
Abstract
Genome-wide association studies (GWAS) have successfully identified genetic variants associated with risk for breast cancer. However, the molecular mechanisms through which the identified variants confer risk or influence phenotypic expression remains poorly understood. Here, we present a novel integrative genomics approach that combines GWAS information with gene expression data to assess the combined contribution of multiple genetic variants acting within genes and putative biological pathways, and to identify novel genes and biological pathways that could not be identified using traditional GWAS. The results show that genes containing SNPs associated with risk for breast cancer are functionally related and interact with each other in biological pathways relevant to breast cancer. Additionally, we identified novel genes that are co-expressed and interact with genes containing SNPs associated with breast cancer. Integrative analysis combining GWAS information with gene expression data provides functional bridges between GWAS findings and biological pathways involved in breast cancer.
Introduction
Breast cancer is one of the leading causes of death among women in the United States and around the world. 1 In 2009, an estimated 192,370 new cases of invasive breast cancer were diagnosed among women, as well as an estimated 62,280 additional cases of carcinoma in situ. 1 At the same time, an estimated 40,170 women died from breast cancer. 1 While considerable progress has been made in reducing mortality rates due to increased screening, digital mammography, specialized care, and the widespread use of therapeutic agents such as selective estrogen receptor modulators, aromatase inhibitors, trastuzumab and others, identifying genetic markers remains an important long-term goal for the development of more effective therapeutic strategies and early interventions. Over the last decade, considerable effort and financial resources have been directed at identifying molecular signatures for breast cancer using gene expression profiles.2,3 At least two of these signatures have proven useful for prognostic purposes in the clinic.4,5 However, although these primary analyses have made great strides in deciphering the molecular basis of breast cancer, they have been unsuccessful in determining which genes have causative roles as opposed to being consequences of the breast cancer state.
Recent advances in genotyping and reduction in genotyping costs have made possible the use of genome-wide association studies to identify single nucleotide polymorphisms (SNPs) associated with risk for breast cancer.6–11 Results from these studies are providing valuable information about the genetic susceptibility architecture of breast cancer. However, to date, data generated from GWAS have not been combined with gene expression data to identify biologically relevant associations beyond the ones that meet the stringent genome-wide significance threshold.
12
In addition, the single SNP analysis widely used currently could potentially miss important genes and biological pathways. Moreover, the molecular mechanisms through which the identified variants confer risk or influence phenotypic expression remains poorly understood. From current GWAS findings, relatively few SNPs have
While GWAS can effectively map loci conferring risk for breast cancer, they offer limited insights about the mechanisms by which the SNPs exert their effects or influence phenotypic expression. In addition, GWAS findings do not always lead directly to the gene or genes because some SNPs as evidenced in this study and other studies6–11 map to intergenic regions close to nearby genes. Consequently, identified SNPs do not typically inform the broader context in which the disease-associated genes operate.
12
Genes may be regulated in
The objectives of this study were (a) to investigate the power of combining GWAS information with gene expression data to identify functionally related genes and biological pathways enriched by SNPs associated with risk for breast cancer, and (b) to identify genes and biological pathways that could not be identified using traditional single-SNP GWAS analysis. We hypothesized that genes containing SNPs associated with risk for breast cancer are functionally related and interact with each other and other genes not identified by traditional GWAS in intricate biological pathways. We have tested this hypothesis using GWAS information from 43 genome-wide association studies and three different gene expression data sets representing the Caucasian and the Asian populations. Throughout this analysis, we defined the genes containing SNPs associated with risk for breast cancer as candidate genes, and the genes identified from gene expression data but not containing SNPs reported in GWAS as novel genes. Our analysis assumed the gene and the pathway as the units of association. This holistic approach allowed us to account for all the SNPs including rare variants and those with small to moderate effects mapped to genes in our analysis.
Methods
Source of SNP data
SNP data and gene information were obtained by mining data from published reports on GWAS in breast cancer through Pub Med searches and web-sites containing supplementary data. Our search included terms (GWAS, GWA, WGAS, WGA, genome-wide, genomewide, whole genome, all terms + association, or + scan) in combination with breast cancer from the primary published reports through November, 2010. We catalogued all the SNPs that showed significant (
Sources of gene expression data
Publicly available gene expression data was downloaded from GEO; http://www.ncbi.nlm.nih.gov/geo/. The data included three data sets derived from Caucasian and Asian populations. The first data set derived from the Caucasian population involved gene expression data derived from RNA extracted from 143 histologically normal breast tissues obtained from patients harboring breast cancer who underwent curative mastectomy and 42 invasive ductal carcinomas (IDC) of various histological grades (1–3). The samples were obtained from breast cancer patients at Moffitt Comprehensive Cancer Center, Florida United States. The data set has been fully described by the originators. 15 Briefly, this data set consisted of histological data. Histologically-normal breast has the potential to harbor pre-malignant changes at the molecular level and thus provides an opportunity for identifying risk markers. We postulated that a histologically-normal tissue with tumor-like gene expression patterns might harbor substantial risk for future cancer development. Thus genes associated with these high-risk tissues would be considered to be malignancy-risk genes. “Normal” breast cancer tissue included histologically normal and benign hyperplasia. The data set was generated using the Affymetrix platform on U133 Plus 2.0 Array containing ~54,000 probes. The data set was downloaded from GEO accession number GSE10780. 16
The second data set involved a multi-ethnic Asian population, consisting of Malaysian breast cancer patients (Malays, Chinese and Indian). The data set involved invasive ductal carcinomas and was very similar to the first data set. The data set has been fully described by. 17 Briefly, the data set consisted of a total of 43 IDCs with histological grades 1–3 and 43 patient-matched normal tissues collected from Kuala Lumpur, UKM and Putrajaya Hospitals in Malaysia. The data set was generated using the Affymetrix platform's U133A Chip containing ~22,000 probes, and was downloaded from GEO accession number GSE15852. 16 Population of Asian descent provide special opportunities for this research because of the emerging evidence that genetic variants may confer population-specific risk.18,19 It is conceivable that expression of genes containing SNPs associated with risk for breast cancer could equally be population-specific. Therefore, the rationale for using this data set was to determine whether genes containing SNPs associated with risk for breast cancer would exhibit similar patterns of expression in the Caucasian and Asian populations. Unfortunately no similar data was found on the Africans or African-American population, therefore, no analysis were attempted in those populations.
To determine the clinical utility of genes containing SNPs associated with risk for breast cancer, we used a third data set involving two disease states. The third data set involved 286 lymphnode-negative primary breast cancer patients from the Caucasian population. 20 The data set consisted of 209 ER+ and 77 ER– breast cancer patients. The data set was generated using RNA extracted from fresh-frozen breast cancer tissues. The estrogen receptor alpha (ERα) is a master transcriptional regulator of breast cancer phenotype and the archetype of a molecular marker and therapeutic target. ER+ tumors respond to endocrine therapy. 21 Conversely, ER- tumors are generally treated with chemotherapy and do not respond to endocrine therapy. Thus, the objective of this analysis was to determine whether candidate genes could distinguish ER+ from ER- breast cancer patients. The data set was generated using the Affymetrix platform using U133A Gene Chip containing ~22,000 probes. The data set was downloaded from GEO under accession number GSE2034. 16
In each of the data sets described above, entries in the data matrix were expression values generated by Affymetrix's Microarray Analysis Suite 5.0 (MAS5) statistical algorithm. 21 Following normalization and scaling, MAS5 signal values were summarized by Turkey's biweight estimation of the probe level intensities within each probe set. This was followed by a global normalization (linear scaling) to give all chips the same average intensity. These procedures yield robust weighted means called average-scaled differences that are proportional to the amount of a particular RNA transcript present in the sample after background correction, which we used in this analysis.
Data analysis

where
Correlations among
To evaluate the pathway as a unit of association we used the hypergeometric test (Fisher's exact test) to search for an overrepresentation of significantly associated genes in the pathway.22,23 Briefly, let

Using the above equation, the total effect of the pathway as a unit of association was computed by direct enumeration of the
To determine whether genes containing SNPs associated with risk for breast cancer are co-expressed and to assess their relationships with each other and with novel genes, we performed unsupervised analysis using hierarchical clustering based on the complete linkage model. We used the correlation coefficient to assess the level of co-expression and to identify genes with similar patterns of expression among SNP-containing genes and novel genes. The correlation coefficient (

where
Finally, we performed pathway prediction to determine whether SNP-containing genes interact with each other and with novel genes in biological pathways. For pathway prediction, the input were the genes containing SNPs and novel genes that were found to be associated with cancer or to distinguish disease states in the case of ER+ and ER–. We performed pathway prediction and network modeling using the Osprey System. 33 Additional information and validation of predicted pathways and gene regulatory networks was obtained through the literature mining module built in the Osprey System which also provides biological and experimental information about the genes under study and identifies other functionally related genes interacting with input genes. In pathway prediction, genes were represented by nodes and the interactions by vertices. Two genes were considered to share a genetic susceptibility architecture and network properties if they were interconnected as represented by the vertices and were co-expressed as determined by pattern recognition analysis using hierarchical clustering based on the correlation distance as explained in the preceding paragraph. Additional functional assessment was performed using the Gene Ontology (GO) nomenclature. 34 Genes with spurious interactions and without interactions were removed from the networks. The rationale being that such genes could be less informative or could distort the reliability of pathway prediction and network modeling.
Results
Characterization of candidate genes and genetic variants
We mined publicly available data on 43 reported GWAS through November 2010 and the accompanying supplementary data posted on websites to identify genetic variants and genes associated with risk for breast cancer. The results of data mining for GWAS information used in this study are summarized in Table A provided as supplementary data. The search yielded 525 SNPs with
Interestingly, the
Association of candidate genes with gene expression
To determine whether genes containing SNPs associated with risk for breast cancer can distinguish breast cancer patients from cancer-free controls, and ER+ from ER–, we analyzed three separate gene expression data sets. We asked the question do genes containing SNPs associated with risk for breast cancer differ in their expression profiles between cancer-free controls and breast cancer patients in the Caucasian and Asian populations; and between ER+ and ER– breast cancer patients? ER+ and ER– data set was not available in the Asian population, therefore this analysis was restricted to the Caucasian population. Secondly, we asked the question does expression in genes containing SNPs differ between Caucasian and Asian populations?
The results showing a list of significantly differentially expressed genes between cases and controls in the Caucasian and Asian populations for genes containing SNPs with the smallest
List of genes containing SNPs with the largest effect sizes (

EU indicates Caucasians, ER+ and ER– indicate estrogen positive and negative, respectively,—indicates that the gene was not represented on the Chip, thus no estimate of
List of genes containing significantly associated (

Indicates the number of times the SNP has been replicated in multiple independent studies.
A comparison between cancer patients and controls in the Caucasian population produced 69 significantly (
A major concern about GWAS reported thus far, is that majority of these studies have been performed on the Caucasian population. Emerging evidence in the published literature tends to suggest that genetic variants may confer population-specific risk.18,19 To test this hypothesis we compared the expression of candidate genes between cancer patients and cancer-free controls in the Asian population. We asked the question, do genes containing SNPs associated with risk for breast cancer significantly differ in their expression profiles between cancer patients and cancer-free controls in the Asian population? Out of the 111 candidate genes examined, only 35 genes were significantly (
As expected, not all candidate genes associated with gene expression identified in the Caucasian population were found in the Asian population and vice versa. There were 12 genes containing SNPs that were found to be significantly differentially expressed between cases and controls in both the Caucasian and Asian populations. Five of those genes which overlapped between the two populations including
To determine the clinical utility of genes containing SNPs associated with risk for breast cancer, we compared gene expression between ER+ and ER– breast cancer patients. We asked the question do genes containing SNPs associated with risk for breast cancer differ in their expression between ER+ and ER– breast cancer patients? This analysis produced 70 significantly (
The variation in gene expression observed in this study is not unique. The results of variability in gene expression among populations observed here are consistent with previous reports. For example, genome-wide association of gene expression variation in humans has been reported, 35 though the study used cell lines and did not focus on any particular disease. Variation in gene expression within and among natural populations has also been reported. 36 Inconsistent expression of SNP-containing genes has also been reported in prostate cancer. 37
Interestingly, majority of the genes distinguishing cases from controls and ER+ from ER– were those which contained SNPs with moderate
Evaluation of functional relationship of candidate genes
To determine whether genes containing SNPs associated with risk for breast cancer are functionally related, we used the Gene Ontology (GO) information and co-expression analysis using gene expression data. The first, GO analysis allows characterization of genes according to the GO nomenclature. The GO Consortium has developed three separate ontologies-molecular function, biological process and cellular component to describe the attributes of gene products. Molecular function defines what a gene product does at the biochemical level without specifying where or when the event actually occurs; biological process describes the contribution of a gene product to a biological objective; and cellular component refers to where in the cell a gene product functions. Here, we have characterized the candidate genes according to all three GO categories. The results are provided in Tables B1 for the Caucasian population, in Table B2 for the Asian population and Table B3 for the ER+/ER– Caucasian population, provided as supplementary data. In all the three cases studied, we found that candidate genes are functionally related and are involved in multiple related functions and biological processes.
The second approach co-expression analysis was conducted to identify candidate genes with similar patterns of expression profiles. We hypothesized that genes containing SNPs associated with risk for breast cancer are co-regulated and have similar patterns of expression profiles. The results showing patterns of expression profiles for candidate genes are shown in Figure A1 and Figure A2 for the Caucasian and Asian population, respectively in the Appendix. The results showing patterns of gene expression profiles for candidate genes in ER+ and ER– breast cancer patients are shown in Figure A3. Only the candidate genes with the most consistent patterns of expression profiles are represented in the figures. In all the three cases, genes containing SNPs associated with risk for breast cancer were co-expressed and produced clusters of genes with similar patterns of expression. Interestingly, genes containing SNPs with moderate
Combining GWAS information with gene expression data to identify novel genes
To determine whether genes containing SNPs associated with risk for breast cancer are functionally related and co-expressed with other genes which have not been identified using single-SNP GWAS analysis, we performed supervised followed by unsupervised analysis using hierarchical clustering, as described in the methods section. We asked two fundamentally distinct questions. First, we asked, are there significantly differentially expressed novel genes not identified by GWAS, which distinguish breast cancer from cancer-free controls and ER+ from ER–? Second we asked the question are the novel genes distinguishing breast cancer patients from cancer-free controls functionally related or co-expressed and have similar patterns of expression with candidate genes distinguishing breast cancer from cancer-free controls and ER+ from ER– breast cancer patients?
In all the three cases we identified significantly differentially expressed novel genes not reported in GWAS. For the Caucasians and Asian populations, we identified 62 and 73 highly significant genes which distinguished breast cancer patients from cancer-free controls. We identified 85 highly significantly differentially expressed genes, which distinguished ER+ from ER– breast cancer patients. The novel genes were functionally related. The results showing
The results showing patterns of gene expression profiles for candidate genes and novel genes are shown in Figure 1 for the Caucasian population, Figure 2 for the Asian population and Figure 3 for the ER+ and ER– breast cancer patients. In all the three cases, genes containing SNPs associated with risk for breast cancer were found to be functionally related, co-expressed and exhibited similar patterns of expression with novel genes not identified in GWAS analysis. These results demonstrate and unequivocally confirm our hypothesis that integrating GWAS information with gene expression data provides a complementary approach to identify novel genes that could not be identified using traditional single-SNP GWAS analysis. Interestingly, genes containing SNPs with moderate

Patterns of gene expression profiles for candidate and novel genes in the Caucasian population. The rows represent genes, columns represent 143 cancer-free controls and 42 breast cancer patients. The red and blue colors indicate up and down regulation, respectively.

Patterns of gene expression profiles for candidate and novel genes in the Asian population. The rows represent genes, columns represent 43 (N) controls and 43 (C) breast cancer patients. The red and blue colors indicate up and down regulation, respectively.
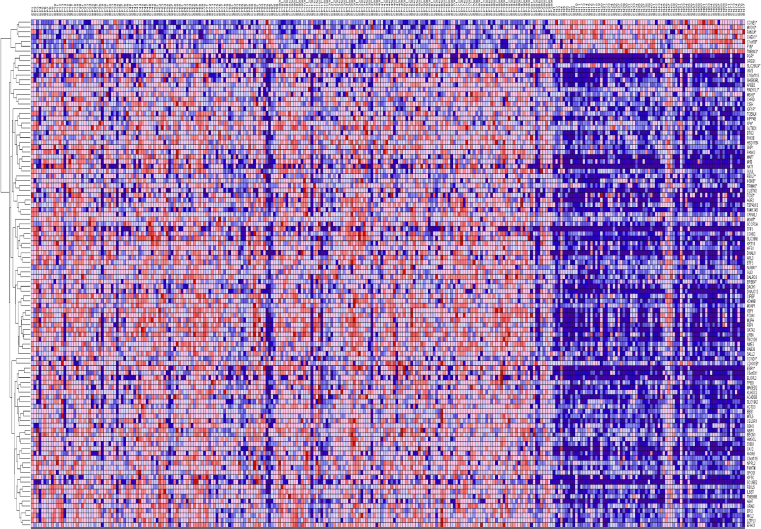
Patterns of gene expression profiles for candidate and novel genes in the ER+ and ER–Caucasian population. The rows represent genes, columns represent 209 ER+ and 77 ER– breast cancer patients, respectively. The red and blue colors indicate up and down regulation, respectively.
Predicted pathways and gene regulatory networks
To determine whether genes containing SNPs associated with risk for breast cancer interact with each other and with novel genes in biological pathways and gene regulatory networks, we performed pathways analysis and gene network modeling. We hypothesized that candidate genes and novel genes are functionally related, interact with each other in putative biological pathways and gene regulatory networks associated with breast cancer. Figure 4 presents the color codes showing the biological process in which the genes are involved as depicted in predicted pathways and gene regulatory networks. The results showing predicted pathways and gene regulatory networks using candidate and novel genes are presented in Figure 5 for the Caucasian population, in Figure 6 for the Asian population and in Figure 7 for the ER+/ER– Caucasian population.

Color code indicating the biological process in which the genes in predicted biological pathways and the regulatory networks are involved.
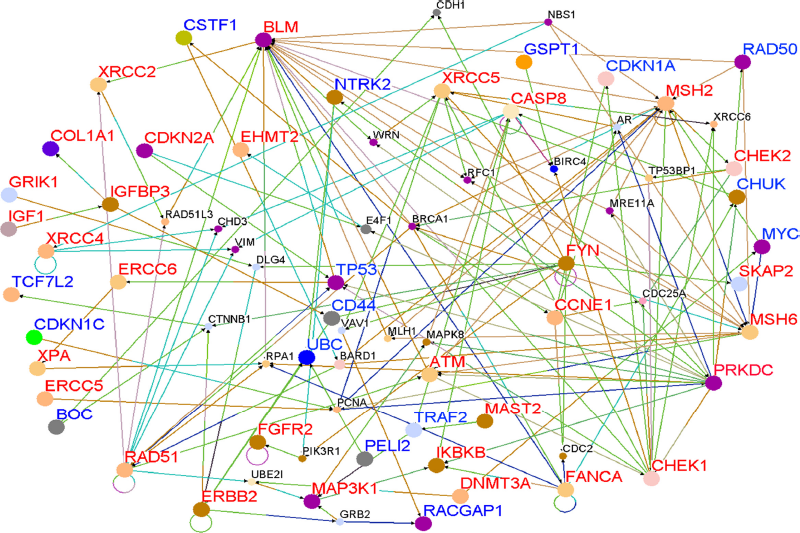
Gene interaction networks for genes containing SNPs (Red) and novel genes (Blue) identified using a threshold (

Gene interaction networks for genes containing SNPs (Red) and novel genes (Blue) identified using a threshold (

Gene interaction networks for genes containing SNPs (Red) and novel genes (Blue) identified using a threshold (
In all the three cases studied, genes containing SNPs associated with risk for breast cancer (shown in red) and novel genes (shown in blue) were found to interact with each other in intricate biological pathways and gene regulatory networks. Additionally, genes experimentally confirmed to interact with genes containing SNPs and novel genes were also identified (genes marked in black font) through co-expression and pathway analysis and network modeling. Our analysis also revealed, as depicted by the color codes, that genes containing SNPs associated with risk for breast cancer are involved in the same biological processes with novel genes identified by gene expression analysis that could not be identified using traditional GWAS alone. Pathway prediction and network modeling confirmed that integrating genetic with gene expression data is a powerful approach to identifying putative biological pathways and gene regulatory networks that could not be identified using traditional GWAS alone.
Interestingly, genes containing SNPs with moderate
Consistent with our analysis, a wealth of clinical and preclinical information exists on the functional correlations between the genes and pathways identified in this study.
Discussion
We report a novel integrative genomics approach that combines GWAS information with gene expression data to identify functionally related genes and biological pathways enriched by SNPs associated with risk for breast cancer. Our results demonstrate that integrative analysis combining gene expression data with GWAS information has the ability to identify novel genes not identified in single-SNP GWAS analysis. The integrative genomics approach presented here offers several remarkable features.
First, the novel paradigm associates genes containing SNPs associated with risk for breast cancer with expression and demonstrates the predictive value of SNP-containing genes by distinguishing breast cancer patients from controls in the Caucasian and Asian populations; and distinguishing ER+ from ER– breast cancer patients. This is a critical step to translating GWAS findings into clinical practice. Second, the results demonstrate unequivocally that an integrative genomics approach can add structure to data by combining GWAS information with gene expression data, allowing us to gain insights about the broader biological context in which SNP-containing and novel genes operate, and a deeper understanding of the functional basis of GWAS findings. Third, the approach demonstrates that genes containing SNPs associated with risk for breast cancer are functionally related and interact with each in putative biological pathways and gene regulatory networks. This is a significant finding given that traditional single-SNPs GWAS analysis is underpowered to uncover complex biological interactions. The results provide insight into the biological processes underlying breast cancer. Fourth, the integrative approach identified novel genes and established their functional relationship with genes containing SNPs with small and moderate
Although recent findings tend to suggest that common variants could explain most of the variation, 38 almost all the known studies reported on breast cancer thus far 13 have documented only a small number of loci and provide no putative functional bridges between GWAS findings and genes and biological pathways associated with breast cancer. The presence of SNPs in genes of similar biological functions interacting in biological pathways and gene regulatory networks as demonstrated in this study gives a degree of confidence that the associations could potentially be genuine even if none of SNPs individually is highly significant. Importantly, identification of genes not identified by GWAS could partially explain the missing variation from GWAS findings “also coined as the missing heritability”. 13
Firth, genes containing SNPs replicated in multiple independent studies were found to be functionally related and co-expressed with genes containing SNPs not replicated. Because replication is difficult to achieve in current GWAS analysis, this approach may help overcome that limitation. Replication of association findings at the gene or pathway level is potentially much easier than replication at the SNP level. In the published literature, meta-analysis has been carried out as a means of increasing sample size and power.39,40 However, meta-analysis provides no information about the functional basis of GWAS findings or the biological mechanisms underlying the disease. The practical application of the approach and results produced using this approach lies in the fact that it could guide future experimental designs. For example, genes in the pathways identified by this approach could be prioritized for targeted sequencing. Alternatively, SNP-containing genes in the identified pathways could be prioritized for allele-specific expression profiling to understand how genetic variants regulate gene expression and
Our results strongly challenge the single-SNP GWAS analysis paradigm, that focuses on SNPs producing the most highly significant
Many studies have now attempted pathway-based approaches to dissect the genetic susceptibility architecture of common diseases. This approach has been used in inflammatory diseases, 42 bipolar disorder, 43 multiple sclerosis, 44 breast cancer,45,46 and seven other common diseases. 47 To our knowledge, this is the first study to demonstrate the power of combining GWAS information with gene expression data and biological knowledge to identify genes, pathways and gene regulatory networks that could not be identified using traditional GWAS alone.
As mentioned in the preceding sections, the integrative approach has many attractive features. However, limitations must be acknowledged. First, our approach relies on using gene expression data and pathway prediction. Although this holistic approach accounts for all the SNPs in the genes, it provides no information about allele-specific expression. Therefore, it is difficult to discern the effects of individual SNPs on gene expression. The effect sizes (overall
Considerable investments have been directed to GWAS over the past five years. The studies contain rich information that could be turned into knowledge to identify targets for early therapeutic interventions. Traditional single-SNP GWAS analysis does not provide the insights about the function basis of GWAS findings and the biological mechanisms underlying the disease. An integrative approach that combines gene expression data with GWAS information provides a complementary approach to single-SNP GWAS analysis and offers the better prospect of facilitating translation of GWAS findings to the bedside. More work is need to determine allele-specific expression and to elucidate the impact of SNPs on gene and protein functions, and to identify
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Appendix
Gene expression patterns in the three cases studied for the most highly expressed candidate genes only.
Acknowledgements
This project was support by the University of Mississippi Cancer Institute and the Office of Research, to whom the authors are very thankful.