
Abstract
The purpose of our study is to identify epigenetic markers that are differently expressed in the stomach adenocarcinoma (STAD) condition. Based on data from The Cancer Genome Atlas (TCGA), we were able to detect an age-related difference in methylation patterns and changes in gene and miRNA expression levels in young (n = 14) and old (n = 70) STAD subjects. Our analysis identified 323 upregulated and 653 downregulated genes in old STAD subjects. We also found 76 miRNAs with age-related expression patterns and 113 differentially methylated genes (DMGs), respectively. Our further analysis revealed that significant upregulated genes (n = 35) were assigned to the cell cycle, while the muscle system process (n = 27) and cell adhesion-related genes (n = 57) were downregulated. In addition, by comparing gene and miRNA expression with methylation change, we identified that three upregulated genes (ELF3, IL1β, and MMP13) known to be involved in inflammatory responses and cell growth were significantly hypomethylated in the promoter region. We further detected target candidates for age-related, downregulated miRNAs (hsa-mir-124-3, hsa-mir-204, and hsa-mir-125b-2) in old STAD subjects. This is the first report of the results from a study exploring age-related epigenetic biomarkers of STAD using high-throughput data and provides evidence for a complex clinicopathological condition expressed by the age-related STAD progression.
Introduction
Stomach adenocarcinoma (STAD), the most common type of stomach cancer, is one of the major malignancies worldwide, and generally affects older subjects (50–70 years). Although rarely occurring in the younger subjects, young subjects represent between 2 and 8% of all subjects with stomach cancer.3,4 In spite of recent improvements in the medical screening techniques used in the diagnosis of multiple cancer types, stomach cancer in young people still remains a serious diagnostic challenge. The prognosis in young subjects has shown considerable variability, and clinicopathological features of stomach cancer are reported to differ between young and old subjects.5,6 STAD is histologically classified into two categories based on Lauren's criteria: the intestinal type and the diffuse type.7,8 The intestinal type of STAD arises from chronic atrophic gastritis and is associated with Helicobacter pylori infection. This type is common in Asia, particularly in Japan and Korea, and its incidence rate increases with age.9–11. The diffuse type often occurs in young subjects with a positive familial history and includes aggressive clinical forms, such as linitis plastica, that have poor prognosis. 12
Recent studies have improved our understanding of the molecular mechanisms underlying tumorigenesis, proliferation, and progression in STAD.13–16 Several tyrosine kinase receptors, such as ERBB2, EGFR, FGFR2, and MET, have been shown to be activated in STAD tissues. 17 Expression of c-Myc, which plays a role in the induction of cell proliferation, is upregulated in STAD subjects. 18 Furthermore, cytokine-induced inflammation plays an important role in the development of STAD, and it is well established that H. pylori infection induces gastric mucosal inflammatory responses, resulting in the upregulation of IL1β, which in turn promotes inflammation-associated carcinogenesis.19,20
With the recent advances in the field of biotechnology, a number of high-throughput studies have been conducted to determine the biological basis of STAD tumorigenesis.21–23 However, to our knowledge, there are no reports that describe the genetic comparison of tumor tissues from young and old STAD subjects to identify genetic and epigenetic biomarkers. In this study, we analyzed gene and miRNA expressions, methylation changes derived from RNAseq and miRNAseq, and methylation chip data sets of the STAD samples to discover age-related signatures at transcriptomic and epigenomic levels. Our data were able to document significant age-related molecular signatures in STAD subjects. The significance of our study is that the gene alterations involving the cell cycle, the muscle system process, and cell adhesion were meaningfully distinguishable in young and old STAD subjects; furthermore, these changes had not been previously identified as important molecules in aging and cancer processes.
Materials and Methods
STAD data Collection
All data were retrieved from The Cancer Genome Atlas (TCGA). The TCGA is a comprehensive and coordinated project to characterize the genomic data of about 25 different types of cancers by the National Cancer Institute (NCI), the National Human Genome Research Institute (NHGRI), and more than 24 participating institutions. From the TCGA, we downloaded RNAseq (Illumina HiSeq), miRNAseq (Illumina HiSeq), and methylation chip (Infinium HumanMethylation450 BeadChip) data of 184 samples from age-identified STAD subjects between 34 and 90 years old (Supplementary Table 1). We divided the subject groups as old (≥70) and young (≤49). The samples in each age group used in this study are described in Table 1. The available number of samples of the stage and histological types of cancer were insufficient to allow analysis of genetic alteration in young and old STAD subjects.
The number of STAD samples used in this study.

Identification of Age-Related mRNA and miRNA Expression and DNA Methylation
We used mRNA and miRNA expression data sets (TCGA level 3) processed by the Broad Institute's TCGA workgroup. The RNAseq and miRNAseq level 3 data comprise reads per kilobase per million mapped reads (RPKM), which is the numerical value of the gene expression generally used for RNAseq normalization. 24 Using the EBSeq program of the R package, we computed age-related differentially expressed genes (DEGs) and miRNAs in the old and young groups. 25 Unsupervised two-dimensional hierarchical clustering was applied by using the MeV program. 26 Owing to irregular variation in mRNA expression across cancer samples, hierarchical clustering used median RPKM from binned groups, each bin covering samples in 10–year increments. Median RPKM data were normalized using the Normalize Genes/Rows function of MeV software, and hierarchical clustering was performed using average linkage and Pearson correlation. Significant differential expressions of mRNA and miRNAs were defined by an absolute fold change threshold of 1.5 and a posterior probability of differential expression (PPDE) threshold of 0.95. To identify age-related methylation differences, we used the Illumina Methylation Analyzer (IMA). 27 Differentially methylated regions (DMRs) were identified as sites with the significantly different methylation levels (beta difference) between the young and old groups. The cutoff was defined at a beta difference of 0.14 and a P-value of 0.05.
Gene Set Enrichment Test and Functional Categorization of Age-Related Genes in STAD
To characterize the biological pathways associated with the age-related STAD genes, DEGs were analyzed in the context of several databases such as Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.ad.jp) and BioCarta (http://www.biocarta.com) using the Database for Annotation, Visualization and Integrated Discovery (DAVID). 28 The cutoff was defined at a P-value of 0.05. Additionally, to elucidate the biological processes associated with age-related DEGs, we analyzed gene ontology (GO) terms associated with each age-related DEG using the DAVID database (P < 0.05). To identify the previously reported age-related DEGs, we used AGEMAP, which is a public database categorizing changes in gene expression as a function of age in mice. The AGEMAP database includes expression changes for 8,932 genes in 16 tissues as a function of age. 29
Identification of Age-Related miRNA-Target Interactions in STAD
To identify the target genes of the differentially expressed miRNA, we merged two public miRNA databases, miRTarBase 30 and miRecords. 31 miRTarBase contains the largest amount of validated miRNA-target interactions, while miRecords includes predicted miRNA targets produced by 11 established miRNA-target prediction programs and validated miRNA-target interactions. Using the merged miRNA-target database, we identified age-related miRNA-target interactions.
Results
Detection of Altered Gene Expression in Young and Old STAD Subjects
We computed the age-related, genome-wide mRNA expression profiles for old (n = 70) and young subjects (n = 14) using EBSeq and identified 323 upregulated and 653 downregulated genes whose expression levels were altered by 1.5–fold or more (Supplementary Table 2). In order to see the overall age-related gene expression patterns in each STAD sample, we conducted unsupervised two-dimensional hierarchical clustering on mRNA expression data from the whole cohort (n = 184), using 976 mRNAs identified as differentially expressed between the young and old subjects. The hierarchical clustering showed clear patterns of age-related gene alteration in young (≤49), intermediate (50–69), and old (≥70) groups (Supplementary Fig. 1). Both cancer type and cancer stage are known to effect mRNA levels; therefore, we examined the distribution of mRNA expression differences for the 976 DEGs identified according to the histological type of STAD (eg, diffuse, intestinal), the stage of STAD (eg, I, II, III), and the age group (eg, young and old). Figure 1 shows that the age-related DEGs have low impact in cancer progression and histological type.
Genes in pathways significantly changed by the aging-related stomach cancer.


Expression value distributions of DEGs in each cancer stage: (
Gene Set Enrichment Analysis
To classify the DEGs using the DAVID database, we analyzed the predefined biological pathways of genes that showed significant differences in expression levels between young and old STAD subjects in the gene set enrichment analysis. 32 Representative terms for biological pathways were used as defined in the KEGG (http://www.genome.ad.jp) and BioCarta (http://www.biocarta.com) databases. In KEGG and BioCarta terminologies, the genes of five pathways were found to be upregulated, while the genes of 14 pathways were found to be downregulated (filtered at P < 0.05; Table 2). In the STAD samples belonging to the old subject group, several pathways, including the cell cycle, free radical-induced apoptosis, and mitotic spindle regulation pathways, were found to be upregulated. Downregulated pathways included vascular smooth muscle contraction, calcium signaling, neuroactive ligand–receptor interaction, focal adhesion, and cell adhesion pathways. GO analysis revealed that the upregulated genes were involved in cell cycle-related processes, while the downregulated genes were involved in the cell adhesion and the muscle system processes (Supplementary Table 3). We selected 178 genes, which were assigned to the cell cycle, the muscle system, and cell adhesion, and conducted unsupervised two-dimensional hierarchical clustering using samples from the whole cohort (n = 184, aged 34–90 years). Figure 2 shows clear differences in age-related gene alteration.

Hierarchical clustering analysis of 178 genes assigned by the cell cycle, the muscle system, and cell adhesion in GO terminologies. Rows represent genes, and columns represent age groups, which grouped age at every 10 years. Red and green blocks, respectively, represent high and low median RPKM values relative to each age group, while black blocks indicate midpoint values. Magenta blocks represent genes assigned to the cell cycle in GO terms; blue blocks represent cell adhesion; and cyan blocks represent the muscle system in GO terms.
Identification of Age-Related miRNA Expression and miRNA-Target Interactions
To identify age-related miRNA expression, we analyzed the miRNA expression profiles of samples from old (n = 75) and young subjects (n = 14) using EBSeq. We identified 8 upregulated and 22 downregulated miRNAs whose expression levels were altered by 1.5-fold or more (PPDE ≥0.95) in the old group of STAD subjects (Supplementary Table 4). Next we used validated miRNA-target databases (miRTarBase and miRecords) to identify potential miRNA-target interactions. We found 105 potential age-related miRNA-target interactions in STAD subjects. Among them, we identified several miRNA targets in DEGs (39 upregulated and 4 downregulated genes), whose expression levels were inversely correlated with the levels of their target miRNA (2 upregulated and 6 downregulated miRNAs; Fig. 3).
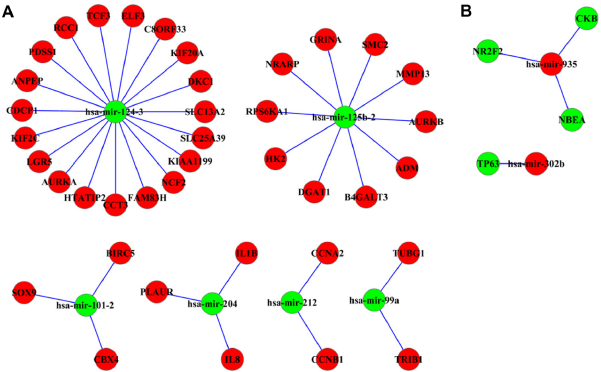
Age-related miRNA-target interactions: (
Identification of Age-Related Changes in Methylation Patterns
We compared the methylation levels (beta value) of STAD in young and old subjects and identified 3,630 age-related DMRs using the IMA. Among them, 2,800 DMRs were hypomethylated and 830 DMRs were hypermethylated in the old group of STAD subjects. We separated the DMRs by their genetic regions (3'-UTR, 5'-UTR, exon1, gene body, TSS1500, and TSS200) and identified 2,652 differentially methylated genes (DMGs; 2,124 hypomethylated and 528 hypermethylated; Supplementary Table 5). A comparison of the DMGs and DEGs revealed that 107 DEGs showed an inverse correlation with DNA methylation in their genetic region in the old group. Upregulated genes (n = 54) were hypomethylated, while downregulated genes (n = 53) were hypermethylated (Supplementary Table 6). We also identified 146 age-related DMRs in genome-wide miRNA regions (Supplementary Table 7). Three upregulated (hsa-miR-105-1, hsa-miR-512-1, and hsa-miR-512-2) and two downregulated miRNAs (hsa-miR-124-2 and hsa-miR-124-3) showed an inverse correlation with DNA methylation in the old group of STAD subjects.
Discussion
In order to explore the differences in the molecular basis of STAD progression between young and old STAD subjects, our analysis investigated high-throughput genetic and epigenetic data. We identified age-related expression changes in genes and miRNAs and DMGs and revealed that significant gene alterations were involved in the cell cycle, the muscle system process, and cell adhesion in STAD subjects. Furthermore, by comparing DEGs, miRNA expressions, and DMGs, we detected genetic and epigenetic correlations in age-related gene alterations from STAD subjects.
To identify previously reported age-related DEGs, we examined the overlap between the DEGs we found in STAD subjects and those from previous research. When comparing the AGEMAP database, which is an age-related gene database of mice, we detected that out of the 976 DEGs in old STAD subjects, 765 genes (79%, P < 2.6 x 10-14) were identified as novel genes associated with STAD aging. In addition, we compared our data with previous STAD studies that reported cancer-related DEGs in disease-free tissue and cancer tissue.33–35 Our analysis showed a variability of gene expression among public studies, including age-related DEGs (Supplementary Fig. 2). To find the gene alteration mediated by a histological type of cancer, we investigated a previous report that compared diffuse-type with intestinal-type STAD.36–38 Among 976 DEGs, we found that 942 age-related DEGs (P < 3.0 x 10-4) do not share histology-related DEGs (n = 1502) with STAD subjects. This finding indicates that the gene alterations we identified are specifically affected in old STAD subjects, and thus may be good target molecules in identifying the correlation between young and old STAD subjects.
Our analysis detected that a significant number of genes (n = 35), which were assigned to the cell cycle (P < 8.1 x 10-8) of GO, were upregulated in old STAD subjects. In contrast, a number of genes involved in the muscle system process (n = 27, P < 4.0 x 10-11) and cell adhesion (n = 57, P < 4.8 x 10-10) were downregulated. Interestingly, in pathway analysis, a number of genes involved in vascular smooth muscle contraction (P < 1.3 x 10-8) and heart muscle disease (dilated cardiomyopathy: P < 1.8 x 10-7, arrhythmogenic right ventricular cardiomyopathy: P < 4.7 x 10-6, and hypertrophic cardiomyopathy (HCM): P < 1.5 x 10-5) were downregulated in old STAD subjects as assigned by the KEGG database. We mentioned that muscle-related gene alteration may be a reason for the aged stomach tissue or histological characteristics of cancer. In physiological aging process, the progressive loss of an organ's ability to function is a general feature of the aging process because of an inefficient cellular repair system. Furthermore, it is well known that the precise control of somatic stem cell proliferation ensures maintenance of tissue homeostasis in damaged tissue intestinal stem cells. 39 In terms of tissue homeostasis, upregulation of genes involved in cell proliferation can also be described as being the cause of reduced gastric motility function. Alternatively, the observed downregulation in muscle-related genes may be related to the greater proportion of diffuse-type STAD in the young group, rather than age-related differences per se. Owing to insufficient histologically diverse STAD samples in some age ranges, we could not analyze RNAseq data based on histological STAD type. In addition, we detected the upregulation of inflammation-related genes (n = 16, P < 6.2 x 10-3) in old STAD subjects. Furthermore, XDH and NOX1 genes, which are involved in the production of reactive oxygen species (ROS), 40 and IL8, which can be activated by ROS, 41 were also found to be upregulated in old STAD subjects. It is well known that chronic inflammation plays a key role in oxidative stress-induced aging. Inflammatory processes, activated by ROS, result in a chronic inflammatory condition during aging, as predicted by molecular inflammation.42–44
A recent study suggests that DNA hypomethylation of genes increases with age. 45 Consistent with this observation, our studies also revealed a large number of age-related changes in DNA hypomethylation patterns. Figure 4 shows DNA methylation changes between young and old STAD subjects. We calculated the distribution of absolute beta value difference and found that old STAD subjects had a lower DNA methylation content in gene regions (Fig. 4A) and miRNA regions (Fig. 4B) compared to young STAD subjects. Approximately 80% of the DMGs in tissues from the old STAD subjects were hypomethylated. Interestingly, a comparison between DMGs and DEGs revealed that 54 upregulated genes (17%) were hypomethylated (P < 4.6 x 10-9, Fisher's exact t-test), whereas 53 downregulated genes (8%) were hypermethylated (P < 2.2 x 10-16, Fisher's exact t-test) in old STAD subjects. Notably, AZU1, ELF3, NOX1, IL1B, and S100A12, which are associated with inflammatory responses, were found to be upregulated and hypomethylated in old STAD subjects.

DNA methylation changes between young and old STAD subjects: (
In our miRNA analysis, we identified several significantly changed miRNA in old STAD subjects. hsa-mir-124–1 (84–fold), hsa-mir-124–2 (80–fold), and hsa-mir-124–3 (76–fold) were significantly downregulated in old STAD subjects. mir-124 is already a well-known tumor-suppressive miRNA, and downregulation of mir-124 was previously reported in various cancer types, such as colon, lung, and central nervous system.46–48 Furthermore, the loss of mir-124 in Caenorhabditis elegans is known to accelerate the aging process, resulting in an increase in ROS. 49 On the other hand, mir-302a (140–fold) and mir-105c (106–fold) were greatly upregulated in old STAD subjects. It is known that mir-105 has a different action at different phases of the cell cycle, such as affecting the increase of PCNA, which suppresses cyclin D1 during the cell cycle process in the S-phase and decreases cyclin B1 in the G2-phase, 50 and this miRNA can inhibit prostate cancer growth by suppressing CDK6 in the G1-phase. 51 Furthermore, miR-302a is known to promote an increase in the S-phase activity and a decrease in the G1-phase activity by targeting cyclin D1 in the human embryonic stem cell. 52 The majority of the upregulated cell cycle genes in the present study were related to the M-phase rather than other phases in the cell cycle process, and may be regulated by miRNA expression.
Importantly, through integrated analysis of DEGs, DMRs, miRNA expression, and miRNA-target interactions, we identified significant age-related genes, such as ELF3, IL1β, and MMP13, which were upregulated (Supplementary Fig. 3) and hypomethylated in the promoter region but inversely correlated with miRNA expression in the old STAD subjects (hsa-mir-124-3, hsa-mir-204, and hsa-mir-125b-2). Notably, these data also show that hsa-mir-124-3, the putative interacting partner of ELF3, was downregulated and hypermethylated within the TSS1500 regions in old STAD subjects. ELF3 is an epithelial specific transcription factor and plays an important role in epithelial cell differentiation and tumorigenesis. 53 ELF3 can directly bind to the MMP13 promoter; its expression is enhanced by IL1β stimulation in chondrocytes under proinflammatory stress. 54 These results show that these genetic and epigenetic alterations may be specifically associated with old STAD subjects and can be affected in STAD processing.
Conclusion
In summary, our study provides the first comprehensive genetic and epigenetic analysis of STAD through the examination of the DEGs, DMGs, and miRNAs that are known to characterize the underlying age-related biological differences in STAD tumorigenesis. Although the present study was unable to secure sufficient data from young and old STAD subjects to perform a comparative analysis of tumor tissues, it provides valuable data at molecular levels on the cell cycle and muscle system-related gene alterations that can be important distinguishing factors in age-related STAD. Furthermore, our results indicate that three genes (ELF3, IL1β, and MMP13) are correlated in the STAD aging process, and therefore may play a significant role in age-related STAD. Our studies provide insights into the mechanisms of age-related STAD progression and may help identify potential biomarkers in STAD subjects.
Author Contributions
Conceived and designed the experiments: BCK, HOJ, HYC. Analyzed the data: BCK, HOJ, DR Wrote the first draft of the manuscript: BCK. Contributed to the writing of the manuscript: HOJ, CHK, EKL, DHK, EI, NDK. Agreed with manuscript results and conclusions: SL, BPY, JB, HYC. All authors were involved and jointly developed the structure and arguments for the paper. All authors reviewed and approved of the final manuscript.
supplementary Materials
Supplementary Figure 1
Hierarchical clustering analysis of age-related DEGs in STAD subjects.
Supplementary Figure 2
Venn diagram of DEGs comparing previous study.
Supplementary Figure 3
A scatter plot of upregulated ELF3, IL1B, and MMP13 genes.
Supplementary Table 1
The list of 184 samples from age-identified STAD subjects.
Supplementary Table 2
The significantly changed genes during the aging process in STAD subjects.
Supplementary Table 3
The result of GO enrichment test.
Supplementary Table 4
Differentially expressed miRNAs during the aging process in STAD subjects.
Supplementary Table 5
DMGs during the aging process in STAD subjects.
Supplementary Table 6
The comparison result of the DMGs and DEGs.
Supplementary Table 7
DMGs during the aging process in STAD subjects.
Footnotes
Acknowledgments
We thank Robin Alasdair McGregor for editing and the Aging Tissue Bank for providing research information. All data used in this manuscript were collected by TCGA.