
Abstract
Background:
Though the development of targeted cancer drugs continues to accelerate, doctors still lack reliable methods for predicting patient response to standard-of-care therapies for most cancers. DNA methylation has been implicated in tumor drug response and is a promising source of predictive biomarkers of drug efficacy, yet the relationship between drug efficacy and DNA methylation remains largely unexplored.
Method:
In this analysis, we performed log-rank survival analyses on patients grouped by cancer and drug exposure to find CpG sites where binary methylation status is associated with differential survival in patients treated with a specific drug but not in patients with the same cancer who were not exposed to that drug. We also clustered these drug-specific CpG sites based on co-methylation among patients to identify broader methylation patterns that may be related to drug efficacy, which we investigated for transcription factor binding site enrichment using gene set enrichment analysis.
Results:
We identified CpG sites that were drug-specific predictors of survival in 38 cancer-drug patient groups across 15 cancers and 20 drugs. These included 11 CpG sites with similar drug-specific survival effects in multiple cancers. We also identified 76 clusters of CpG sites with stronger associations with patient drug response, many of which contained CpG sites in gene promoters containing transcription factor binding sites.
Conclusion:
These findings are promising biomarkers of drug response for a variety of drugs and contribute to our understanding of drug-methylation interactions in cancer. Investigation and validation of these results could lead to the development of targeted co-therapies aimed at manipulating methylation in order to improve efficacy of commonly used therapies and could improve patient survival and quality of life by furthering the effort toward drug response prediction.
Keywords
Introduction
Recent years have seen an expansion in availability of cancer drugs targeting specific genes, but the standard of care for most cancers remains a set of older, less targeted therapies for which few reliable biomarkers exist. The ability to predict drug efficacy in each patient requires identifying the molecular factors involved. One promising but under-utilized source of drug response biomarkers is DNA methylation. 1 DNA methylation has been shown to be involved in cancer development, progression, and drug response. It is of particular interest in cancer drug research because changes in methylation are heritable and will persist in new cells as the cancer grows, 2 but unlike genetic mutations, changes in methylation can be reversed under the right conditions.3,4 An additional benefit of methylation-based biomarkers is that, in many cases, tumor methylation patterns can be detected with minimally invasive liquid biopsies. 5 Identifying and understanding methylation patterns involved in tumor drug response therefore has the potential of expanding the number of drugs for which we can predict patient-specific responses and of revealing potential therapeutic targets for manipulating methylation to prevent or reverse tumor drug resistance, thus improving patient survival and quality of life.
The use of methylation-based biomarkers of drug response is not new; there exist several that are already in use clinically.6-9 For example, several tests exist to detect hypomethylation in the promoter region of MGMT (O-6-methylguanine-DNA methyltransferase), which is a biomarker of poor response to temozolomide.10,11 Most known drug-methylation interactions in the literature were identified the same way most of the existing body of knowledge on DNA methylation has been studied: one gene at a time, measuring overall methylation levels in the promoter region of the target gene, with the assumption that methylation affects cell function by suppressing transcription of the downstream gene. 12 However, this understanding of methylation and its role in gene expression has been increasingly challenged with the wider adoption of whole-genome, site-specific methylation sequencing technologies. It is now clear that methylation at individual CpG sites can increase or decrease expression of nearby genes13-15 and even distal genes. 16 Analysis of genome-wide, site-specific methylation data has helped elucidate much about cancer, especially about carcinogenesis,17,18 molecular characterization,19-21 and molecular indicators of prognosis,21-23 but so far, methylation studies on cancer drug efficacy have been limited in scope, for example, focusing on a single cancer.24,25 As of yet, there have been no systematic efforts to identify drug-methylation interactions in most cancers despite mounting evidence that methylation often plays a key role in the development of cancer drug resistance.26-28
The Cancer Genome Atlas (TCGA) has collected tumor samples from a wide range of cancers for molecular characterization. TCGA is an excellent data resource for studying methylation because of the large number of patient samples with DNA methylation data as well as thorough clinical annotations with drug treatment data and survival data available for most patients. It is especially suited for identifying biomarkers predictive of drug response because the molecular assays are performed on pre-treatment samples, representing the state of a tumor at the point when treatment decisions are made. Our group has previously identified molecular features associated with drug-specific survival using the TCGA gene expression, 29 copy number variation,30,31 protein, 32 and miRNA 33 datasets. While the high dimensionality of the methylation dataset makes it the most challenging to analyze, the promise that DNA methylation holds as a source of biomarkers as well as our success with these other molecular data types make it a critical dataset to explore for molecular features related to drug efficacy.
In this analysis, we performed a systematic analysis across many cancer types and drugs using the methylation data and clinical data from TCGA to identify methylation-drug interactions and potential biomarkers of drug response. Survival analyses of patients grouped by cancer and drug exposure identified individual CpG sites whose pre-treatment methylation status is significantly associated with drug-specific survival. We then explored these drug-specific CpG sites for meaningful biological patterns. We identified CpG sites with the same drug-specific survival effects in different cancers, and we used a clustering method to identify sets of these CpG sites that tended to be methylated in the same patients, which revealed even stronger associations with drug-specific patient outcomes. Our results included known methylation-drug interactions and many novel interactions that could be investigated further for new insights into drug mechanisms or validated for clinical use as biomarkers of patient drug response.
Results
CpG sites with drug-specific associations with survival
To identify potential methylomic biomarkers of drug response, we performed survival analyses using clinical data and binarized pre-treatment methylation data from TCGA. The basic workflow is summarized in the diagram in Figure 1 and detailed in the Methods section. For each of 82 cancer-drug combinations, we defined a target patient group that included all patients with that cancer who received that drug, and we also defined a corresponding control group of all patients with that cancer but no exposure to the drug. Within the target patient groups, we performed survival analyses for 396 065 CpG sites to identify those where methylation status (methylated or unmethylated) is associated with significant survival differences. We tested a total of 9.2 million cancer-drug-CpG combinations, identifying 155 554 combinations with significant survival effects. To remove survival signals general to the cancer but not specific to the drug, we then excluded 104 454 of these cancer-drug-CpG combinations that showed similar survival effects in the corresponding control group, leaving 51 100 cancer-drug-CpG combinations with significant drug-specific effects on survival. We found these drug-specific survival markers in 38 of the 82 tested cancer-drug patient groups, including representation from 15 cancers and 20 drugs. Table 1 summarizes our results for each cancer-drug combination and lists the number of patients in both the target patient group and the corresponding control group. For survival analysis results of all cancer-drug-CpG combinations with drug-specific survival effects, see Supplemental File 1.
Summary of individual CpG site analysis results.

Table showing the 38 cancer-drug combinations that had CpG sites associated with survival, along with the number of patients in the target patient group (patients with the indicated cancer who were treated with the indicated drug) and the corresponding control group (patients with the indicated cancer with no exposure to the indicated drug) for each combination. Also listed are the total number of CpG sites tested for survival differences in the target patient group, the number of CpG sites found to have significant (q ⩽ .1) survival differences in the target patient group, the number of these that were also significant (P ⩽ .1) in the corresponding control group, and the final number of CpG sites with drug-specific survival differences after removal of cancer-related effects. Cancers are listed according to their TCGA acronyms (https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations).

Flow chart outlining the steps taken in the primary analysis in this study.
Promising examples from a variety of cancers are highlighted in Table 2, which lists the relevant drug for each example, the specific CpG site, and nearby genes along with survival analysis statistics and Kaplan-Meier survival curves illustrating the survival differences we observed between patients with and without methylation at the indicated CpG site in both the target cancer-drug patient group and the corresponding control group. In the top row, for example, we highlight a CpG site near the HOXB3 (homeobox B3) gene where methylation status is associated with survival in bladder urothelial carcinoma (BLCA) patients who took cisplatin. Log-rank survival analysis showed that there is a significant difference in survival between methylated and unmethylated patients in the target patient group, as illustrated by the first Kaplan-Meier curve, which shows much better survival for patients with methylation at that site (orange) than for patients without methylation (blue). In the control group of BLCA patients who did not take cisplatin, methylation at this site was slightly negatively associated with survival, meaning that the positive association between methylation and survival in BLCA patients is specific to cisplatin.
Examples of top results from each cancer.
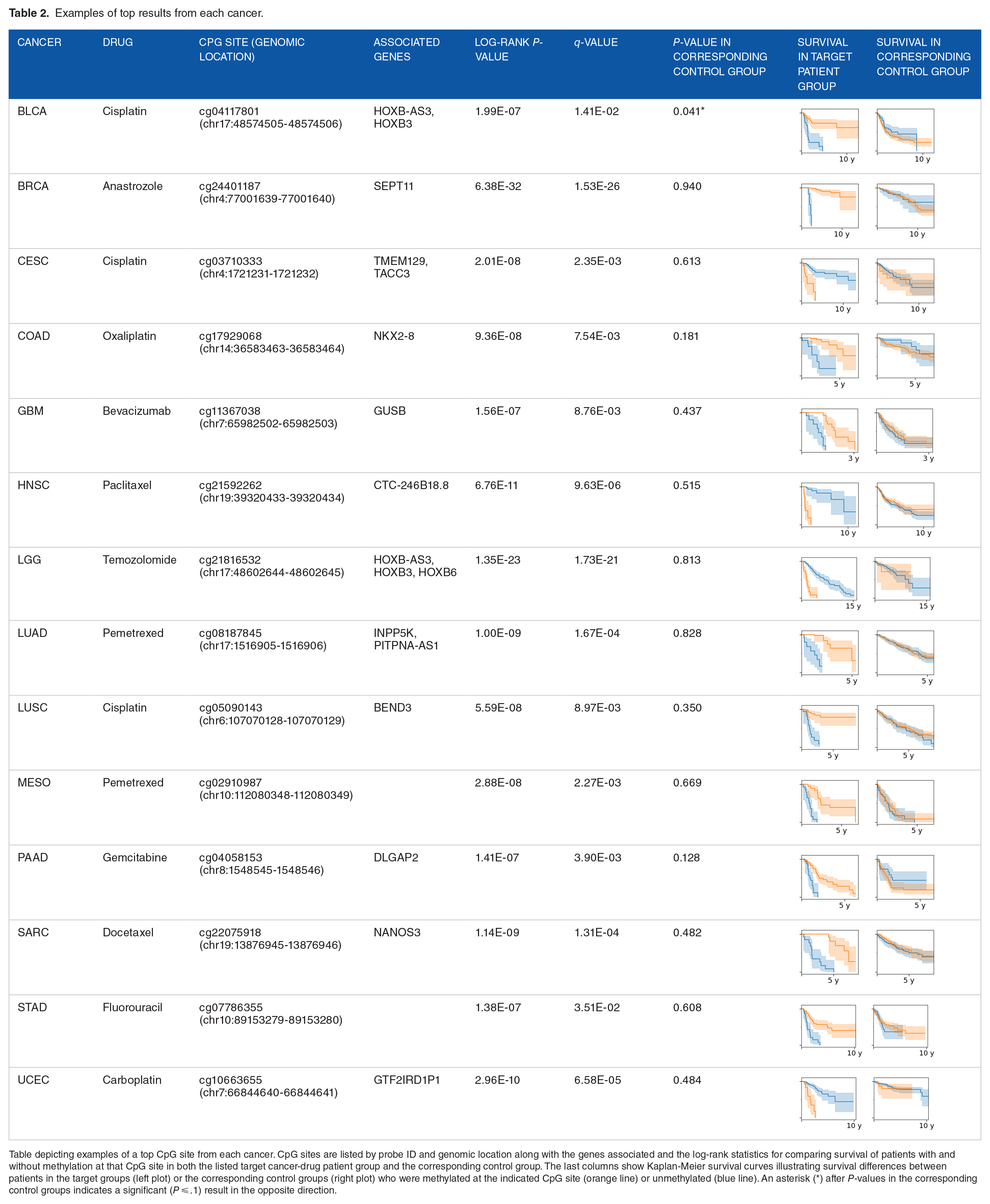
Table depicting examples of a top CpG site from each cancer. CpG sites are listed by probe ID and genomic location along with the genes associated and the log-rank statistics for comparing survival of patients with and without methylation at that CpG site in both the listed target cancer-drug patient group and the corresponding control group. The last columns show Kaplan-Meier survival curves illustrating survival differences between patients in the target groups (left plot) or the corresponding control groups (right plot) who were methylated at the indicated CpG site (orange line) or unmethylated (blue line). An asterisk (*) after P-values in the corresponding control groups indicates a significant (P ⩽ .1) result in the opposite direction.
To investigate the biological implications of the identified drug-specific survival markers, we first compared our results to known drug-methylation interactions in the literature. Our analysis strongly identified the relationship between the drug temozolomide and methylation in the MGMT promoter, a well established interaction that is one of the few methylation-based biomarkers of patient drug response currently in clinical use. Hypermethylation within the MGMT promoter region has been reported to increase the efficacy of temozolomide by decreasing the expression of MGMT, a DNA-repair enzyme which works to repair the DNA damage that temozolomide causes in cancer cells to kill them. 12 We found 5 CpG sites within the MGMT promoter region with significant survival differences in brain lower grade glioma (LGG) patients who took temozolomide, and, consistent with previous reports, in all 5 cases, patients with these CpG sites methylated had significantly better survival. Another example of a well characterized drug-methylation interaction is between taxanes and methylation of the gene CHFR (checkpoint with forkhead and ring finger domains). While this interaction has yet to be adopted clinically as a biomarker of drug response, it has been widely reported that methylation and subsequent transcriptional inactivation of CHFR is associated with increased sensitivity to taxanes.34-36 In our results, we found 2 CHFR-associated CpG sites with drug-specific survival differences in breast invasive carcinoma (BRCA) patients treated with paclitaxel (a taxane), both of which showed better patient survival associated with a methylated state, which is consistent with previous reports. These widely reported examples of drug-methylation interactions corroborate our results.
Although methylation patterns are often tissue-specific,13,37 we found 11 CpG sites that had similar drug-specific effects across multiple cancer types from different tissues. These CpG sites were found to have drug-specific survival differences in 2 groups of patients with 2 different cancers but treated with the same drug. For example, in both sarcoma (SARC) and BRCA, patients with methylation at a specific CpG site (cg18009000, chr10:70052171-70052172) in the H2AFY2 (H2A histone family member Y2) promoter region had longer survival when taking docetaxel than patients who were unmethylated at that site, but there was no difference in survival in BRCA or SARC patients who did not take docetaxel. All such CpG sites are highlighted in Table 3, which shows the log-rank statistics comparing methylated and unmethylated patients in each cancer-drug group and the corresponding control group, confirming these CpG sites were drug-specific survival markers. Table 3 also shows Kaplan-Meier survival curves illustrating the observed survival differences for each CpG site in the 2 cancer-drug contexts, which were in the same direction in all cases.
CpG sites with similar drug-specific survival effects across multiple cancers.

Table showing drug-CpG site combinations with significant, drug-specific effects in the same direction (in all cases, methylated patients had better survival) in multiple cancer-drug patient groups that shared the same drug. CpG sites are listed by probe ID and genomic location along with associated genes. Log-rank P-values and q-values in the target patient groups are indicated along with log-rank P-values in the corresponding control groups. An asterisk (*) after P-values in the corresponding control groups indicates a significant result in the opposite direction. The last column shows Kaplan-Meier survival curves illustrating survival differences between patients in the 2 indicated target patient groups who were methylated (orange line) or unmethylated (blue line) at that CpG site.
Patterns of Methylation With Survival Effects
Another way to investigate these results is by finding larger methylation patterns that are related to survival. Within each cancer-drug patient group, we clustered CpG sites based on their tendency to be methylated in the same set of patients. Using this strategy, we identified 76 CpG clusters in 18 cancer-drug patient groups. The CpG clusters had a minimum size of 5 CpG sites and ranged in size up to 5705 CpG sites (for lists of CpG sites in each cluster, see Supplemental File 2). For each CpG cluster, we stratified patients by the percent of CpG sites that were methylated into “hypermethylated” or “hypomethylated” strata and applied the log-rank test to examine differential survival between them. Table 4 reports the results of example clusters from each of the groups, and the statistics from all clusters are available in Supplemental Table 1 (Table 4 clusters correspond to Supplemental Table 1 clusters labeled A). Figure 2 shows Kaplan-Meier survival curves of 4 of the example clusters shown in Table 4 illustrating the survival differences between patients with cluster hypermethylation and those with cluster hypomethylation.
Highlights from cluster-based analysis of methylation and drug-specific survival.
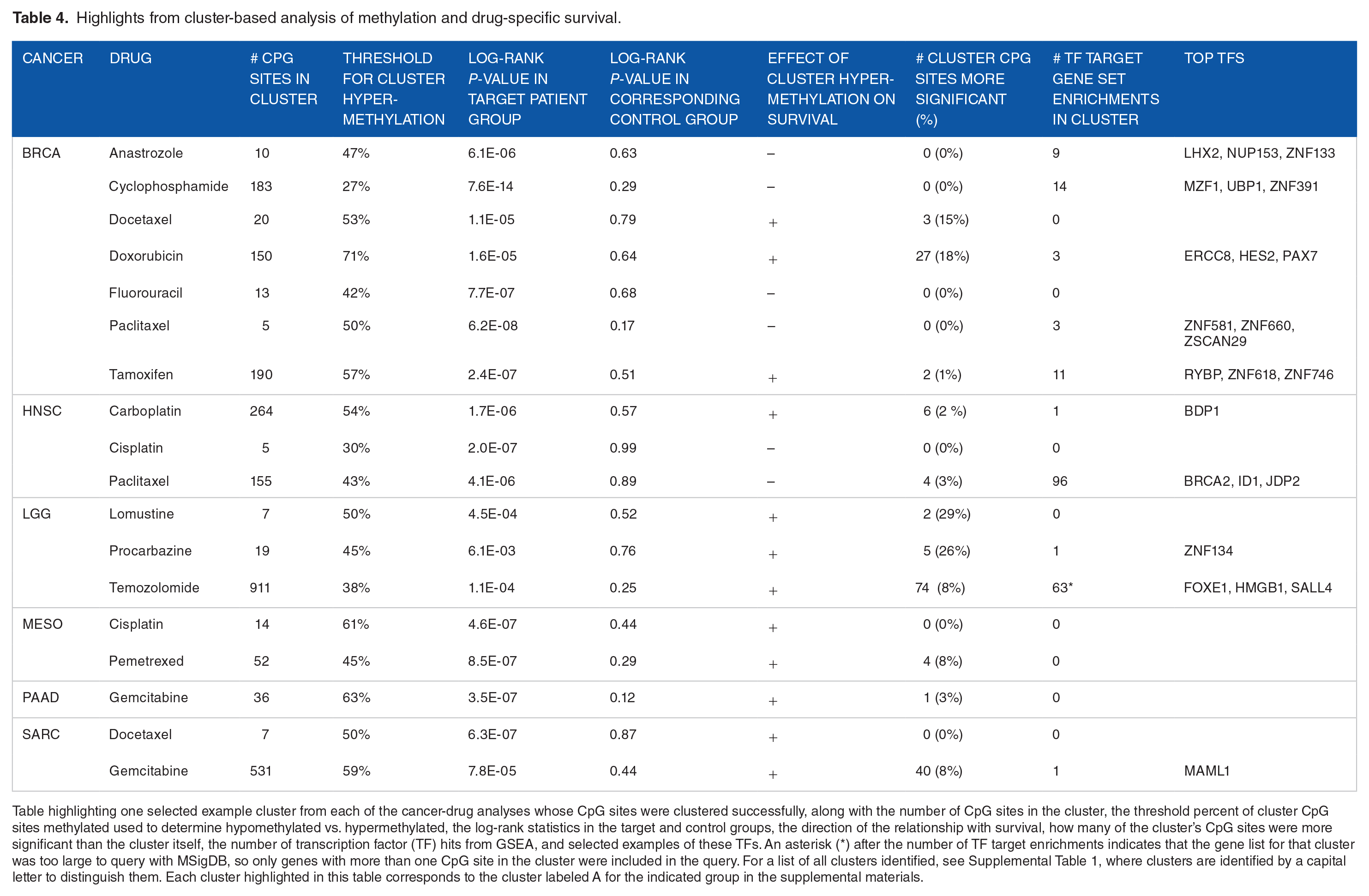
Table highlighting one selected example cluster from each of the cancer-drug analyses whose CpG sites were clustered successfully, along with the number of CpG sites in the cluster, the threshold percent of cluster CpG sites methylated used to determine hypomethylated vs. hypermethylated, the log-rank statistics in the target and control groups, the direction of the relationship with survival, how many of the cluster’s CpG sites were more significant than the cluster itself, the number of transcription factor (TF) hits from GSEA, and selected examples of these TFs. An asterisk (*) after the number of TF target enrichments indicates that the gene list for that cluster was too large to query with MSigDB, so only genes with more than one CpG site in the cluster were included in the query. For a list of all clusters identified, see Supplemental Table 1, where clusters are identified by a capital letter to distinguish them. Each cluster highlighted in this table corresponds to the cluster labeled A for the indicated group in the supplemental materials.
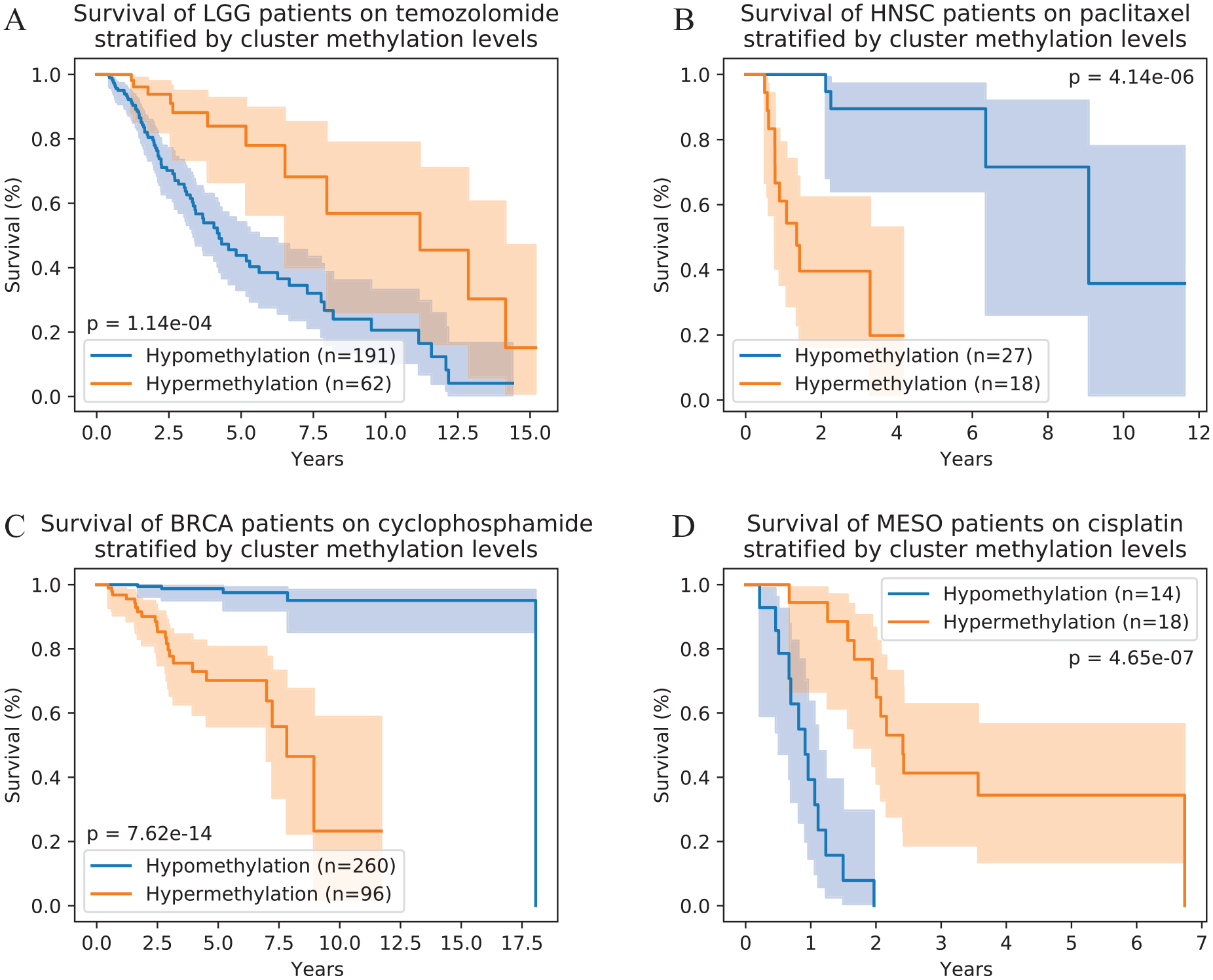
Methylation levels of clusters of co-methylated CpG sites are associated with post-treatment survival. Here we show Kaplan-Meier survival curves illustrating survival differences between patients with hypermethylation (orange) and hypomethylation (blue) of the example clusters from the following groups: (A) LGG patients treated with temozolomide, (B) HNSC patients treated with paclitaxel, (C) BRCA patients treated with cyclophosphamide, and (D) MESO patients treated with cisplatin. The clusters shown correspond to the example clusters in Table 4 for the indicated groups and to the clusters labeled A for these groups in the full cluster list in Supplemental Table 1. Survival differences in (C) and (D) are more significant than those observed in any single CpG site in their respective cancer-drug groups.
Our strategy of grouping CpG sites based on co-methylation in patients tended to produce clusters that successfully separated patients who responded well to the drug from patients who responded poorly: all clusters we identified showed statistically significant differences in survival, most of which were not observed in the corresponding control group. In fact, for more than half of the clusters, the survival differences observed based on the clusters were more significant than those of any individual CpG site belonging to those clusters, and in the 2 examples shown in Figure 2C and D, cluster methylation level was a better predictor of survival than any other measure (individual CpG site or CpG cluster) for their respective groups. Figure 3 shows the log-rank P-values of each cluster vs. the median P-value of the individual CpG sites in each cluster, with error bars to show the distribution of log-rank P-values of the individual CpG sites in the cluster. Almost all of the points are above the equivalence line (shown in gray), indicating that clusters are more predictive of drug-specific survival outcomes than their median CpG sites. Furthermore, for the majority of the CpG clusters, the entirety of the error bars was also above the equivalence line, illustrating that the clusters tend to outperform most or all of the individual CpG sites they contain.

CpG clusters more accurately stratify patients into responders and non-responders than their constituent CpG sites. This scatter plot shows the log-rank P-value distribution of CpG sites in each cluster compared to the P-value of the cluster itself. Blue circles plot the cluster P-value versus the median P-value of CpG sites in that cluster, with error bars extending to the 5th and 95th percentile P-values of CpG sites in each cluster. The gray dashed line indicates equivalence; the region above the line is where clusters are more significant than their median CpG site, and the region below the line is where clusters are less significant.
In addition to their utility as biomarkers, these CpG clusters can be mined for insights into potential underlying biological mechanisms. Each CpG site in the methylation data is identified by the array probe that measures it, and the raw data files include reference information about the CpG site, including genomic location and any associated genes. We found no obvious patterns in the genomic locations of the CpG sites within each cluster. It is difficult to search for meaningful biological information using the array probe identifier, so we used the reference information to convert the set of CpG sites in each cluster to a set of associated genes to investigate. We then performed gene set enrichment analysis (GSEA) on our gene sets to look for unifying regulatory features that could be related to methylation. We queried MSigDB transcription factor (TF) target gene sets and found a total of 553 instances of TF target enrichment in our methylation gene sets.
Because the MSigDB gene sets were of genes with TF binding sites in their promoter regions, we then looked at how often the cluster CpG sites associated with the overlapping genes were located in the promoter region in each of these instances. We did not see any relationship between the significance of gene set enrichment matches and the percentage of overlap genes whose associated cluster CpG sites were in their promoter region nor any other relationship with the percentage of overlap genes with promoter CpG sites in the cluster. However, we observed that the distribution of these percentages was bimodal, with 93% being either below 40% or above 60% (data not shown). Therefore, we considered only matches with at least 50% of the overlapping genes having promoter CpG sites in the respective cluster as potentially reflecting an interaction between methylation of the cluster CpG sites and the TF binding to these promoters. This narrowed our TF target enrichment results to 284 instances of TF target enrichment in our methylation-based gene sets.
We found that the gene sets derived from 21 of our CpG site clusters from 10 cancer-drug patient groups had significant enrichment for gene targets of one or more TFs, some of which are known to interact with the respective drug. For example, Figure 2A illustrates the survival of LGG patients who took temozolomide, grouped by methylation levels in one of this group’s CpG clusters wherein the associated genes are enriched for targets of SALL4 (spalt-like transcription factor 4). SALL4 has been shown to decrease efficacy of temozolomide in killing cancer cells, 38 and hypermethylation of CpG sites in the promoter regions of its targets could prevent SALL4 binding, thus inhibiting this effect and maintaining sensitivity to temozolomide. Consistent with this proposed mechanism, hypermethylation within this cluster was associated with better survival in LGG patients treated with temozolomide. According to the canonical understanding of how methylation can mediate TF function, we expect this kind of CpG cluster-TF relationship to be common among our results: hypermethylation in a cluster enriched with CpG sites near TF binding sites may block TF binding and inhibit the effect a TF might otherwise have on cellular drug response. However, other mechanisms exist whereby methylation could enhance the effect of an associated TF. For example, Figure 2B shows significantly shorter survival in head and neck squamous cell carcinoma (HNSC) patients on paclitaxel who have hypermethylation in a cluster with significant enrichment of ID1 (inhibitor of DNA-binding 1) targets among the genes associated with the cluster’s CpG sites. Paclitaxel has been reported to downregulate 39 and degrade 40 ID1, which is involved in cell growth, 41 while overexpression of ID1 has been shown to block crucial pathways in the anti-cancer effects of paclitaxel.42-45 ID1 acts by blocking certain regulatory proteins from binding DNA; likewise, DNA-binding proteins can also be blocked by methylation near protein-binding regions, like many of this cluster’s CpG sites that are associated with ID1 target genes. Our observations of this cluster are consistent with what we would expect to see if hypermethylation of its CpG sites could block some of the same proteins as ID1 and thus have similar effects: paclitaxel treatment decreases growth-inducing ID1, but patients with hypermethylation that imitates the effect of ID1 would still have poor outcomes. These examples illustrate mechanisms by which methylation levels in CpG clusters could interact with the TFs identified using GSEA.
While these examples use previously reported literature to illustrate potential relationships and mechanisms that could be found in our analysis, many of the drug-TF relationships identified in our analysis have not been reported previously and may provide novel insights into pathways involved in drug efficacy. More example TFs with targets enriched in cluster gene sets are listed in Table 4, and complete lists are included in Supplemental Table 1. The CpG clusters we identified in many of these groups were strongly associated with survival outcomes in patients taking the respective drugs, and the biological implications of these clusters, including putative TF-cluster interactions, may point to mechanisms of cellular drug response in tumors.
Discussion
Our analysis has identified many individual CpG sites with drug-specific effects on survival in certain cancers. The identified drug-specific CpG sites include ones that are consistent with previously reported drug-methylation interactions, including the only methylation-based biomarker for drug response in current clinical use. The corroboration of our results by the best-known drug-methylation interactions in the literature suggests our results, the majority of which are novel, likely contain useful drug-methylation interactions. Our top results, such as those highlighted in Table 2, are excellent candidates for validation as biomarkers of drug efficacy. Additionally, among the CpG sites identified as drug-specific survival markers, we found 11 CpG sites that had a similar impact on survival on a particular drug in 2 different cancers. These results are especially promising as biomarkers because the CpG sites were linked to the same drug in 2 different cancer contexts, so they are more likely to be important in the processes involved in drug response and could be more widely applicable as biomarkers than the results that are only found in one cancer. Interestingly, 8 of the 11 drug-CpG site relationships found in multiple cancers involved the drug gemcitabine, and the remaining 3 were taxanes. This limited variety suggests that some drugs may have mechanisms that can be impacted by similar methylation patterns in a variety of cancer contexts, suggesting they may be prime candidates for developing possible co-therapies that alter methylation states to improve efficacy or to reverse or prevent the development of drug resistance.
In addition, the clusters of CpG sites identified in this study could also be valuable tools for predicting drug response given their high accuracy in separating patients into drug responders and non-responders. In fact, the cluster-based survival differences we observed often surpassed that of individual CpG sites within the clusters, which suggests that the clusters may be more useful as biomarkers than the individual CpG sites. Moreover, the CpG clusters may have an advantage as potential biomarkers in that using multiple CpG sites to predict drug response may be less vulnerable to the individual CpG sites’ biological and technical variability. The CpG clusters also may reveal informative biological relationships, as suggested by our gene set enrichment analysis. We queried our cluster-associated gene sets against sets of genes with shared TF binding sites in their promoter region because methylation within protein binding sites in promoter regions is still considered the primary mechanism by which methylation impacts gene expression and cell function. We found numerous instances of TF target enrichment within clusters and highlighted 2 that are consistent with known TF-drug interactions. While we cannot draw firm conclusions from this type of analysis without experimental evidence, these findings indicate potential mechanistic relationships between our CpG clusters and the GSEA-implicated TFs. The clusters identified in this analysis provided additional informative avenues for biological interpretation of our results and are themselves strong candidates for use as predictive biomarkers.
We devised our analysis strategy to be sensitive enough to identify modest survival effects and to strictly exclude survival effects not specific to the drug. We defined significance in survival differences using a 10% false discovery rate (FDR) threshold in target cancer-drug patient groups, which is a relatively lenient standard that increases the sensitivity of our analysis, although it increases false positives. In contrast, when considering log-rank test results from corresponding control groups for exclusion from our significant results to delineate drug-specific effects, we used a raw P-value ⩽ .1 for our significance threshold. This threshold is very lenient for significance, but because it is used to determine exclusion from our proposed drug-specific survival effects, a lenient threshold increases stringency for considering observed survival differences to be drug-specific effects. As Table 1 shows, in most cancer-drug combinations, the majority of our significant CpG sites in each cancer-drug group were also drug-specific after considering the corresponding control group. The major exception is in LGG, which frequently involves major molecular changes impacting disease-specific survival 46 and even our stringent exclusion step may not be able to remove all of them. However, overall, although many of our identified drug-CpG site relationships are only found in one cancer, the strict exclusion of survival effects in corresponding control groups ensures that our results reflect drug-specific relationships.
While the potential drug-specific survival biomarkers we have highlighted in this study warrant further investigation, our analysis has several limitations. Due to the inconsistent reporting of drug response in the TCGA clinical data, we could not study drug efficacy directly and we had to use overall survival as a proxy measure. Also, the granularity of our analysis required stratifying patients by both cancer type and treatments received. Stratification by drug allowed us to narrow our results to those specific to the drug; stratification by cancer was also necessary to avoid confounding the analysis with tissue-specific methylation differences or survival differences between cancers. However, despite the large number of total patients in the TCGA dataset, this stratification yielded many patient groups that were too small to analyze, so we were not able to identify drug-related CpG sites for all drugs in the dataset. In addition, our results are based on exploration of one dataset, and have not been validated experimentally. We were unable to perform computational validation on an independent dataset because we did not find other datasets that provide information on methylation, survival, and drug treatment for a common cohort of patients. Another challenge we faced was in the biological interpretation of our results because little is known about most of these loci and their effects on gene expression and cell function. The reference information provided by TCGA lists associated genes for each CpG site based on the genomic distance between the CpG site and the gene, but the actual relationship between CpG sites and the listed genes is often unknown, and CpG sites may also affect distal genes that were not listed. Thus, while we have used the location-based gene annotations of the CpG sites to explore our findings for recognizable biological processes, any conclusions based on these assumed relationships are inherently weak. To mitigate this, when doing the gene set enrichment analysis, we verified potential gene set matches based on the co-location of cluster CpG sites and the identified TF’s binding sites, where methylation could reasonably affect the ability of the TF to bind. This minimized our reliance on assumptions of a direct relationship between a CpG site and the expression of nearby genes, but it still assumes an indirect relationship. Because of these limitations, as with any computational study, our most promising results should be validated before clinical adoption as biomarkers of drug response.
Our results show promise for both potential clinical use and informing biological understanding of drug response, but further studies are needed. Computational validation of our findings in other methylation datasets from similar cohorts to those in TCGA could help remove false positives and clarify the most important methylation features to study. Exploring these results experimentally could elucidate underlying biological mechanisms involved in tumor drug response. Additionally, since clustering of binarized methylation data was able to define clusters of CpG sites that accurately separated responders and non-responders in the cancer-drug patient groups, another future research direction would be to apply this analysis strategy across multiple TCGA data types to identify multi-omics relationships associated with patient drug response, which could contribute valuable insights in developing more complete system-level understanding of cellular drug response across multiple levels of molecular interactions.
This study represents the first systematic search for drug-methylation interactions across multiple cancers and drugs and the first large scale effort to link drug efficacy with individual methylation sites. We identified many sites of DNA methylation and larger methylation patterns with strong associations with drug-specific survival. While any of our putative drug-methylation relationships would require further research, by linking our results to known molecular mechanisms that impact drug efficacy, we have demonstrated the power of our analysis to capture drug-specific effects and to implicate potential molecular mechanisms in drug response pathways, providing compelling support for future studies examining these results. Validation of these putative drug-methylation relationships as biomarkers of patient drug response could help doctors avoid prescribing ineffective treatments. In addition, experimental inquiries into these results will undoubtedly lead to expansion of our understanding of drug-methylation interactions and by extension, wider drug response mechanisms in tumors, and could help explain variation in drug efficacy among patients. This could, in turn, inform future drug development by identifying DNA methylation targets for potential co-therapies to increase efficacy and decrease resistance to common therapies.
Methods
Data acquisition and cleaning
TCGA methylation beta values were downloaded from the NCI Genomic Data Commons (GDC) database using their Data Transfer Tool to download files based on a file manifest acquired using the GDC API, querying using the parameter return_type: manifest. To acquire methylation data, we used the filters files.data_type: Methylation Beta Value and files.platform: Illumina Human Methylation 450; for drug exposures, we used the filters files.data_format: BCR Biotab and files.data_type: Clinical Supplement. Survival data were obtained via direct query to the GDC API.
A total of 485 577 methylation probes (CpG sites) were listed in the raw methylation data. We removed 89 512 of these where no beta values were available for any samples (where the raw data contained only N/A values). All available samples were used in calculating CpG site binarization threshold; for survival analyses, only primary tumor samples (sample IDs ending in -01A) were used. In cases where there were 2 raw methylation data files from the same sample, we used the average of the beta values. We used a manually curated drug name mapping (available at https://gdisc.bme.gatech.edu/Data/DrugCorrection.csv) to standardize drug names in the clinical data. We used TCGA study acronyms as cancer type; full cancer names are available at https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.
Binarizing Methylation Beta Values
We binarized the beta values of the remaining 396 065 CpG sites using a threshold calculated for each locus as previously described. 47 Briefly, we ordered (lowest to highest) the beta values of all samples in the TCGA methylation dataset, and then we fit a step function to these data that minimizes the sum of the mean square error within the “methylated” and “unmethylated” subsets. To accomplish this, we tested 200 thresholds evenly distributed along the ordered beta values plus 200 thresholds distributed along the range of the beta values (0 -1), testing a total of 400 thresholds and choosing the threshold with the lowest variance in the 2 stratified groups. Since these binarization thresholds were computed based on all patient samples across various cancer types and tissue types, these thresholds were able to robustly define the on/off status of methylation. All beta values were binarized; missing data were kept as N/A to be handled later in the analysis as described.
Survival Analysis
Survival analyses were performed on patient groups defined by cancer type and drug exposure. For each cancer-drug combination, we defined the target patient group as all patients in the dataset with the respective cancer who had exposure to the respective drug and the corresponding control group as all patients in the dataset with the respective cancer who were never exposed to the respective drug. For each cancer-drug patient group, we analyzed all CpG sites where the target patient group and corresponding control group each had at least 10 patients with methylation and 10 patients without methylation. Because of these parameters, the minimum number of patients in the target and control groups was effectively 20 patients; a total of 82 cancer-drug combinations in the dataset met these criteria and were included in our analysis.
For each survival analysis, we used a log-rank test to compare survival between patients with and without methylation at a given CpG site (patients with missing values at that site were excluded) within a group. We then used the Benjamini-Hochberg procedure, as implemented in the statsmodels Python package, to adjust the P-values from these tests for a given group to obtain q-values. We used a significance threshold of q ⩽ .1 to identify CpG sites whose methylation was associated with survival differences in that group. We then tested all significant CpG sites for differential survival in patients in the corresponding control group using the same statistical tests, which identified CpG sites with significant survival effects not specific to the drug of interest. We considered CpG sites to be drug-specific if the corresponding control group showed no significant survival differences (log-rank P ⩾ .1) or showed the opposite effect on survival (eg, if the methylation status associated with better survival in the target cancer-drug patient group is associated with worse survival in the corresponding control group). By removing from consideration CpG sites that are prognostic for the cancer overall unrelated to treatment, we narrowed down our findings to identify CpG sites with drug-specific survival effects. The lifelines Python package was used for log-rank tests and generating Kaplan-Meier curves.
Clusters were tested in a similar way, comparing patients in the appropriate cancer-drug patient group with hypomethylation and hypermethylation in each cluster. For each cluster, we scored patients in the relevant group based on the percent of the cluster’s CpG sites that were methylated and binarized these scores to categorize patients as having either hypomethylation or hypermethylation in that CpG cluster. We then used a log-rank test to determine survival differences between the patients with hypomethylation and hypermethylation in that CpG cluster. Clusters were considered significant with a log-rank P ⩽ .1.
Co-occurrence Clustering
We obtained CpG clusters for each group by clustering all CpG sites with drug-specific survival effects that had no missing values in the group. The CpG sites were clustered based on the similarity of their methylation patterns across patients in the respective cancer-drug patient group. We used a previously described clustering algorithm called co-occurrence clustering, 48 which constructs a graph of CpG sites based on a chi-squared pairwise association measure and uses the Louvain algorithm to identify CpG clusters. Patients are then clustered in the same way based on their methylation levels within these CpG clusters, and then the algorithm runs iteratively over its resulting clusters. This algorithm yielded sets of CpG sites that tend to be co-methylated in patients in the group. No survival information was used in the clustering process.
GSEA
We performed gene set enrichment analysis (GSEA) to look for promoter-region transcription factor (TF) binding sites that might be related to our CpG clusters. This first required creating a list of genes, which were the set of genes listed as associated with the CpG sites in the original data documents. We then used the Molecular Signatures Database (MSigDB)49,50 v7.5.1 to perform GSEA on our cluster-related gene sets, querying against the GTRD 51 subset of the transcription factor targets (TFT) collection (C3) in MSigDB. The output indicated TFs with binding sites in the promoter regions of a significant (hypergeometric test, FDR 5%) number of genes in our gene set. To narrow our results to TF binding sites co-located with the CpG sites in our clusters, we only considered matches where at least 50% of the overlapping genes were associated with cluster CpG sites that were in their promoter region.
Supplemental Material
sj-csv-2-cix-10.1177_11769351221131124 – Supplemental material for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer
Supplemental material, sj-csv-2-cix-10.1177_11769351221131124 for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer by Bridget Neary, Shuting Lin and Peng Qiu in Cancer Informatics
Supplemental Material
sj-csv-3-cix-10.1177_11769351221131124 – Supplemental material for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer
Supplemental material, sj-csv-3-cix-10.1177_11769351221131124 for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer by Bridget Neary, Shuting Lin and Peng Qiu in Cancer Informatics
Supplemental Material
sj-xlsx-1-cix-10.1177_11769351221131124 – Supplemental material for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer
Supplemental material, sj-xlsx-1-cix-10.1177_11769351221131124 for Methylation of CpG Sites as Biomarkers Predictive of Drug-Specific Patient Survival in Cancer by Bridget Neary, Shuting Lin and Peng Qiu in Cancer Informatics
Footnotes
Acknowledgements
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the National Science Foundation (CCF2007029). PQ is an ISAC Marylou Ingram Scholar and a Wallace H. Coulter Distinguished Faculty Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors were involved in method development. B.N. and S.L. acquired and cleaned data. B.N. performed the analysis and drafted the manuscript. P.Q. revised the manuscript. All authors have read and approved the final manuscript.
Abbreviations
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.