
Abstract
Apoptosis is physiological cell death required for the cellular maintenance of homeostasis, and caspases play a major role in the execution of this process. Numerous disorders occur when levels of apoptosis within an organism are excessive, and several studies have explored the possibility of using caspase inhibitors to prevent these disorders. Q-VD-OPh (quinolyl-valyl-O-methylaspartyl-[2,6-difluorophenoxy]-methyl ketone), a novel pan caspase inhibitor, has been used because of its efficacy to inhibit apoptosis at low concentrations, its ability to cross the blood–brain barrier, as well as being nontoxic in vivo. This review examines Q-VD-OPh's ability to inhibit apoptosis in several animal models of human disease.
Introduction
Apoptosis is an energy-dependent cell death process that helps maintain homeostasis as well as regulate tissue involution. 1 The characteristic features of apoptosis are caspase activation, intact plasma membrane, oligonucleosomal DNA ladder fragmentation, formation and phagocytosis of apoptotic bodies, and the lack of an inflammatory response.2,3 When a cell dies apoptotically, it undergoes a number of morphological changes, including condensation of the nucleus and cytoplasm as well as cell shrinkage. During embryogenesis, apoptosis is particularly important for immune and neuronal development. 4 A number of diseases can occur when excessive apoptosis occurs, and this is associated with a number of autoimmune and neurological diseases.3–6
The Role of Caspases in Apoptosis
Caspase activation plays a major role in apoptosis. Caspases, which are cysteine aspartyl proteases, are normally present as procaspases and are inactive zymogens. 7 In order for a cell to undergo apoptosis, procaspases must become activated via cleavage or dimerization (Fig. 1).8–11 There are two classes of apoptotic caspases; initiator caspases, which are associated with the initiation of apoptosis (caspases 2, 8, 9, 10), and effector caspases, which cleave cellular substrates needed for the cell's survival (caspases 3, 6, 7) (Table 1).8,9,11 Initiator caspases are activated by dimerization via different cell stressors that can be triggered either from within the cell or externally. The initiator caspases then cleave and activate their substrates, the effector caspases. Effector caspases subsequently cleave essential survival proteins and DNA. What makes caspases unique is a tripeptide amino acid sequence immediately preceding an aspartic acid reside where cleavage occurs. Importantly, only substrates with sequence-specific, tripeptide-aspartyl residues that are structurally exposed can be cleaved by a particular caspase.8,11 Two predominant apoptotic pathways exist: the extrinsic (death receptor) pathway and the intrinsic mitochondrial (cytochrome c-dependent) pathway.
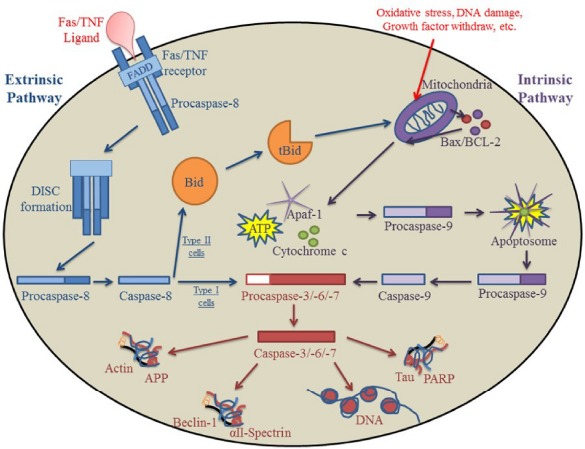
Extrinsic and intrinsic pathways of apoptosis. One of the major pathways for caspase activation is the extrinsic pathway. After apoptosis is initiated via death signal ligation in the cell membrane, a complex (DISC) is formed after FADD and procaspase-8 are recruited to the site. In type I cells, the DISC directly activates caspase-8; caspase-8 then activates effector caspases that go on to cleave substrates essential for survival. In type II cells, caspase-8 cleaves Bid, transforming it into the truncated and active form (tBid). tBid migrates to the mitochondria where it triggers the release of Bax/Bcl-2 and subsequently activates the intrinsic pathway. The other major apoptotic pathway for caspase activation is the intrinsic pathway. After toxic insults or DNA damage, the mitochondria releases cytochrome c, Apaf-1, and ATP into the cytosol of the cell, which lead to the formation of the apoptosome and the recruitment of procaspase-9. The apoptosome then activates procaspase-9, triggering the effector caspases and leading to the apoptotic destruction of the cell.
Two classes of human apoptotic caspases. Caspases involved in apoptosis are classified as initiators (2, 8, 9, 10) or effectors (3, 6, 7).

Extrinsic Death Receptor Pathway
The extrinsic pathway is triggered by the binding of Fas ligand (FasL), tumor necrosis factor (TNF)-α, or TNF-related apoptosis-inducing ligand (TRAIL) to its cognate death receptor located on the plasma membrane of the cell (Fig. 1).12,13 When death receptors bind to their ligand, the receptor becomes activated and triggers the recruitment of apoptotic adaptor proteins, such as Fas-associated death domain (FADD), among others (Fig. 1).3,13 Activated receptors undergo a conformational change and recruit initiator procaspase-8 to the intracellular death domain by binding to the death effector domain (DED) of procaspase-8. This formation creates a complex known as the death-inducing signaling complex (DISC). 14 Within the DISC, caspase-8 is activated by autocleavage and can then trigger effector procaspase-3. 14 Two types of cells have varying reactions to the activation of caspase-3. In type I cells, which are generally lymphoid in origin, caspase-3 is directly activated by caspase-8, which then cleaves prosurvival substrates (Fig. 1).6,15,16 In type II cells, which include most cell types, caspase-8 is unable to directly activate caspase-3. Caspase-8 cleaves a direct but inactive target, BH3 interacting-domain death agonist (Bid), into a truncated and active protein (tBid).15–18 tBid migrates to the mitochondria, resulting in the oligomerization and activation of Bax/Bak, thereby inducing the intrinsic pathway and subsequently triggering apoptotic death of the cell.16,19
Intrinsic Mitochondrial Pathway
The intrinsic pathway is triggered by DNA damage, growth factor withdrawal, oxidative stress, and/or toxic insults that are recognized by the mitochondria of an injured cell (Fig. 1).4,7 Apoptotic stimuli that disrupt cellular homeostatic conditions, normally regulated by the prosurvival B-cell lymphoma domain-2 (BCL-2) family of proteins, result in an increase in mitochondrial membrane permeability and the subsequent loss of ATP.15,20,21 One of the critical proteins that escapes from the mitochondria is cytochrome c, which binds together with Apaf-1 and ATP in the cytosol to form a complex known as the apoptosome. Formation of the apoptosome triggers the recruitment and activation of initiator procaspase-9 that can then cleave and activate effector procaspase-3 (Fig. 1).22,23 Effector caspases subsequently cleave substrate proteins that are essential for cell survival, such as poly (ADP-ribose) polymerase-1 (PARP), used in DNA repair; lamin A, responsible for the condensation of chromatin and the breakdown of the nuclear membrane; as well as αII-spectrin, which contributes to the structural integrity and organization of the cell.23–29
The Pan Caspase Inhibitor, Q-VD-OPh
Several types of caspase inhibitors are commercially available, including Boc-D-fmk and Z-VAD-fmk, which work by inhibiting the activation of some, but not all, initiator or effector caspases.27,28 The concern with these types of inhibitors is that they require much higher doses to be effective, are not true pancaspase inhibitors, and are toxic at higher concentrations (50 μM).27,28 Additionally, Z-VAD-fmk has been linked to the endogenous production of fluoroacetate, which is toxic to the cells, especially in the liver. 28 Q-VD-OPh (quinolyl-valyl-O-methylaspartyl-[-2,6-difluorophenoxy]-methyl ketone) is a true pancaspase inhibitor that is effective at significantly lower doses, is capable of crossing the blood–brain barrier, and is nontoxic in vivo.27,29,30 Q-VD-OPh has a significant advantage over other caspases and has an effective dose of 5 μM in cell culture in numerous cell types and 20 mg/kg in vivo. 27 The valine–aspartate amino acids allow Q-VD-OPh to function as a broad-spectrum caspase inhibitor with IC50 values ranging from 25 to 400 nM for recombinant caspases 1, 3, 8, and 9. 27 The quinolyl and phenoxy moieties are believed to enhance cellular permeability and substrate access. Owing to the clear superiority of Q-VD-OPh as a pancaspase inhibitor, this review will focus on its use in animal models of human disease.
Q-VD-OPh Inhibition of Apoptosis in Models of Human Disease
Ischemic stroke
Ischemic stroke is caused by a decrease in blood flow because of an obstruction of a cerebral artery, leading to a localized blood clot. 31 The decrease in blood flow triggers hypoxic restriction and energy depletion that results in the initiation of apoptosis and necrosis in neuronal cells in the blocked region or the core. 4 Tissue around the core, known as the penumbra, is also partially damaged by the decrease in blood flow, triggering inflammation and apoptosis.4,32
The involvement of caspase-1 and -3 in the damage caused by stroke has been supported by research in knockout mice.33,34 Absence of caspase-1 causes a reduction in the production of IL-1β, resulting in neuroprotective effects.33,34 The absence of caspase-3 leads to cortical neurons that are resistant to death after oxygen-glucose deprivation.33,34 During brain infarction, different pathways are triggered in different areas; caspase-8 is activated because of ligand binding in neuronal cells in the core, whereas caspase-9 induces neuronal death in the penumbra. 35
Stroke has recently been reported to be a sexually dimorphic condition. 36 In male 129S6SvEv mice following middle cerebral artery occlusion and reperfusion, Q-VD-OPh increased the survival rate post stroke. Interestingly however, neuronal cells that were caspase-3 positive in the ischemic core were not significantly inhibited. 37 In contrast, Q-VD-OPh dramatically reduced the number of caspase-3 positive cells in the penumbra. 37
The effectiveness of Q-VD-OPh during reperfusion was also tested by inducing stroke in both male and female PARP knockout mice (PKO). 38 The absence of PARP in stroke-induced males resulted in smaller infarct sizes but no change in the levels of cytosolic cytochrome c. In PKO, stroke-induced females, however, the infarct size was much larger and the amount of cytosolic cytochrome c release was higher than in the wild type, stroke-induced female controls. 38 Administration of Q-VD-OPh was able to significantly reduce the infarct size in PKO and wild type, stroke-induced females, whereas no effect was observed in males. 38
Perinatal stroke
Perinatal stroke differs from ischemic stroke because it occurs in newborns before or around the time of birth.39,40 The causes of perinatal stroke include clot formation, blood vessel wall injury, and the lack of blood flow and can occur because of placental infections or changes in the concentration of procoagulant and anticoagulant proteins during the transition from gestation to neonatal life.40–42
Seven-day-old male and female rats with unilateral focal ischemia followed by reperfusion were administered Q-VD-OPh, in order to test their efficacy in providing neuroprotection. 43 Q-VD-OPh was neuroprotective in females and reduced cell death while significantly increasing the animals’ survival rate between 48 hours and 21 days. 43 However, no significant effect was observed in males. 43 While ischemia can activate caspases after reperfusion, the mechanisms that activate caspase-3 appear to differ between genders because of the mitochondrial response. In males, cytochrome c release was extensive and occurred quickly. In contrast, cytochrome c release in females was large but constant during the first 24 hours of recovery. 43
The effectiveness of Q-VD-OPh, in response to short-and long-term treatments, was also examined in male neonatal C57Bl/6 mice following the induction of stroke by unilateral hypoxia-ischemia (HI). 44 Q-VD-OPh administered acutely at 12 and 36 hours was able to reduce apoptosis, specifically casapse-3 activation, and reduce levels of inflammatory chemokines but not inflammatory cytokines. 44 Male mice injected with Q-VD-OPh chronically over a 14-day period exhibited improved motor skills when examined a few weeks after the induction of stroke; however, examination a few months later showed that the improvements gained where no longer observed. 44 The reasons for these findings are currently unclear but warrant further investigation.
Spinal cord injury
Spinal cord injuries are caused by a primary trauma that can trigger secondary damage and lead to permanent tissue loss through insults such as mechanical compression, bleeding, and edema. 45 Secondary damages can include hemorrhaging, an inflammatory reaction, ischemia, interference with energy metabolism, as well as the induction of apoptosis. 45 A number of studies suggest that both necrosis and apoptosis occur after spinal cord injury, with necrosis occurring first, followed by apoptosis. 46
In order to test the efficacy of Q-VD-OPh in spinal cord injury, Wistar albino rats with a spinal cord injury at T8-T10 were administered Q-VD-OPh intraperitoneally, immediately after the injury. 45 Hemorrhaging, necrosis, vascular thrombi, as well as edema were found within the cells of the spinal cord 24 hours later. 45 Apoptosis was substantially reduced in animals treated with Q-VD-OPh, and the surviving animals had significantly better neurologic function than control animals. 45
Ischemic renal failure
Acute ischemic renal failure occurs when blood flow to the kidneys is severely reduced.47,48 This triggers the depletion of cellular ATP and leads to cell injury caused by inflammation and neutrophil infiltration.47,48 Tissue damage caused by the activation of inflammatory cytokines, such as IL-1β and IL-18, in acute renal failure has been shown to be reduced by caspase inhibitors. 48 In a surgically induced model of renal failure, C57BL/6 mice treated with Q-VD-OPh 60 minutes before the induction of renal failure was able to significantly inhibit the activation of caspase-1, leading to the decreased levels of IL-18 and thus preventing neutrophil infiltration of the kidneys. 49
Burn injury and cardiac dysfunction
Major burn injuries, which cover over 20-40% of the body's surface, can cause a number of complications, one of which is cardiac dysfunction.50,51 The occurrence of cardiac dysfunction caused by major burn injuries is more likely to occur in elderly individuals or young children who have a suppressed immune system.52,53 Cardiac dysfunction occurs when apoptosis is triggered in the myocardial cells by neutrophils that become activated after a major burn injury. 53 Sprague-Dawley rats that received third-degree burns and were treated with Q-VD-OPh had decreased levels of caspases-1, -3, -8, as well as a decrease in other cellular factors that contributed to cardiac dysfunction when compared to controls. 54
Alzheimer's disease
Alzheimer's disease (AD) is a disorder that results in progressive dementia caused by the deposition of amyloid-β (Aβ) peptides in senile plaques, neurofibrillary tangles (NFTs), and the subsequent loss of neurons.4,55 Activation of caspase-3 leads to the cleavage and formation of Aβ and NFTs. 56 Aβ peptides are formed after the amyloid precursor protein (APP) is cleaved by enzymatic secretases, and these newly made Aβ peptides then self-aggregate into insoluble β-sheet structures, leading to the formation of extracellular plaques.56,57 NFTs are formed when paired helical filaments become hyperphosphorylated on the microtubule-binding protein tau.56,58 Tau normally helps maintain microtubule stability; however in AD, tau becomes impaired after it is abnormally phosphorylated. 59 Tau's impairment is believed to occur after the protein is cleaved by caspases-1, -3, -6, and 8; these caspases are activated when apoptosis is triggered by the formation of Aβ peptide. 58 Once caspase-3 becomes active, it cleaves a number of substrates, including beclin-1, which is normally responsible for autophagy in cells as well as actin, fodrin (spectrin), and tau, which are all responsible for maintaining cytoskeletal structure.25,58,60–63 Tau has three structurally exposed cleavage sites for caspase-3, and when cleaved, it can impair axonal transport that leads to neuronal cell death.
In order to test the effectiveness of Q-VD-OPh, TgCRND8 mice that have a double mutation in APP and display AD pathology were treated and analyzed.61,63 The results indicated that APP and tau were cleaved by caspases and that tau accumulated with Aβ in plaque-rich areas; however, caspase-7 and not caspase-3, was observed to be the predominate effector caspase. 63 Q-VD-OPh administration was able to reduce caspase cleavage and activation; however, no changes in the levels of Aβ deposition were observed. 63
Parkinson's disease
Parkinson's disease (PD) is a neurodegenerative disease that is characterized by motor disorders, including resting tremors, slowness of movement and rigidity, as well as depression, apathy, dementia, and psychosis.64,65 These symptoms are caused by the loss of dopaminergic neurons located in the substantia nigra, resulting in the reduction of striatal dopamine and the formation of fibrillar cytoplasmic inclusions or Lewy bodies. 66 Caspase-3 has been identified as one of the primary caspases involved in the cleavage of proteins associated with the symptoms of PD. 66
Male Swiss Webster mice injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a mitochondrial toxin known to produce symptoms of PD, undergo apoptosis in dopaminergic neurons.67,68 Q-VD-OPh administration inhibited caspase-induced apoptosis and inhibited dopamine depletion in mice that received a low level of MPTP (10 and 15 mg/kg); however, this protection did not extend to mice that received a higher dose of MPTP.68,69 This may be because of the induction of necrotic as opposed to apoptotic cell death.68,69 Q-VD-OPh was also able to prevent the apoptotic cell death of immunoreactive neurons expressing tyrosine hydroxylase (TH), a dopamine precursor in the substantia nigra, thus preventing apoptosis and ensuring that dopamine can still be produced.68,69
Huntington's disease
Huntington's disease (HD) is a disorder that affects the central nervous system and symptoms include twitching, behavioral disturbances, and the development of dementia. 70 HD is an autosomal dominant disease that affects medium spiny striatal and cortical neurons and is caused by a mutation in the gene that encodes for the huntingtin (Htt) protein. 71 The inherited mutation causes an abnormal expansion of a trinucleotide CAG repeat that encodes a polyglutamine tract expansion in the N-terminus. This mutation alters protein folding, resulting in the generation of aggregates in neurons that are involved in the neurodegeneration process and are early pathological findings in the brains of HD individuals. 72
3-nitropropionic acid (3NP) is a mitochondrial toxin that can reproduce the symptoms of HD. 68 In male Lewis and Sprague-Dawley rats, treatment with 3NP leads to a significant increase in the activation of caspases 8 and 9 and the cleavage of Bid. 68 Q-VD-OPh administration was able to inhibit caspase activation as well as the truncation of Bid, and dramatically reduced striatal lesions. 68
Marfan's syndrome
Individuals with Marfan's syndrome are unusually tall, have very long arms and legs, and are prone to aortic root aneurysm. 73 Marfan's syndrome is a connective tissue disorder that affects the heart, eyes, and skeleton. Fibrillin-1 is a glycoprotein that plays a critical role in the formation of elastic fibers, which help make up the connective tissue that supports and strengthens organs and other structures throughout the body. Marfan's syndrome is caused by a heterozygous mutation in fibrillin-1 gene (Fbn1), and this mutation results in elevated levels of transforming growth factor beta that further contribute to the overall pathogenesis. 73
To assess the role of apoptosis in the formation of early aortic aneurysm, Fbn1C1039G/+ mice, which serve as a mouse model of Marfan's syndrome, were treated daily with Q-VD-OPh from two to six weeks. 74 Q-VD-OPh administration significantly reduced aortic diameter and aortic wall elastin fragmentation. Although localized early aneurysm growth was inhibited by caspase inhibition, Q-VD-OPh could not prevent the eventual development of aortic aneurysms in Fbn1C1039G/+ mice. 74
Discussion
A number of studies have used Q-VD-OPh as a therapeutic agent to treat animal models of human disease that occur because of the activation of apoptosis.4,35,43,44,49,54,61,67 Q-VD-OPh is a true pancaspase inhibitor that has been shown to bind only with caspases. Since Q-VD-OPh is nontoxic to cells, it provides a significant advantage over other caspases inhibitors that have been shown to interact with cysteine proteases such as cathepsins B, H, and L and cause cellular toxicity.26,28,75
It should be noted that most studies that use Q-VD-OPh only use DMSO as the vehicle control.35,43,44,49,54,61,67 Q-VE-OPh is a recently developed negative control and was altered by replacing the aspartic acid (D) of Q-VD-OPh with glutamic acid (E). 28 This minimal amino acid change results in Q-VE-OPh being unable to inhibit caspases while retaining its amino acid negativity and structure and providing a true control for comparison in studies assessing Q-VD-OPh and apoptotic cell death.27,28,76,77
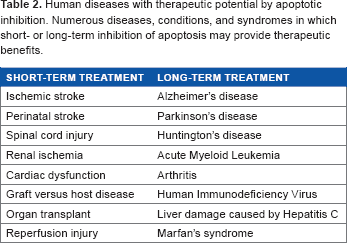
There have been some concerns expressed over the long-term use of caspase inhibitors. In particular, this long-term use may disrupt the homeostatic balance by causing cells that would naturally undergo apoptosis to remain in the body and eventually lead to immunological compromise, autoimmune disease, tumors, or cancer (Table 2).35,43,44,49,54,61,67,78 In animal models, however, daily administration of Q-VD-OPh for up to four months had no reported side effects.63,74 These findings suggest that daily or intermittent administration of Q-VD-OPh may be suitable for long-term treatment; however, further studies are needed.
Human diseases with therapeutic potential by apoptotic inhibition. Numerous diseases, conditions, and syndromes in which short- or long-term inhibition of apoptosis may provide therapeutic benefits.
Conclusion
The pancaspase inhibitor Q-VD-OPh is the preferred option for the inhibition of apoptosis because of its superior structure, effectiveness at lower concentrations, exclusive caspase specificity, and more importantly, its ability to remain nontoxic in cells, even during long-term administration. In summary, Q-VD-OPh is the prototype pancaspase inhibitor that provides a clear examination of the effects of apoptosis in animal models of human disease. Identifying the control and regulation of apoptotic cell death in disease models is essential to developing new therapeutic regimens as well as obtaining a better understanding of disease-causing mechanisms.
Footnotes
Acknowledgments
We would like to thank Melissa R. Kaufman and Renee E. Albers for critical analysis of the manuscript.
Author Contributions
Analyzed the data: CK and TLB. Wrote the first draft of the manuscript: CK. Contributed to the writing of the manuscript: CK and TLB. Agreed with manuscript results and conclusions: CK and TLB. Jointly developed the structure and arguments for the paper: CK and TLB. Made critical revisions and approved the final version: CK and TLB. All authors reviewed and approved the final manuscript.