocytes, impaired mitochondrial DNA lysis in neutrophils, which leads to the release of a large amount of these molecules into the extracellular space, as well as increased production of ROS.
Footnotes
Author Contributions
writing—original draft preparation, AVB, ADZ; writing—review and editing, VNS,MAP, AVG, ANO.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Russian Science Foundation (Grant # 23-25-00,339).