
Abstract
Binary tree classification has been useful for classifying the whole population based on the levels of outcome variable that is associated with chosen predictors. Often we start a classification with a large number of candidate predictors, and each predictor takes a number of different cutoff values. Because of these types of multiplicity, binary tree classification method is subject to severe type I error probability. Nonetheless, there have not been many publications to address this issue. In this paper, we propose a binary tree classification method to control the probability to accept a predictor below certain level, say 5%.
Introduction
A classification tree is a rule for predicting the class of an object from the values of its predictors. A tree is built by recursively partitioning and splitting it up further on each of the branches. The first tree-structured approach was the Automatic Interaction Detection (AID) program. 1 In this program, recursive partitioning was used as an alternative to the least squares regression for model fitting. Breiman et al. 2 developed the Classification and Regression Trees (CART) method of selecting tree of appropriate size for classification and regression. CART, THAID, 3 and C4.5 4 search exhaustively for a split of a node by minimizing a measure of node heterogeneity. These methods may cause a selection bias, because variables with more distinct values have a higher chance to be chosen. Loh and Vanichsetakul 5 proposed a Fast Algorithm for Classification Trees (FACT) by recursive application of linear discriminant analysis. Quick, Unbiased, Efficient Statistical Trees (QUEST) 6 and Classification Rule with Unbiased Interaction Selection and Estimation (CRUISE) 7 use similar approach to test for the split without an exhaustive search. CART and QUEST use binary split, while FACT, C4.5, CHAID, 8 and FIRM 9 use multiway split.
In a classification tree, each partition is represented by a node in the tree. The process starts with a training set with known classes or with a cross-validation. A node in a tree splits recursively with the goal of making the data within each node more homogeneous according to a splitting criterion until the tree is fully grown. A measure of node impurity given a node can be defined by the Gini diversity index. 2 For growing a large initial tree, the nodes continue splitting until a terminal node is either pure or it contains a small number of observations. Pruning is used to remove nodes and branches to avoid overfitting.
In CART, after the initial large tree is constructed, a nested sequence of subtrees is obtained by progressively deleting branches according to the pruning method. Breiman et al. 2 defined the cost-complexity measure by imposing penalty (complexity parameter) to the number of terminal nodes. To choose the best tree among the nested subtrees, they recommend estimating the optimal complexity parameter by minimizing the cross-validation or the holdout sample error. Besides cost-complexity pruning, various pruning algorithms including reduced-error pruning, 10 minimum description length pruning,11–13 and minimum error pruning 14 have been developed.
There have been other works aiming at correcting the classification bias due to large number of candidate predictors in a more general context not restricted to the tree classification. Bernau et al. 15 developed a bias-correction method based on a decomposition of the unconditional error rate of the tuning procedure. Tibshirani and Tibshirani 16 proposed a biased correction method for the minimum value of the cross-validation error. Ding et al. 17 introduced a biased correction method based on learning curve fitting by inverse power law.
In this paper, we propose to control the type I error accounting for the multiplicity due to many candidate predictors and possible cutoff values for each of them. Employing the family-wise error rate concept that is used for multiple testing, our type I error rate is defined as the probability to have at least one split when none of the candidate predictors are associated with the clinical outcome. We also propose two permutation-based procedures, called single-step procedure (SSP) and step-down procedure (SDP), to control the multiplicity-adjusted type I error. We perform extensive simulations to show that the two procedures control the type I error accurately and have reasonable power with moderate sample sizes. We apply the proposed methods to the classification with real microarray data. Through simulation and real data analysis, we observe that SDP tends to make larger trees than SSP.
Methods
Suppose that there are
Let
In a binary classification tree, we will continue classifying each sub-sample if there exists any predictor with a splitting point classifying the current subset with a 
Single-step procedure
Once we have the significance level øα (or critical value ζα) in (1), we will continue the classification until we do not find any splitting variable whose
Since the splitting variables as well as the test statistics (or the
Permutation Procedure for øα
Conduct the following process for
At the
Randomly match response variables
Calculate
Approximate øα by the [α
At the
Randomly match
Calculate
Approximate ζα by the [(1 - α)
We conduct
When presenting a classification tree, we may want to show how significant the classification is for each node with a split. To this end, we propose to calculate the 
An approximate
Step-down procedure
Although SSP is simple to implement, it may not have a high power to identify significant prognostic predictors since it applies the same strict critical value (or significance level) at all partitioning of subtrees that have smaller numbers of possible cutoff values than the original data. In this section, we propose a multistep procedure, called SDP, that is expected to discover more prognostic predictors than SSP while controlling the type I error rate α at the same level.
We define node 
where
As the partitioning continues, the size of current data set for each node decreases and the statistical tests to determine a partition suffer less multiple testing burden, so that the critical values for SDP are less strict than those of SSP for the subtrees. Hence, SDP may grow a larger tree than SSP. The significance level øl = ø
Permutation procedure for øl,α
At node
At the
Randomly match response variables
Calculate
Approximate ø
Permutation procedure for ζ
At the
Randomly match response variables
Calculate
Approximate ζ
Note that, while SSP requires permutations only once with the entire data, SDP requires a new set of permutations with the current data for each node. We repeat above procedure until all the terminal nodes have no more splits for a specified α level. Since
For node 
The adjusted
Simulations
In this section, we investigate the performance of the proposed tree classification method using a survival outcome variable.
For each subject, we generate the splitting variables from a multivariate normal distribution and the survival time from a log-normal distribution. Under 
Note that the survival time
Empirical type I error probability for nominal α = 5% with

For power analysis, we assume that the first 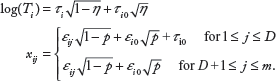
Note that the first
Empirical power under

Empirical power under

The SDP method gives more splits yielding a larger tree than the SSP method in general. For both methods, the number of splits increases as
Real Examples
The proposed method is applied to gene imprinting data 20 and lung cancer data. 19 The former have a binary outcome variable and the latter have a censored survival outcome variable to represent a wide range of genomic data.
Gene imprinting data
Imprinted genes are unusually predisposed to causing disease due to the silencing of expression of one of the two homologues at an imprinted locus, requiring only heterozygosity for a mutation affecting the active allele to cause complete loss of gene expression. It would be valuable to know which genes in the human genome undergo imprinting. Greally 18 described the first characteristic sequence parameter that discriminates imprinted regions – a paucity of short interspersed transposable elements (SINEs).
The genomic data collected to study imprinted genes were from the UCSC Genome Browser (http://genome.ucsc.edu). Annotation data were downloaded for the human genome (hg16, July 2003 freeze). The data contain 131 samples and 1446 predictors. Among the 131 samples, 43 are imprinted and 88 are non-imprinted (control) genes. The current dataset has been made available by Greally, and downloadable from http://www.ams.sunysb.edu/~hahn/research/CERP/imprint.txt. Before applying the methods, we removed the predictors that had identical values for more than 98% of the samples. For the gene imprinting data, 1248 out of 1446 predictors were selected using this criterion.
Both SSP and SDP yielded the same size of tree at the type I error level α of 0.05. The corresponding critical value is ζα = 12.27. Figure 1 displays the tree generated by SSP. The number in each circle or square is the sample size for the node. The first split occurred on Gene ALU. DNSC550 (

Classification tree for the gene imprinting data generated by SSP with α= 0.05.
It is difficult to find one measure for classification accuracy, because there are several splits in one tree and the classification accuracies in nodes at different levels cannot be weighed equally. However, the trees obtained in this paper show that the classifications are quite accurate. For the gene imprinting data, one terminal node contains only one imprinted gene out of 52 genes, while the other two nodes dominantly contain imprinted genes.
Lung cancer data
Shedden et al. 19 studied gene expression-based survival prediction in lung adenocarcinoma. The data from this multisite study contain survival information for 445 lung cancer patients with expression of 20,000 genes. Since the analysis of these high-dimensional data by our method is heavily computer intensive, we selected 1,000 genes using the ratio of the between-group to within group sums of squares (BW) ratio 22 before applying the method. A predictor variable containing the indicator of the centers is added to the 1,000 covariates.
Figure 2 shows the tree obtained using SSP. The type I error level α was chosen to be 0.2, and the corresponding critical value ζ
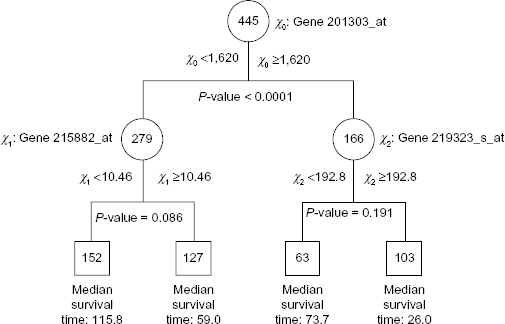
Classification tree for the lung cancer data generated by SSP with α = 0.2. The median survival time is in months.

Kaplan–Meier survival curves for the samples in the terminal nodes of the tree in Figure 2 for the lung cancer data.
As shown in Figure 4, the SDP approach at the type I error level of 0.05 gave two more splits to the tree generated by SSP. Regarding the splits at the children nodes of the root, the adjusted

Classification tree for the lung cancer data generated by SDP with α = 0.2. The median survival time is in months.
For lung cancer data, the type I error level was chosen to be 0.2 in order to provide more information on classification using SSP. Because the
Discussion
We have proposed a method to control the type I error rate for tree classification by adopting the multiple testing concept. Our method allows us to avoid overfitting issues in tree classification when there are a large number of candidate predictors as in a prediction problem based on microarray data. Also proposed are two procedures, called SSP and SDP, to control the type I error rate using permutation method. Through simulations, we observe that both procedures control the type I error rate well. While SDP requires more computing time than SSP, the former tends to generate a larger tree than the latter when there exist prognostic predictors. To reduce the computing time, we may use SSP to construct a tree and then apply SDP to the terminal nodes derived from SDP.
Although SDP requires more computing time than SSP, SDP is preferred because it might provide more informative results and the computing power keeps improving. The type I error level was preassigned in this paper. To determine the type I error probability, we may search an optimal type I error probability in cross-validation at the training phase.
Matlab was used for the simulation and real data analysis. In the simulation, the computing time for generating a tree was approximately 20 seconds if there was no split. For trees with several splits, it took 45 seconds to 1 minute. For the gene imprinting data, it took approximately 9 hours to generate the tree by SSP on a Windows Vista 2.0 GHz machine. It will be much more efficient if SAS, R, or C is used. Using parallel computing might be practical for users.
We present an analysis of computing time under general settings. Assume
Once we find a splitting node, the
Author Contributions
Conceived and designed the experiments: SHJ. Analyzed the data: YC, HA. Wrote the first draft of the manuscript: SHJ. Contributed to the writing of the manuscript: HA. Agree with manuscript results and conclusions: SHJ, HA, YC. Jointly developed the structure and arguments for the paper: SHJ, HA. Made critical revisions and approved final version: SHJ, HA. All authors reviewed and approved of the final manuscript.