
Abstract
The broad-spectrum apoptosis (caspase) inhibitor, Q-VD-OPh, has been shown to have no side effects and is effective at a much lower concentration than other FMK-type caspase inhibitors. However, an appropriate negative control to use with this inhibitor has not been available. In this study, we developed and analyzed a new compound, based on the Q-VD-OPh backbone, which acts as a cognate negative control. To create the negative control, we substituted a glutamate residue for the aspartate residue to create Q-VE-OPh, thereby retaining the identical charge and molecular properties with only the addition of an extra -CH2 group. The purity and quality were assessed by ion trap mass spectrometry and verified by nuclear magnetic resonance. We determined the effectiveness of Q-VE-OPh, in comparison to Q-VD-OPh, to prevent DNA fragmentation in human Jurkat T leukemia cells that were induced to undergo apoptosis. DNA fragmentation was analyzed by agarose gel electrophoresis for the presence of DNA laddering, the hallmark indicator of apoptosis. Our results indicate that apoptosis was potently inhibited by Q-VD-OPh. In stark contrast, Q-VE-OPh did not inhibit apoptosis at a similar dose but required at least 20 times greater concentration than Q-VD-OPh to have any inhibitory effect. Western blot analysis showed that Q-VE-OPh was similarly less effective at inhibiting the activation of the extrinsic (caspase 8) and intrinsic (caspase 9) initiator caspases. Cell proliferation and viability studies further demonstrate that Q-VE-OPh is non-toxic, even at high concentration. Our data indicate that the specificity, effectiveness, and absence of toxicity of Q-VE-OPh provides the appropriate and superior negative control for in vitro and in vivo studies when analyzing the effects of o-phenoxy caspase inhibitors.
Introduction
Apoptosis is an active process of cell death characterized by the lack of an inflammatory response, crosslinking of the plasma membrane, and formation of apoptotic bodies. 1 Apoptosis is mediated by specific initiator and effector cysteine proteases (caspases) that are unique in cleaving substrates specifically following aspartate residues.2–5 In humans, the apoptotic process is defined by the activation of caspases that are specific for two major pathways. The intrinsic pathway (caspase 9/3) is activated by the release of cytochrome C from the mitochondria into the cytosol. 6 The extrinsic pathway (caspase 8/10) is activated via ligand binding to death receptor systems of tumor necrosis factor alpha, Fas/CD95, or Trail. 7
Over the past several years, inhibitors have been formulated that prevent caspase activation. Specifically, broad-spectrum caspase inhibitors consist of O-methylated mono- to tetra-peptide amino acids that enhance cell permeability and are conjugated to carboxyterminal groups such as chloromethyl ketone (CMK), fluoromethyl ketone (FMK), or aldehyde (CHO) that enable them to act as competitive inhibitors. These cell permeable inhibitors reversibly (CHO) or irreversibly (CMK/FMK) block apoptosis by preventing caspase activation, substrate cleavage, and DNA ladder formation. Although fluoromethylketone (FMK)—conjugated caspase inhibitors have been effective in inhibiting caspase-mediated events in vitro and cell culture, they have been reported to be toxic in vivo and the relatively high doses required limit their usefulness.8–10
In contrast to these caspase inhibitors, Q-VD-OPh (quinolyl-valyl-O-methyl aspartyl-[-2,6-difluorophe noxy]-methyl ketone) has been previously described as a broad-spectrum caspase inhibitor that can prevent all major apoptotic pathways. 10 Q-VD-OPh, consists of the dipeptide amino acids, Val-Asp, coupled to a C-terminal O-phenoxy group. Q-VD-OPh is capable of preventing apoptosis at concentrations as low as 5 μM and is at least 10 times more effective than current fluoromethyl ketone (FMK) inhibitors in cell culture. 10 In addition, Q-VD-OPh has been shown to be nontoxic and highly effective in numerous in vivo studies, and is capable of crossing the blood brain barrier.10–18
An important factor in determining the specificity of any pharmacological inhibitor is the use of an appropriate cognate negative control with predominantly similar properties that can be used at similar doses, but demonstrates no adverse effects. For studies using caspase inhibitors, either with—FMK or —O-phenoxy conjugation, the only commercially available negative control has been carboxybenyzl-FA-fmk (Z-FA-fmk). This is an inappropriate negative control, as aspartate (D) is larger and has a significant negative charge, whereas alanine (A) is small and uncharged and thus, does not approximate an appropriate substitution. In addition, Z-FA-fmk on its own has been reported to be a specific inhibitor of cathepsins, thus obviating its use as a negative control for caspases. 19 Furthermore, the conjugation of—FMK to Z-FA negates its use as a negative control for in vivo studies where O-phenoxy-type of inhibitors are used, since it is not an analogue. In all reported uses of —FMK conjugated or Q-VD-OPh caspase inhibitors, no appropriate negative control to confirm specificity of the results, other than vehicle, have been reported.
In this study, we developed and determined the effectiveness of a novel, cognate negative control, Q-VE-OPh, to the broad-spectrum caspase inhibitor Q-VD-OPh. Q-VE-OPh was found to be nontoxic and 20 times less active than the caspase inhibitor, Q-VD-OPh, by DNA ladder assay and Western blot analysis. Our data indicate that the specificity and effectiveness, using an optimal negative control for O-phenoxy inhibitors, provides a new and useful tool to confirm the specificity of O-phenoxy caspase inhibitors both in vitro and in vivo.
Materials and Methods
Materials
Jurkat T cells, clone E6-1, were obtained from ATCC. Caspase 8 antibody was purchased from Cell Signaling, Inc. and caspase 9 antibody from Santa Cruz Biotechnology. Actinomycin D was purchased from Calbiochem, Inc. Immobilon P was purchased from Millipore. Supersignal chemiluminescence reagent was purchased from Pierce, Inc.
Production of Q-VD-OPh and Q-VE-OPh
The caspase inhibitor Q-VD-OPh and corresponding negative control Q-VE-OPh were produced by Apoptrol, LLC (Beavercreek, Ohio). Briefly, amino terminally-protected aspartyl or glutamyl groups were converted to—OPh conjugates and then reacted to elicit the methylated forms of Q-VD(OMe)-OPh and Q-VE(OMe)-OPh. This was followed by HPLC purification. Production of the non-methylated compounds was achieved by saponification. Q-VD-OPh and Q-VE-OPh were solubilized in DMSO prior to use and were found to be stable for greater than six months.
Ion trap mass specrtometry
Q-VE-OPh and Q-VD-OPh were purified by size exclusion HPLC (GBC, Australia), at 0.5 ml/min in 90% acetonitrile/0.1% TFA. Fractions were collected at one minute intervals, dried by speed vacuum, and weighed. An aliquot of each fraction was collected, solubilized in 50% acetonitrile/0.1% formic acid, and injected into a Bruker HCTUltra Ion Trap mass spectrometer in positive ion mode. Dry gas was set at 10 L/min, heat at 200°C and capillary set at 129.6 volts. Spectra were captured for 3 minutes and analyzed using Bruker Data Analysis Software.
Cell culture
Jurkat human T cells were cultured at 1 × 106 cells/ml, unless otherwise indicated, in RPMI 1640 containing 10% FBS and antibiotic/antimycotic. All cells were cultured at 37°C and 95% O2/5% CO 2 .
Apoptosis assays
Jurkat cells (1 × 106 cells/ml) were preincubated 1 hr with vehicle, drug alone, or various concentrations of Q-VE-OPh or Q-VD-OPh, prior to treating with actinomycin D for 4 hrs. Cytoplasmic DNA was isolated and analyzed for DNA laddering. Briefly, apoptosis was measured by analysis of an oligonucleosomal DNA ladder in agarose gels as previously reported.2,10,20,22 Cells (1 × 107) were centrifuged at 1,000 rpm for 5 min and lysed in HL buffer for 15 min. The lysate was extracted with an equal volume of phenol, phenol:chloroform:isoamyl alcohol (25:24:1), and precipitated 18 h at −20°C with an equal volume of isopropanol and 0.1 volume of 5 M NaCl. The precipitated DNA was resuspended in Tris/EDTA, pH 8.0, containing DNase-free RNase A and incubated at 37°C for 30 min. The DNA was analyzed on a 1.2% agarose gel containing ethidium bromide.
Western blotting
Protein concentrations and Western blotting were performed as described previously.2,10,22,23 Briefly, 100 μg of whole cell lysate was separated by SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride membrane. Protein transfer was empirically determined by staining with 1.0% Ponceau S.2,10,22 The membrane was incubated in blocking buffer (60 mM Tris, 200 mM NaCl with 0.05% Tween 20 containing 5% nonfat dry milk, pH 7.4) and incubated with a 1/1,000 dilution of primary antibody for 2 h at room temperature. The blot was washed and then incubated with a 1/50,000 dilution of goat anti-rabbit horseradish peroxidase secondary antibody for 1 h at room temperature and processed using the Supersignal chemiluminescence reagent according to manufacturer's instructions.
Cell proliferation and viability assay
Jurkat human T cells (1 × 105 cells/ml) were treated for 72 hrs with vehicle (V), cell death inducer 1 μg/ml actinomycin D, 100 μM Q-VD-OPh or 100 μM Q-VE-OPh. Cell viability was determined by cell count and trypan blue staining at 24 hr intervals.
Results
The main objective of this study was to produce a cognate dipeptide amino acid analog similar to the broad-spectrum caspase inhibitor, Q-VD-OPh, which could be used in experiments as an appropriate negative control. Q-VE-OPh substitutes a single amino acid, glutamic acid (E) for the aspartic acid (D) in Q-VD-OPh, thus retaining a nearly identical size and charge. Corresponding, qualitative, ion trap mass spectrometry data are shown for Q-VE-OPh and Q-VD-OPh to indicate molecular mass and level of purity (Fig. 1). The data indicate that Q-VE-OPh is present as a 528 dalton peak and Q-VD-OPh is identified as a 514 dalton peak (Fig. 1). Contaminating molecules are minimal, if present at all, and represent less than 1% of the total ion species. These data were confirmed by NMR-analysis immediately following synthesis (data not shown).
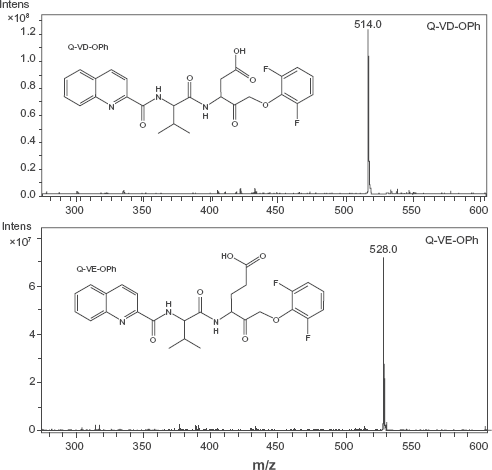
Chemical Structure and Mass Spectrometry of Q-VD-OPh and Q-VE-OPh Chemical structures for Q-VE-OPh and Q-VD-OPh “No Methyl” were generated with CS ChemDraw Pro (Cambridge Software) and mass isotopes determined based on the chemical formula. Q-VD-OPh (upper panel) and Q-VE-OPh (lower panel) were analyzed by ion Trap mass spectrometry. The expected mass for Q-VD-OPh was 514 daltons and for Q-VE-OPh was 528 daltons, and with a charge of 1 (m/z). The scale on the Y-axis indicates relative intensity. The major peak in the upper panel following synthesis was 514 daltons, representing Q-VD-OPh. The major peak in the left panel following synthesis is a 528 dalton peak representing Q-VE-OPh.
To determine the ability of Q-VE-OPh to inhibit DNA fragmentation, apoptosis was induced by treatment of human Jurkat T leukemia cells for 4 hours with actinomycin D and evaluated by DNA ladder formation, the hallmark of apoptosis (Fig. 2). In the absence of caspase inhibitors, actinomycin D induced substantial apoptosis after 4 h, as determined by the presence of a classical DNA ladder (Fig. 2). Incubation with Q-VE-OPh or Q-VD-OPh or the vehicle alone did not induce apoptosis, even at high concentrations (Fig. 2, data not shown). Q-VE-OPh, upon co-incubation with actinomycin D, was unable to prevent apoptosis from occurring within the effective 5-20 μM range for Q-VD-OPh (data not shown). At higher concentrations of Q-VE-OPh, partial inhibition began to be observed beginning at 100 μM (Fig. 2A). In contrast, under the same conditions, Q-VD-OPh completely prevented apoptosis at concentrations as low as 5.0 μM (Fig. 2B). This indicates that Q-VE-OPh is at least 20 times less effective at inhibiting apoptosis than the caspase inhibitor Q-VD-OPh and well beyond the effective concentration range of Q-VD-OPh of 5-20 μM, i.e. concentrations that are normally used in experimental situations.
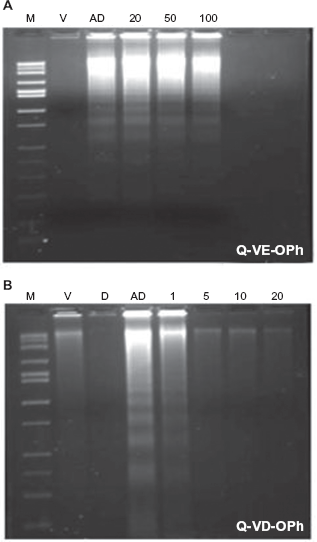
Q-VE-OPh and Q-VD-OPh Effects on Apoptotic DNA laddering in Jurkat Cells. Jurkat cells (1 × 106 cells/ml) were treated for 4 hrs with vehicle (V), 1 μg/ml actinomycin D (AD), Drug alone 100 uM (D), or various concentrations (1,5,10,20,50, or 100uM) of Q-VE-OPh (
Apoptosis is dependent upon caspase activation. Caspase activation is dependent on the cleavage of specific caspase enzymes.24–29 To further confirm the inability of Q-VE-OPh to inhibit caspase activation at relevant concentrations, Western blot analysis was performed to examine the cleavage (and thus activation) of the two main human initiator caspases, i.e. 8 and 9 (Fig. 3A and B, respectively). We observed cleavage of both caspase 8 and 9 protein in cells treated with the apoptotic inducer, actinomycin D, and Q-VE-OPh (Fig. 3A), while in cells treated with actinomycin D and Q-VD-OPh there was no caspase 8 or 9 cleavage at the same concentrations (Fig. 3B). This indicates that Q-VD-OPh inhibited cleavage and activation of caspase 8 and caspase 9, whereas Q-VE-OPh did not. The Western blot results confirm our DNA ladder findings that Q-VE-OPh is a superior cognate negative control for Q-VD-OPh and O-Phe-noxy caspase inhibitor studies; when used within the correct and recommended concentration ranges.
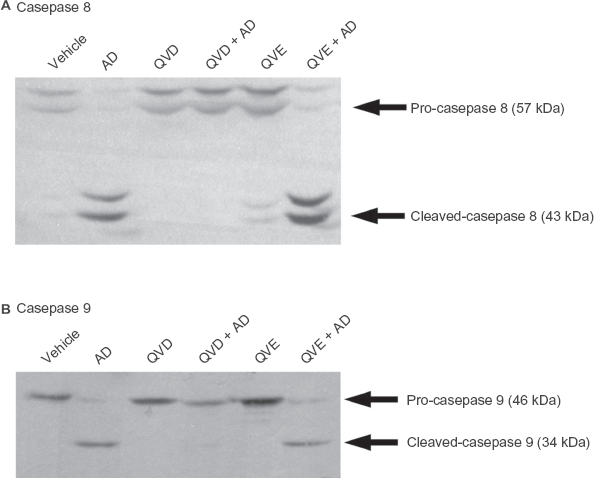
Q-VE-OPh does not prevent initiator caspase 8 or caspase 9 activation. Jurkat human T cells (1 × 106 cells/ml) were treated for 4 hrs with vehicle (V), 1 μg/ml actinomycin D (AD), 20 μM Q-VD-OPh (QVD), 20 uM Q-VD-OPh preincubated 1 hr prior to addition of 1 μg/ml actinomycin D (QVD + AD), 20 μM Q-VE-OPh (QVE), 20 μM Q-VE-OPh, preincubated 1 hr prior to addition of 1 μg/ml actinomycin D (QVE + AD). 100 μg of whole cell lysate was separated by SDS-PAGE and Western blotting was performed using rabbit polyclonal antibody to caspase 8 (Upper panel) and caspase 9 (Lower panel), as described in the Materials and Methods, and developed using Supersignal chemiluminesence. Arrows indicate the molecular weights of the inactive proforms as well as the cleaved and activated forms of caspase 8 and 9. Faint cleavage fragments seen in the vehicle and Q-VE-OPh lanes in Figure 2A are due to a low level of background apoptosis. Equal protein loading was present in all lanes (data not shown).
Q-VD-OPh has previously been shown to be nontoxic. 10 To determine if Q-VE-OPh was also non-toxic, cells were treated with Q-VE-OPh for 72 hrs and cell viability was assessed (Fig. 4). As anticipated, Q-VE-OPh demonstrated no signs of toxicity and allowed undisrupted cell proliferation to occur over the entire time course evaluated.
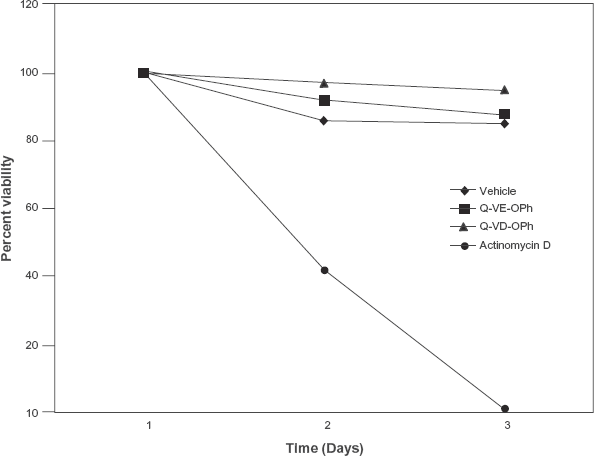
Q-VE-OPH is not toxic. Jurkat human T cells (1 × 105 cells/ml) were treated for 72 hrs with vehicle (V), 1 μg/ml actinomycin D (AD), 100 μM Q-VD-OPh (QVD) or 100 μM Q-VE-OPh. Cell viability was determined by trypan blue staining of cells at 24 hr intervals.
Discussion
The present study was designed to identify and characterize an analog of the widely used in vivo, broad-spectrum caspase inhibitor, Q-VD-OPh. Based on a previous report in Drosophila that demonstrated that DRONC caspase activity cleaves equally well after an aspartate or a glutamate,30–32 it was anticipated that Q-VE-OPh might act similarly to Q-VD-OPh and act as a caspase inhibitor at low concentrations in mammals.
To create this analog, a glutamate residue was substituted for the aspartic acid residue in Q-VD-OPh. To our surprise, analysis of Q-VE-OPh indicated that it is 20 times less effective than Q-VD-OPh, based on concentration, and provided an optimal negative control at effective concentrations, where Q-VD-OPh would normally inhibit apoptosis in cell culture or in vivo. Q-VE-OPh was unable to inhibit DNA fragmentation or caspase activation at relevant concentrations. Further evaluation indicated that Q-VE-OPh is not toxic over time, even at high concentrations.
In contrast to the only currently commercially available “negative control” for caspase inhibitors, Z-FA-fmk, our study suggests that Q-VE-OPh is the appropriate and cognate negative control for Q-VD-OPh. In fact, Z-FA-fmk has been reported to inhibit effector caspases as well as act as a cathepsin B inhibitor and therefore obviates its use as a negative control.19,33,34 Furthermore, Z-FA-fmk, as with all “fmk” containing caspase inhibitors, has been shown to be toxic in vivo due to the accumulation of fluoroacetate.9,10,35 Alternatively, Q-VD-OPh has been widely used in vivo for extended periods of time and is recommended for use due to its lack of toxicity. It is likely that the—OPh moiety eliminates in vivo toxicity and that the use of Q-VE-OPh as a negative control for caspase inhibitors in vivo will also have no associated toxicity.
This study provides strong support that optimal negative controls could be produced for each of the specific O-phenoxy caspase inhibitors, such as Q-IETE-OPh, Q-LEHE-OPh, and Q-DEVE-OPh for use in vivo, which should have no functional inhibitory or cytotoxic capacity within the effective dose ranges, thus serving to define and validate an entire panel of compatible and appropriate cognate negative controls for O-phenoxy caspase inhibitors. Finally, while Q-VE-OPh serves as an optimal negative control for in vitro, cell culture and in vivo mammalian studies, it is anticipated that it may possibly function as an inhibitor of DRONC caspase in Drosophila, based on a previous report. 30
In conclusion, we have produced and determined the effectiveness of a novel and appropriate negative control, Q-VE-OPh, for the broad-spectrum caspase inhibitor Q-VD-OPh. The aspartic acid residue of Q-VD-OPh has been substituted with a glutamic acid residue, thereby maintaining the same negative charge. The substitution of the glutamate residue causes the Q-VE-OPh compound to be significantly less active in mammals, suggesting that the binding pocket for caspases must be highly specific to the size of the side chain. In addition, their molecular sizes and structures are nearly identical. Our data indicate that the specificity, effectiveness, and lack of toxicity, using an optimal negative control for O-phenoxy inhibitors, provides a new and useful tool to confirm specificity of O-phenoxy caspase inhibitors when studying mammalian apoptosis both in vitro and invivo.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. Daniel Ketcha is a scientific advisor for Apoptrol, LLC. Eugene Hern, David Cool, and Thomas L Brown are the founding principles of Apoptrol, LLC. The peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgements
We would like to thank the Proteome Analysis Laboratory at Wright State University for the mass spectrometry analysis. This work was supported in part by a Wright State University Technology Transfer Grant (TLB).