
Abstract
Variable selection methods play an important role in high-dimensional statistical modeling and analysis. Computational cost and estimation accuracy are the two main concerns for statistical inference from ultrahigh-dimensional data. In particular, genome-wide association studies (GWAS), which focus on identifying single nucleotide polymorphisms (SNPs) associated with a disease of interest, have produced ultrahigh-dimensional data. Numerous methods have been proposed to handle GWAS data. Most statistical methods have adopted a two-stage approach: pre-screening for dimensional reduction and variable selection to identify causal SNPs. The pre-screening step selects SNPs in terms of their
Keywords
Introduction
Recently, many high-dimensional datasets have been generated in biomedical science, such as microarrays and single nucleotide polymorphism (SNP) databases. In particular, genome-wide association studies (GWAS), which focus on identifying SNPs associated with a disease of interest, have produced ultrahigh-dimensional data. For theoretical development, we consider data to have high dimensionality if p =
Many efficient approaches have been introduced to overcome these problems. One is the adoption of multistep strategies.1,2 The first stage of this approach reduces the dimensionality
We want to know which of the available pre-screening methods is better for quantitative traits. Although many pre-screening methods are available, we do not know which method performs best in predicting a particular quantitative phenotype. We can find predictors that are jointly associated with the response variable among the parameters that remain after the pre-screening step. When multiple predictor variables exist for a response variable, joint identification becomes a powerful tool. 1
One of the traditional approaches for joint identification is the multiple linear/logistic regression method. However, when we handle high-dimensional data using traditional methods, we experience several problems. First, multiple linear regressions do not work well within high dimensionality, which causes computational complexity. Second, multiple linear regression is very sensitive to multicollinearity among SNPs. To overcome this problem, various penalization methods have been proposed, such as the ridge, bridge, least absolute shrinkage and selection operator (lasso), adaptive lasso, smoothly clipped absolute deviation (SCAD), and elastic-net.5–9 These methods can find jointly associated variables in high-dimensional data. The elastic-net method uses both the ridge and lasso penalties, obtaining the advantages of both approaches. The elastic-net method automatically selects significant variables, and, thus, efficiently resolves the problem caused by multicollinearity. The iterative adaptive lasso (IAL) method 10 retains the appealing property of rapid computation even for ultrahigh-dimensional problems. This method yields a sparse solution by setting certain parameters to zero. Predictor selection is then achieved with the nonzero values.
Many methods have been suggested for pre-screening and the variable selection procedure. However, we do not know which method performs best for quantitative traits. In this paper, we investigate which combination of pre-screening method and penalized regression performs best. To compare the power of pre-screening methods and penalized regressions, we use two GWAS datasets: one from the Korea Association Resource (KARE) project and the other from the Age-Related Eye Disease Study (AREDS). The adjusted
Materials and Methods
Materials
KARE Data
The KARE project began in 2007. 11 Participants in this project were recruited from two community-based cohorts: the rural Ansung cohort and the urban Ansan cohort in Gyeonggi-do province of South Korea. The numbers of people in the Ansung and Ansan cohorts are 5,018 and 5,020, respectively. The age range is from 40 to 69 years. More than 260 phenotypes have been surveyed through physical examinations, epidemiological surveys, and laboratory tests. We focus on the height trait, because height is a highly heritable polygenic characteristic. 11
The KARE data contain 500,568 SNPs. Before analysis, quality control processes are performed following Cho et al. 1 , and missing genotypes are imputed using PLINK software and the Japanese in Tokyo (JPT)/Han Chinese in Beijing (CHB) reference panel in HapMap. 1 After these processes, we obtain a dataset with 327,872 SNPs from 8,842 individuals.
Areds Data
AREDS is a prospective study of 4,757 persons to establish the risk factors of both age-related macular degeneration (AMD) and cataract. 12 The AREDS began in 1992. Ages of participants ranged from 55 to 80 years. Participants have been followed for at least seven years. We used body mass index (BMI) as a quantitative trait. The genotype platform of the AREDS data is an Illumina 100K GWAS chip. A total of 525 individuals were genotyped. Quality control processes were performed using the same criteria as with the KARE data. After quality control, we obtained a dataset with 87,260 SNPs from 462 individuals.
Methods
We formulate a multistage strategy for identifying the significant parameters among an enormous number of explanatory variables. Our strategy consists of three stages. At stage 1, we screen out the variables that are weakly correlated with the response variable via single-variable association tests. We select variables in terms of their
Stage 1. Standardization
Suppose that 
Stage 2. Pre-screening
We use the linear regression model in order to eliminate predictors that are weakly correlated with the response variable to achieve dimensionality reduction.
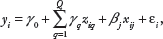
Stage 3. Variable selection
Method 1
Penalized regression. Multiple linear models are fitted for the selected top 
We find the optimal solution by using penalized regressions such as the ridge, lasso, and elastic-net. The penalized regressions find the solution as follows:
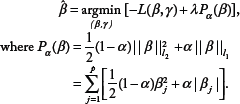
The amount of shrinkage is represented by parameter λ. We can find an optimal λ by using tenfold cross-validation, which accomplishes mean squared error minimization. Ridge regression (α = 0) entails a shrinkage of the least squares estimators.
8
The ridge is a biased estimator. Since the ridge reduces the variance of the estimators, it reduces the mean square error. In cases of high dimensionality, the ridge provides a shrinkage factor that does not accomplish variable selection. The lasso (α = 1) has an
Method 2. IAL
The IAL method is a two-stage procedure. 10 At the first stage, single-variable analysis is implemented to rank the magnitude of the marginal linear regression estimators. At the second stage, a weighted least-squares-type objective function is used to approximate a potential function. This allows us to further define a penalized weighted least square (PWLS) model for moderate-scale selection.
Step 1. Let M = {
to select a subset M1 of
Step 2. For every 
After ordering
Step 3. Apply the PWLS procedure at {M1,
Step 4. Iterate steps 2–3 until |M
Step 5. Finally, we obtain both the predictor set
Stage 4. Ordering
After selecting the significant predictor variables, we rank them in order of importance. For the elastic-net and lasso methods, we use BSS. Joint selection of SNPs via the elastic-net method is performed for the bootstrap samples. Bootstrapping is a resampling technique: a bootstrapping sample is a random sample with replacements from the original dataset. The bootstrap sample size is equal to the original dataset size. 
BSS signifies how many times each selected predictor variable is replicated in
For the ridge and IAL methods, the selected significant predictor variables are ranked in descending order of effect size.
Results
Pre-screening
KARE Data
At this step, we use the linear regression model in order to perform single SNP analysis for 327,872 SNPs. This linear regression model includes adjustment variables such as gender, age, and recruitment area (rural Ansung and urban Ansan).
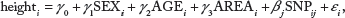
where
Although we use imputation processes, there are still missing values. Therefore, individuals having at least one missing value are eliminated. When data include individuals who have at least one missing value, penalized regressions such as the elastic-net and adaptive lasso cannot be performed. After the elimination process is performed, the number of remaining individuals is 4,183 for the
Areds Data
For the AREDS data, we use the linear regression model in order to perform single SNP analysis for 87,260 SNPs on the AREDs data. The model is given as follows:

where
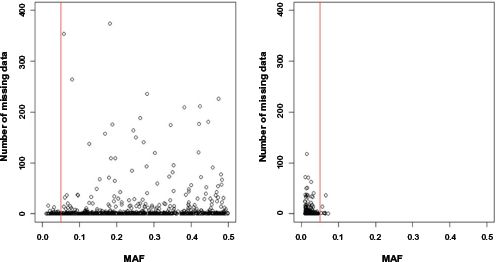
Number of missing data in the top 1,000 variables for each filtering method. The
Top 10 SNPs in each method after coefficient filtering of KARE data by effect size for each method.

Variable Selection
KARE Data
We fit the multiple linear regression model to select the top 944 jointly associated SNPs for the 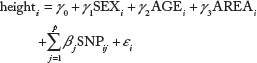
All tuning parameters are determined by 10-fold cross-validation, which minimizes the mean squared error.
We can make eight combinations: (
Top 10 SNPs in each method after
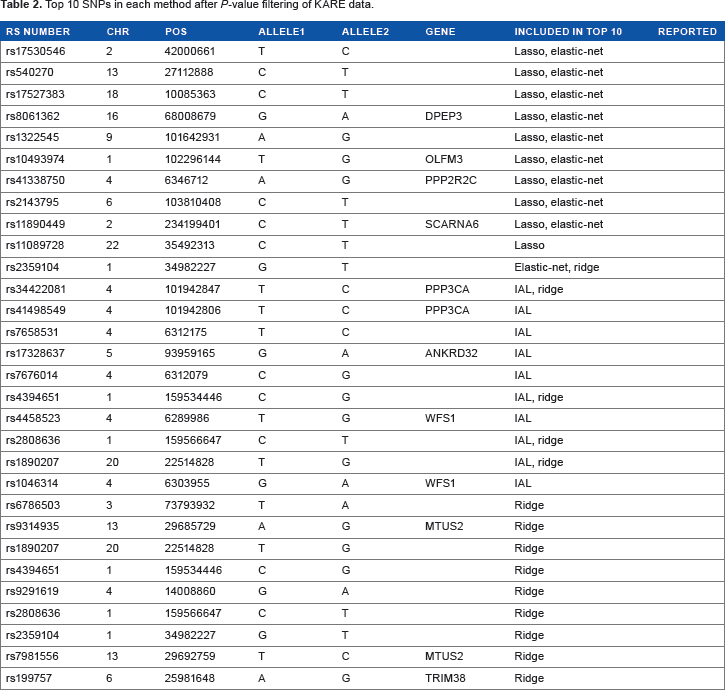
Table 1 shows the results of filtering SNPs with absolute value of coefficients in single variant analysis. Table 1 summarizes the list of SNPs that have the top 10 absolute values of coefficients in each penalized method. Among these SNPs, rs10948187, rs3799977, rs7954185, and rs7969076 were reported in other studies.13,14 Table 2 shows the results of filtering SNPs with
AREDS data.
We fit the multiple linear regression model to select the top 1,000 jointly associated SNPs for the 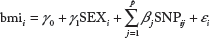
All tuning parameters are determined by 10-fold cross-validation, which minimizes the mean squared error. The combination method identifies 493, 460, 1000, 559, 485, 442, 1000, and 534 SNPs for the eight combinations, respectively, as putative BMI-related genetic variants.
Top 10 SNPs in each method after coefficient filtering of AREDS data.

Top 10 SNPs in each method after

Table 3 shows the results of filtering SNPs with absolute values of coefficients in single variant analysis. Table 3 summarizes the list of SNPs that have the top 10 absolute values of coefficients in each penalized method. Table 4 shows the results of filtering SNPs with
Comparative Study
We calculated the adjusted

Comparison of adjusted

Comparison of adjusted
KARE Data
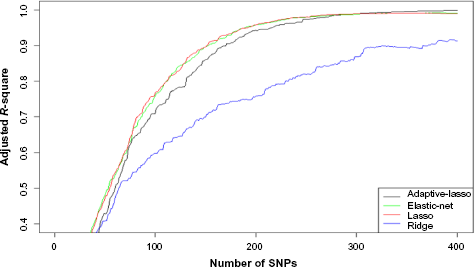
Figures 2–4 show the results of KARE data analysis. Figure 2 shows the adjusted
Figure 3 shows the adjusted

Comparison of adjusted R-squares when SNPs are filtered out by

Comparison of adjusted
Figures 2 and 3 show the consistent results that (1) the
Note that Figures 2 and 3 compare four penalized regression methods for a given pre-screening criterion. Among the IAL, lasso, and elastic-net methods, only the IAL method ranks SNPs by effect size. We wonder whether this difference among these three methods may be because of a different ordering of SNPs. Thus, instead of using BSS for the lasso and elastic-net methods, we use the same ordering of SNPs by effect size. Figure 4 shows the adjusted
Areds Data
Figures 5 and 6 show the results of AREDS data analysis. These figures show very consistent results with those of KARE. Figure 5 shows the adjusted
Comparison of adjusted
Conclusion
Recently many high-dimensional datasets have been generated in biomedical science, such as microarrays and SNP databases. Multistep strategies have been introduced to analyze these data. The first stage is pre-screening, in which the marginally associated response variables are identified, using various criteria. The second stage is variable selection. Various penalization methods have been proposed to analyze high-dimensional data. These include the ridge, bridge, least absolute shrinkage and selection operator (lasso), adaptive lasso, SCAD, and elastic-net methods. However, we do not know which method performs best for quantitative traits. Using an adjusted
In this study, we use only quantitative traits. However, a similar study could be easily conducted using binary traits such as diabetes and high blood pressure.
Because of gaps in the data, we unavoidably eliminate SNPs and individuals who have at least one missing value. This loss of information may reduce the accuracy of the study. We need to improve this accuracy by trialing appropriate imputation methods using simulated datasets.
Author Contributions
Conceived and designed the experiments: TP, SH. Analyzed the data: SH, YK. Wrote the first draft of the manuscript: TP, SH. Contributed to the writing of the manuscript: TP, SH, YK. Agree with manuscript results and conclusions: TP, SH, YK. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgement
The AREDS data used for the analyses described in this manuscript were obtained from the AREDS database, controlled through the database of Genotypes and Phenotypes (dbGaP) accession number phs000001.v2.pl.