
Abstract
Gastric cancer (GC) is one of the most frequent tumors in the world. Stomach adenocarcinoma is a heterogeneous tumor, turning the prognosis prediction and patients’ clinical management difficult. Some diagnosis tests for GC are been development using knowledge based in polymorphisms, somatic copy number alteration (SCNA) and aberrant histone methylation. This last event, a posttranslational modification that occurs at the chromatin level, is an important epigenetic alteration seen in several tumors including stomach adenocarcinoma. Histone methyltransferases (HMT) are the proteins responsible for the methylation in specific amino acids residues of histones tails. Here, were presented several HMTs that could be relating to GC process. We use public data from 440 patients with stomach adenocarcinoma. We evaluated the alterations as SCNAs, mutations, and genes expression level of HMTs in these aforementioned samples. As results, it was identified the 10 HMTs most altered (up to 30%) in stomach adenocarcinoma samples, which are the PRDM14, PRDM9, SUV39H2, NSD2, SMYD5, SETDB1, PRDM12, SUV39H1, NSD3, and EHMT2 genes. The PRDM9 gene is among most mutated and amplified HMTs within the data set studied. PRDM14 is downregulated in 79% of the samples and the SUV39H2 gene is down expressed in patients with recurred/progressed disease. Several HMTs are altered in many cancers. It is important to generate a genetic atlas of alterations of cancer-related genes to improve the understanding of tumorigenesis events and to propose novel tools of diagnosis and prognosis for the cancer control.
Keywords
Introduction
Gastric cancer (GC) is the third leading cause of cancer deaths in the world, according to GLOBOCAN 2018 data. 1 Stomach cancer was responsible for over 1 million of new tumor cases in 2020 and around 769 000 deaths. 2 The high mortality rates are caused, among several factors, by the late diagnosis. Generally, surgical procedures for patient hilling, in advanced stages, are not recommended. The risk of developing gastric adenocarcinoma increases with age, occurring more frequently in ages from 55 to 80 years. 3 Gastric cancer is influenced by several risk factors such as diet, active tobacco smoking, familiar history, and Helicobacter pylori infections.4-6 This cancer is characterized by a wide heterogeneity at the histopathological and molecular levels, turning the prognosis prediction and patients’ clinical management difficult.7,8
However, several studies have shown that genetic alterations as well as dysregulation of specific epigenetic mechanisms play an important role in initiation, progression, and metastasis of different cancers including GC. 9 These alterations include somatic point mutations, somatic copy number alteration (SCNA) and modifications in genes related to histone-modifying complexes. 10 For example, SCNAs, events that involve gains or losses of a large region of tumor DNA, could lead to activation of oncogenes or inactivation of tumor suppressors.11,12 Recent studies have identified common SCNAs in 38 GC samples. In addition, these samples have showed differences in SCNA content between GC samples from young and old patients. 13
Concerning aberrant epigenetics, more than 300 genes have been recently associated with epigenetic alterations in human cancers.14,15 Methylation panels of specific genes can provide a new classification of GC able to predict the prognosis of patients and the risk of metastasis.16,17 In addition, aberrant histone methylation is an important epigenetic alteration seen in tumors, which can occur through misbalance in the histone methyltransferases (HMTs) function.18,19 These enzymes are a group of proteins responsible for the methylation of histones; it includes histone lysine methyltransferases (HKMTs) and histone/protein arginine methyltransferases (PRMTs).20,21 Currently, approximately 51 HMTs have been identified in humans.22,23 Several studies have concentrated their efforts to discover the relationship between histone methylation patterns and tumorigenesis processes. For example, high H3K9me3 levels have been associated with tumor staging, GC recurrence and worse prognosis of a cohort of 261 GC patients. 24 Often, HMTs show alterations such as SCNAs, somatic mutations and changes in their mRNA expression. Often, some HMTs such as EMHT2, NSD3, SMYD3, and SETDB1 are amplified and show different patterns of gene expression in several types of cancer such as in lungs, breast, and colorectal. Changes in their genetic signature could be associated to metastasis and decrease in overall survival rate.25-28
Although advances have been made for the understanding of epigenetic regulation of cancers, the role of HMTs in some cancers, such as GC, remains unclear. Therefore, the objective of this study was to evaluate the most common alterations present in HMT genes such as SCNAs presence, somatic mutations, and gene expression variation in stomach adenocarcinoma samples using bioinformatics analysis. The analysis included the public data from CBioPortal and The Cancer Genome Atlas (TCGA) databases, which allowed providing a genetic atlas of alterations present in HMTs from stomach adenocarcinoma samples to improve the understanding of some GC processes.
Material and Methods
Public data set and patient data collection
The clinical and biological data from patients with stomach adenocarcinoma were obtained through the public repository CBioPortal (www.cbioprtal.org). The data set used was “Stomach Cancer Adenocarcinoma (TCGA, PanCancer Atlas)” with 440 samples. The queries genes used in this study were ASH1L, EHMT1, EHMT2, EZH1, EZH2, KMT2A, KMT2B, KMT2C, KMT2D, KMT2E, KMT5A, KMT5B, KMT5C, MECOM, NSD1, NSD2, NSD3, PRDM1, PRDM10, PRDM11, PRDM12, PRDM13, PRDM14, PRDM15, PRDM16, PRDM2, PRDM4, PRDM5, PRDM6, PRDM7, PRDM8, PRDM9, SETD1A, SETD1B, SETD2, SETD3, SETD4, SETD5, SETD6, SETD7, SETDB1, SETDB2, SETMAR, SMYD1, SMYD2, SMYD3, SMYD4, SMYD5, SUV39H1, and SUV39H2.
Analysis of genetic alterations present in the HMTs
Genetic alteration rates found up to 30% were the values considered to select the HMTs for further analysis. The somatic mutation, SCNA, and differences in the mRNA expression were the genetic alterations evaluated in this work. The mutations were predicted by COSMIC database Catalog of Somatic Mutation in Cancer (https://cancer.sanger.ac.uk/cosmic). The GISTIC 2.0 program identified SCNAs. The program found 5 categories of SCNAs such as deep deletion, deletion, diploid (unaltered samples), gain, and amplification. The mRNA expression data of the HMTs were obtained using the RNASeq V2 Illumina files available in this repository. We accessed the data from “mRNA expression Z-score relative to normal samples (log RNA Seq V2 RSEM)” files. The program PROVEAN (http://provean.jcvi.org/) was used to predict the effect of somatic mutation on the function of selected HMTs gene products. PROVEAN works with similarity search among protein sequences (75% global sequence identity). The program generates 30 clusters of closely related sequences, which will be used to generate the prediction. We used the program at its default parameters. Variants with a score equal to or below −2.5 are considered “deleterious.” Variants with a score above −2.5 are considered “neutral.”
Correlation between mRNA expression and SCNA-type alterations for each HMT
The correlation between the mRNA expression values of each HMTs and their SCNA-type alterations was evaluated sample by sample. In this analysis, the Z-score data of mRNA expression normalized with normal samples and the data of SCNA alterations (log2 copy number values) were used for each HMT. Spearman’s and Pearson’s statistics were used for correlation. For variance analysis, nonparametrical tests such as Kruskal-Wallis and Mann-Whitney were used (P value ⩽ .05).
Stratification of patient data using mRNA expression of HMTs
We evaluated the variance between mRNA expression values for each HMTs relative to several clinical attributes of the patients such as disease-free status (living and deceased status) and ethnicity (Latin or Hispanic and not Latin or Hispanic). Other data used were in according with American Joint Committee on Cancer Code such as metastasis staging (M0, M1, MX), tumor staging (T1A-B, T2A-B, T3, T4A-B, and TX), neoplasm staging (IA-B, IIA-B, IIIA-C, and IV), and lymph node staging (N0, N1, N2, N3A-B, and NX). For overall survival prediction, ethnicity, metastasis staging, and tumor staging 412 samples were used. For neoplasm staging 394 samples were used, and for lymph node staging, 411 samples were used. For all analysis, nonparametrical tests such as Kruskal-Wallis and Mann-Whitney were used (P value ⩽ .05).
Results
On the stomach adenocarcinoma database used, with 440 samples, most patients are not Hispanic or Latin (72.3%); they are predominantly men (64.5%) in contrast to women (35.5%) and the average age found was 67.2 years old in an age range of 40 to 90 years old. These data are summarized in Table 1.
Summary of clinical data from patients with stomach adenocarcinoma (data from CBioPortal www.cbioportal.org, accessed in July 09, 2021).

Abbreviation: NA, not available.
Most altered HMT in stomach adenocarcinoma samples from public repositories
The HMTs that presented the rate of genetic alterations over to 30% were analyzed in this study. In total, we found 10 HMTs most altered in this database. The most altered HMTs found for stomach adenocarcinoma were PRDM14, PRDM9, SUV39H2, NSD2, SMYD5, SETDB1, PRDM12, SUV39H1, NSD3, and EHMT2. These genes are summarized in Table 2. These HMTs are altered in 421 (96%) of 440 stomach adenocarcinoma samples. The PRDM14 and PRDM9 genes are the most altered within the data set presenting rates of 85% and 71%, respectively. In addition, the genes PRDM9, NSD2, EHMT2, and NSD3 are the most mutated with rates of 5.7%, 5.5%, 4.5%, and 4.1%, respectively. In addition, their probable functions in a cellular context, their role in a methylation of lysine residues in the tail of the histones and their chromosomal location are summarized in Table 2.
Histone methyltransferases genes most altered in stomach adenocarcinoma from CBioPortal database (www.cbioportal.org, accessed on July 09, 2021).

Abbreviations: H3 and H4, histones H3 and H4; K4, K9, K20, K21, K27, and K36, lysine residues.
The type of genetic alteration (somatic mutation, SCNA and mRNA expression) and its quantity for each HMT are summarized in the Table 3 and the presentation of their content are divided into the following sections.
Genetic alterations of the HMTs in stomach adenocarcinoma from CBioPortal database (www.cbioportal.org, accessed on July 09, 2021).
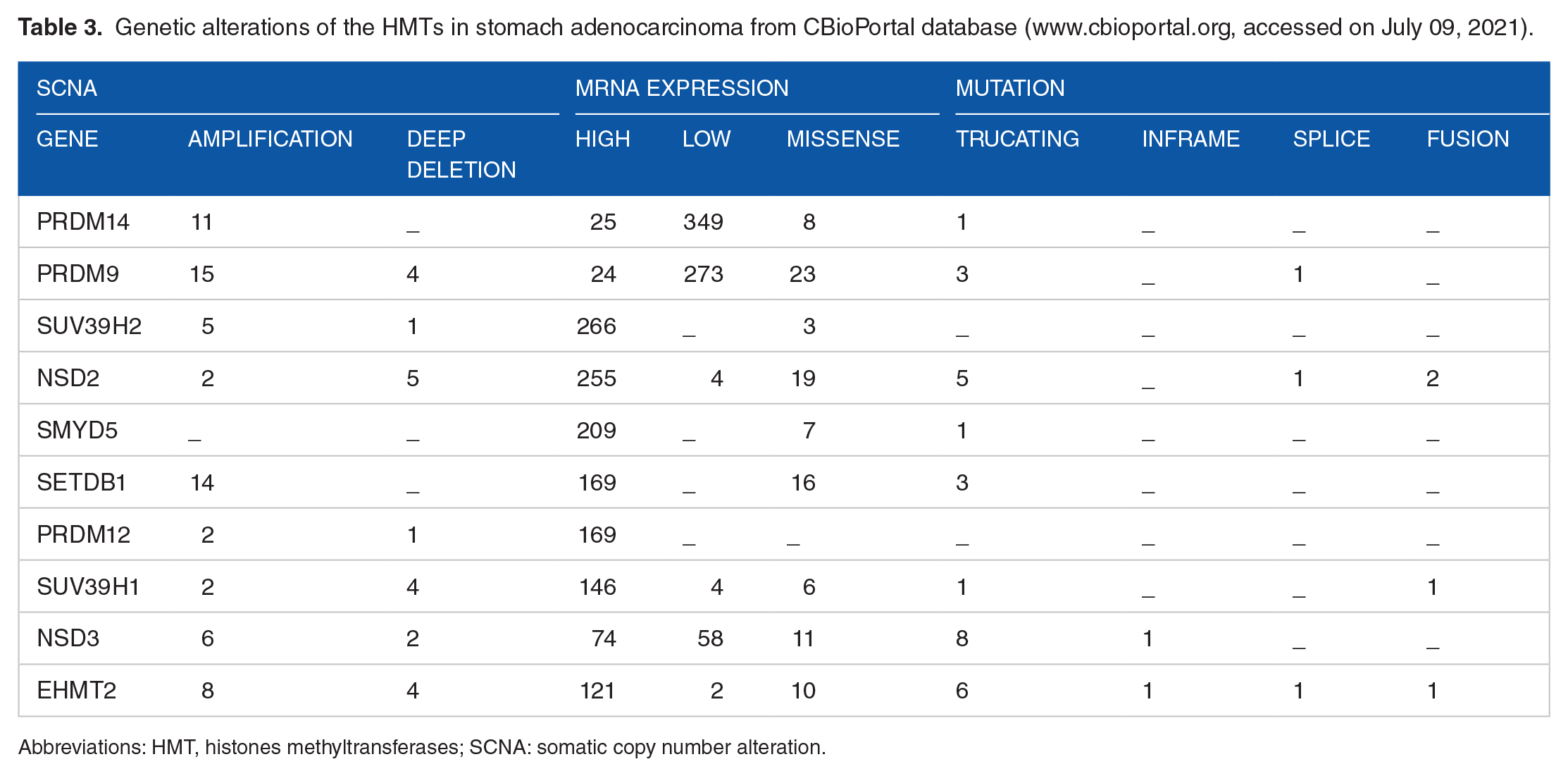
Abbreviations: HMT, histones methyltransferases; SCNA: somatic copy number alteration.
Somatic mutations of HMT in stomach adenocarcinoma
In total, for the 9 HMTs, were described 139 mutations. Except the PRDM12 gene, which has no somatic mutation information available, the other 9 HMTs presented information about somatic mutations. The mutation most common in all HMTs studied was missense type (Table 3) followed by truncating mutations (Table 3). PRDM9 (23), NSD2 (19), SETDB1 (16), NSD3 (11), and EHMT2 (10) presented the major amount of somatic mutation missense type. Almost all HMTs in analysis (except SUV39H2) also presented truncating mutation type, which NSD3 (8), EHMT2 (6), and NSD2 (5) presented the major occurrence of this mutation type. NSD2 (2), SUV39H1 (1), and EHMT2 (1) presented fusion mutations and NSD3 and EHMT2 showed one inframe mutations for each gene. Finally, the genes PRDM9, NSD2, and EHMT2 presented somatic mutation associated to splice process.
In this database, no mutations showed biological or clinical annotation, that is, the probable biological effects of these mutations are not presented (Tables 3 and 4). PRDM14 showed 9 mutations and 2 missense mutations (S291G and S297R) inside its SET domain. PRDM9 presented 27 mutations and 1 missense-type mutation inside the SET domain (G188A). In addition, the missense mutation R337S/T appears in 2 samples. SUV39H2 had 3 missense mutations, and the H331Y mutation appears on its SET domain. NSD2 showed 27 mutations, 1 missense mutation on the SET domain (P1132T), and 2 recurrent mutations (R729C). SMYD5 had 8 mutations, which 4 missense mutations are inside its SET domain (M117I, E203D, and 2 R52W). SETDB1 presented 19 mutations, which 10 are in the SET domain, 2 frameshift deletion, 1 nonsense and 1 missense (G816C, G876Vfs*8, P950Qfs*16, A982T, R1018*, K1162N, V1232F, R1243H, D1241G, and R1256W). SUV39H1 harbors 8 mutations, which one frameshift deletion and 3 missense mutations are inside the SET domain (R144H, A198Rfs*140, G216R, and N223S). NSD3 gene presented 20 mutations, which the majority is, missense type. One missense mutation is located inside its SET domain (G1176S) and the mutation T419Nfs*28/Pfs*8 is recurrent, present in 4 samples. Finally, the EHMT2 gene showed 19 mutations, in which 2 missense mutations (R1135H and R1145Q) are inside the SET domain, 1 mutation inside its pre-SET domain, and 1 frameshift deletion mutation L855Cfs*36 occurred 4 times in the samples. All data are summarized in Tables 3 and 4.
Summary of all mutations type present in the studied HMTs.

Abbreviations: HMT, histones methyltransferases.
Although all somatic mutations seen in the HMTs have no biological effect available, a computational simulation has shown how deleterious could be a specific mutation in these set of proteins. Thirty-five mutations were predicted to be deleterious. SETDB1 has the major highest number of deleterious mutations, and 9 deleterious missense mutations. For example, the mutation S291G (PRDM4) was considered deleterious, and it was within the SET domain of the protein. The same result can be seen for H331Y (SUV39H2), P1132T (NSD2), R52W (SMYD5), G816C, V1232F, D1241G, R1243H, R1256W (SETBD1), D291Y (SUV39H1), G1176S (NSD3), and R1135H (EHMT2). Other likely deleterious mutations are also summarized in the Table 5. A total of 104 mutations seen here were predicted to be neutral, probably with no biological effect on the HMT function.
Prediction of biological effect through somatic mutations in genes HMT-coding of stomach adenocarcinoma samples.

Abbreviations: HMT, histones methyltransferases.
Most of the mutations that are present in stomach adenocarcinoma samples were in advanced stage of cancer. For example, the mutations of PRDM9 are in that show tumor staging between T2* and T4*. The same pattern is seen for NSD2 and SETDB1. SMYD5 mutations are associated to advanced tumor stages such as T3, T4A, T4B, and TX. With regard to cancer staging, this HMT is also associated with advanced stages like IIIB, IIIC, and IV.
SCNA and their linking with mRNA expression
Somatic copy number alterations (high amplification, gain, shallow deletion, and deep deletion) were found for PRDM14, PRDM9, SUV39H2, NSD2, SMYD5, SETDB1, PRDM12, SUV39H1, NSD3, and EHMT2 in stomach adenocarcinoma samples. In this analysis, 372 samples presented data of SCNA and mRNA expression. Almost all HMTs are amplified in the studied data set of stomach adenocarcinoma where the PRDM9 (15), SETDB1 (14), PRDM14 (11), and EHMT2 (8) are those that have presented the highest number of amplifications. However, SMYD5 has no amplifification profile. In addition, several HMTs harbor deep deletion such as, PRDM9 (4), SUV39H2 (1), NSD2 (5), PRDM12 (1), SUV39H1 (4), NSD3 (2), and EHMT2 (4). SMYD5, SETDB1, and PRDM12 have no deep deletion alteration in the samples. Unaltered samples were called diploid. These data are presented in Table 3.
The correlation between mRNA expression and the SCNAs can be seen in Figure 1. When the HMTs genes undergo an alteration of deletion or deep deletion, a decrease in gene expression can be observed. On the contrary, when these same genes showed partial genetic gain or they have high amplification alterations, their mRNA expressions tend to increase (Figure 1A to D). All HMTs presented this behavior between mRNA expression and SCNA; however, this tendency to increase or to decrease the mRNA expression concerning the presence of amplification/deletion were more evident in EHMT2, NSD3, SETDB1, SU39H1, and SUV39H2 genes as show the Figure 1A to E.

Analyses between SCNA and mRNA expression levels of histone methyltransferases (A) EHMT2, (B) NSD3, (C) SETDB1, (D) SUV39H1, and (E) SUV39H2. Graphs of relation between SCNA (amplification, gain, diploid, shallow deletion, and deep deletion, axis X) and mRNA expression of specific HMTs (axis Y). Data obtained from CBioPortal (www.cbioportal.org, accessed in April 16, 2021). Data set: mRNA expression Z-score relative to normal samples of EHMT2, NSD3, SETDB1, SUV39H1, and SUV39H2 and SCNA, P value (<.05).
Finally, the NSD3 genes showed a strong correlation rate Spearman: 0.69 (P = 1.53e−59) and Pearson: 0.77 (P = 2.88e−83) when we correlated the mRNA expression to SCNA type. As results, probably the SCNA change can interfere in mRNA expression levels in this HMT (Figure 2).

Analysis of correlation between SCNA and mRNA expression level of histone methyltransferases NSD3. Graph of correlation between SCNA (amplification, gain, diploid, shallow deletion, and deep deletion, axis X) and mRNA expression of NSD3 (axis Y). Data obtained from CBioPortal (www.cbioportal.org, accessed in April 16, 2021). Data set: mRNA expression Z-score relative to normal samples of NSD3, P value (<.05).
Interestingly, there is a highest tendency for HMT gene amplification in men contrasting women. For example, for PRDM14, almost all samples that have gene amplification occurred in men (81.8%), for PRDM9 the rate was 60%, 78% for SETDB1, and 75% for EHMT2. When the profile of tumor stage and gene amplification was evaluated, it was possible predict a tendency of PRDM14 and PRDM9 to present gene amplification in samples with T3 tumor stage, at rates of 54.5% and 73.3%, respectively. This pattern was not observed for SETDB1 and EHMT2, whose tumor staging was heterogeneous.
Stratification of clinical data from the patients
To analyze the probability of HMTs are involved with specific carcinogenesis processes and clinical features in the patients with stomach adenocarcinoma, we accessed their clinical data from public repository. As previous mentioned, we accessed data such as disease-free status (living and deceased status) and ethnicity (Latin or Hispanic and Not Latin or Hispanic). Other data, in according with American Joint Committee on Cancer Code, also were used such as metastasis data (M0, M1, and MX); tumor staging (T1A-B, T2A-B, T3, T4A-B, and TX); neoplasm staging (IA-B, IIA-B, IIIA-C, and IV) and lymph node staging (N0, N1, N2, N3A-B, and NX). The analysis take into account the mRNA expression of each HMTs studied here and the specific clinical feature. Only the clinical data about disease-free status related to mRNA expression of SUV39H2 was statistical significant (P value: .0286), which the patients, who have presented the status disease free, showed the mRNA expression higher than the patients with status recurred/progressed (Figure 3).

Analysis of relation between mRNA expression level of histone methyltransferases SUV39H2 and disease-free status of patients: graph of relation between mRNA expression level of histone methyltransferases SUV39H2 (axis Y) and disease-free status of patients (axis X). Data set: mRNA expression Z-score relative to normal samples of SUV39H2, P value (<.05).
Regarding the level of mRNA expression, the HMTs PRDM14 and PRDM9 are downregulated in 349 and 273 stomach adenocarcinoma samples, respectively, resulting in rates of 79.3% and 62%. In addition, SUV39H2 (60.4%), NSD2 (57.9%), SMYD5 (47.5%), SETDB1 (38.4%), PRDM12 (38.4%), and SUV39H1 (33.1%) are upregulated in stomach adenocarcinoma (Table 3). Regardless of whether the HMT was upregulated or downregulated, these 2 changes are predominant in men, rates more than 55% for each HMT studied. Furthermore, differential expression whether high or low seem to be a common feature in T2 to T4* tumor staging, in which T3 staging is overrepresented.
Discussion
Currently, novel anticancer drugs are emerging with a focus on HMTs. DNA alterations are not the only factor that lead to carcinogenesis processes but also epigenetics aberrations play a role in tumor development through the regulation of mRNA expression. Furthermore, the emergence of drug resistance is another challenge in cancer treatment, which is closely related to epigenetics chances. 29 Therefore, the HMTs have become an important target group for the development of new cancer control methodology. 30 Here, we present several changes of the genetic signature of 10 HMTs as a genetic atlas to provide new information about HMTs and GC to aim of assisting in the developing of new strategies to control this cancer type.
Histones methyltransferases are often altered in much type of cancers. The HMTs studied harbor about 31% of somatic mutation in the 440 stomach adenocarcinoma samples evaluated, with PRDM9, NSD2, NSD3, SETDB1, and EHMT2 being the most mutated HMTs. According to CBioPortal repository, not all mutations have any biological or clinical annotation. However, even without the biological significance of the 139 mutations seen in these HMTs, for example, it is known that NSD2, is mutated in blood cancers, SETDB1 is altered by amplification, mutation, and fusion in melanoma, lung cancer, and mesothelioma and NSD3, is altered by amplification in several cancers such as breast and colorectal. Furthermore, all evaluated HMTs had mutation in their SET domain, and the EHMT2 gene harbored mutations in its pre-SET domain. Both domains play an important role in the recognition of specific residues in histone proteins and many amino acids residues within these domains need to remain invariant not to affect the function of these enzymes. 31 It may be mutations at these sites could lead to changes in the biological function of HMTs, mainly recurrent mutations such as R337S/T in PRDM9, T419Nfs*28/Pfs*8 mutation in NSD3 and L855Cfs*36 for EHMT2 gene. However, computational simulations found 35 probably deleterious somatic mutations in this stomach adenocarcinoma data set. These mutations were found in 9 HMTs (the PRDM12 has no somatic mutation in this data set). Eleven of them were located within the SET domain and were linked to SUV39H2, NSD2, SMYD5, SETBD1, SUV39H1, NSD3, and EHMT2 HMTs. These mutations are likely to be novel and therefore have no functional annotations. This simulation delivers new information that could lead to future studies to confirm the deleterious role of this type of HMT mutation in GC.
Regarding the SCNA signatures observed for HMTs, several common features are seen in cancers and the amplification appears to be a frequent change that can activate oncogenes and inactivate tumor suppressors. Somatic copy number alteration is the somatic alteration that can affect a large portion of cancer-related genome; in addition, they are extremely frequent in this tissue type. 32 In some cases, focal SCNAs have led to the identification of cancer-causing genes and suggested specific therapeutic approaches.33-36 Genes that can drive the oncogene process in human cancer have overexpression related to their gene amplification, where they present a correlation between gene expression and change in the number of gene copies.22,37 For example, the high amplification is common for NSD3 gene. This HMT is amplified in breast, lung, head and neck, and osteosarcoma cancers leading cell cycle progression. 37
Here, except for the SMYD5 gene, all studied HMTs are amplified, being PRDM9, SETDB1, PRDM14, and EHMT2 the most amplified HMTs. In addition, the EHMT2 and NSD3 genes also harbor deep deletion events. Regarding the high-amplification behavior, for example, it has been reported that SETDB1 is amplified in basal breast cancer, 22 showing that the recurrence of this change in different tumors is common. Overall, SETDB1 activity is correlated with increased aggressiveness and worse disease prognosis and is allocated as an oncogene in the Cancer Gene Census (CGC) database. In colorectal cancer, SETDB1 is overexpressed. 38 Probably the origin of its high expression could also be linked to its amplification. 39 Furthermore, in previous studies, another gene encoding an HMT, NSD3, was reported that to be amplified in colorectal and breast cancer.22,37
Several studies have proposed that there is a correlation between mRNA expression and SCNA.22,37 Here, when the HMTs studied undergo a process of deletion or deep deletion, a decrease in gene expression can be observed. On the contrary, when these same genes show partial gain or show high-amplification alterations, their expressions increase (Figure 1). This same behavior between mRNA expression and SCNA was observed for all HMTs; however, the most evident pattern of increase or decrease of the mRNA expression in relation to presence of amplification/deletion was observed for NSD3, SETDB1, SU39H1, and SUV39H2 (Figure 1). Among them, the NSD3 gene shows strong correlation values Spearman: 0.69 (P = 1.53e−59) and Pearson: 0.77 (P = 2.88e−83) between mRNA expression and SCNA. The SCNA can probably interfere with mRNA expression levels in this HMT, increasing when there is amplification or decreasing when there is deletion (Figure 2). These processes generate a series of histopathological states driven by the accumulation of genetic alterations, which, as note, have repercussions on changes in gene expression and may direct normal cells to states of hyperplasia, dysplasia, invasive cancer, and even metastasis. 40 Changes in NSD3 gene expression can be associated with variation in prognosis depending on the cancer type. In colorectal cancer, the decreased HMT mRNA expression is linked to poor prognosis. 37
Aberrant mRNA expression in multiple HMTs is a common feature in many cancers. In our study, the PRDM14 and PRDM9 are down-regulated in stomach adenocarcinoma. PRDM14 is overexpressed in pancreatic cancer tissues compared to its normal counterparts. 41 In a pan-cancer study, PRDM9 exhibited the highest expression in cancers such as head and neck squamous cell carcinoma and bladder urothelial carcinoma and a tendency to be upregulated in 32 types of cancer. In addition, PRDM9 is transcriptionally active in many cancers, a cancer condition related to nongerm cells. 42 PRDM9 is one of the most mutated genes in our study. Houle et al 42 reported that 45 specific somatic mutations in PRDM9 are associated with aberrant mRNA expression of this HMT. We also report here that SUV39H2 and NSD2 are overexpressed in stomach adenocarcinoma. NSD2 is overexpressed in several types of aggressive solid tumors, including renal cancer, prostate cancer, breast cancer, cervical cancer, and osteosarcoma. This overexpression is associated with poor prognosis and recurrence. 43
These findings reveal the qualitative relationship between genetic variation of HMT and its downstream effect, especially for oncogenes and tumor suppressor genes, which have been critical for prevention, diagnosis, and treatment of cancer. All of the HMTs presented here could be a potential panel of prognostic biomarker, especially the HMTs encoded for PRDM9, SETDB1, PRDM14, EHMT2, and NSD3 genes.
Finally, with regard to the relationship between HMTs alterations and clinical characteristics, we reported here that the HMT SUV39H2 showed reduction in its expression in patients with recurrence and progression of stomach adenocarcinoma. Disease-free patient had increased expression of SUV39H2 gene. Many studies have reported that SUV39H2 acts as an oncogene and is linked to cancer initiation and progression. 44 In breast cancer, SUV39H2 is correlated with metastatic biology and poor survival. Initially, SUV39H2 is primarily responsible for di- and tri-methylation of H3K9 (H3K9me3). Elevated H3K9me3 levels were associated with GC recurrence and was also able to predict a worse prognosis of a cohort of 261 GC patients 45 This result suggests that SUV39H2 plays a role in the progression of stomach adenocarcinoma when its mRNA level decreases. This HMT could be a good target to study the prognosis in stomach adenocarcinoma.
There is evidence that genetic alterations of various HMTs affect the oncogenic or tumor suppressor functions of cancer and influence the cancer initiation and progression. 46 Dysregulation of HMTs could cause an imbalance in transcription, and their abnormal gene expression could promote changes in the cellular environment, resulting in the initiation of the carcinogenesis process. 47 Therefore, abnormalities in HMTs can induce tumorigenesis through of the developmental defects, turning these set of enzymes potential markers for the treatment of cancer based on the development of antitumor drugs. 48
Conclusion
By performing a meta-analysis across independent patient cohorts, we identified 10 HMTs (PRDM14, PRDM9, SUV39H2, NSD2, SMYD5, SETDB1, PRDM12, SUV39H1, NSD3, and EHMT2) that showed various changes in stomach adenocarcinoma. These HMTs are also altered in other types of cancers such as blood, breast, colorectal, and gallbladder. Histone methyltransferases such as PRDM14 and PRDM9 are the most altered in the data set. Furthermore, the genes PRDM9, NSD2, EHMT2, and NSD3 are the most mutated with rates of 5.7%, 5.5%, 4.5%, and 4.1%, respectively. Many of these mutations are recurrent and could lead to loss of biological function; 35 mutations were predicted as deleterious; however, more studies can be carried out to conclude this. All HMTs evaluated have mutations in their SET domain and the EHMT2 gene harbor mutation in its predomain. Both domains play an important role in recognizing specific residues in histone proteins and maintaining invariability within these domains is necessary to maintain the enzyme function. PRDM9, PRDM14, SETDB1, and EHMT2 showed gene amplification, a feature observed in many cancer-related genes. Furthermore, all HMTs genes showed a typical pattern between mRNA expression and SCNA, in which increase or decrease in mRNA expression was observed regarding the presence of amplification/deletion alterations. This pattern was most evident for NSD3 gene.
Finally, HMT SUV39H2 showed a reduction in its expression in patients with recurrence and progression of stomach adenocarcinoma. This result suggests that SUV39H2 play a role in the evolution of stomach adenocarcinoma and alterations in this enzyme, which can interfere in its mRNA expression, may be associated with the prognosis of stomach adenocarcinoma. Our analyses suggest a group of HMTs as potential biomarkers and therapeutic targets in stomach adenocarcinoma. This computational approach provides a new list of genes that can be used as useful tools for therapeutics and diagnosis for GC.
Footnotes
Acknowledgements
The authors are thankful to the Center of Advanced Studies of Maule for the support to perform this work.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
D.A.R. and V.M.S.S.: analysis of data, writing the manuscript. M.S.-V.: Organized the database, analysis of data, and writing the manuscript. V.D’A.: conception of the research, organized the database, analysis of data, and writing the manuscript.